Диагностика миотонической дистрофии у детей. Генетика
Добавил пользователь Дмитрий К. Обновлено: 08.01.2026
Увеличение числа CTG-триплетных повторов (экспансия) в 3'-нетранслируемом участке гена DMPK является причиной развития миотонической дистрофии 1 типа, частого аутосомно-доминантного неврологического заболевания.
Синонимы русские
Миотоническая дистрофия 1 типа (МД1), ген DMPK, экспансия триплетных повторов, генетическое обследование.
Синонимы английские
Myotonic dystrophy type 1 (DM1), gene DMPK, expansion of CTG (cytosine-thymine-guanine) triplet repeats.
Название гена
Локализация гена на хромосоме
Метод исследования
Фрагментный анализ гена DMPK.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Миотоническая дистрофия 1 типа (МД1) - наследственная миотоническая миопатия, ассоциированная со множественными системными нарушениями и вызванная экспансией CTG-повторов в 3'-нетранслируемом участке DMPK гена, расположенного на коротком плече 19-й хромосомы, который кодирует миотонин-протеинкиназу (МПК).
Заболевание наследуется по аутосомно-доминантному типу, то есть имеется 50% риска его развития у потомков. Для МД1 характерен феномен антиципации. Размер экспансии коррелирует с тяжестью симптоматики, уровнем пенетрантности и временем первых проявлений заболевания. При передачи мутантной аллели от матери у потомков наблюдается утяжеление симптоматики.
В норме МПК локализуется в специализированных структурах миоцитов скелетных и сердечных мышц, обеспечивающих внутриклеточную передачу импульса. В основе патогенеза МД1 лежит нарушение регуляции сплайсинга ряда белков из-за аберрации функции CUG-BP и MBNL1, вызванной патологической экспансией CTG-повторов: эффект РНК-токсичности. Белки CUG-BP и MBNL1 обладают высокой аффинностью к экспансионному участку CTG-повторов и накапливаются в так называемых РНК-очагах. В результате этого нарушается транскрипция РНК следующих белков: каналы ионов хлора, рецептор инсулина, тау-белок, ассоциированный с микротрубочками, тропонин Т.
Встречаемость заболевания среди населения с европейскими корнями составляет 1:7000.
Клинические проявления и признаки:
- Мышечные нарушения - дистальная мышечная слабость, миотония (феномен "разогрева").
- Офтальмологические нарушения - задняя субкапсулярная катаракта.
- Кардиальные нарушения - нарушение сердечной проводимости и ритма, внезапная сердечная смерть.
- Дерматологические нарушения - алопеция, пиломатриксомы и эпителиомы.
- Когнитивные и психические нарушения - небольшое снижение интеллектуальных способностей, депрессия.
- Нарушения желудочно-кишечного тракта - желчнокаменная болезнь, дисфагия, запоры, диареи, псевдообструкция.
- Эндокринологические нарушения - инсулинорезистентность, атрофия яичков, гипотиреоидизм, бесплодие, вторичный паратиреоидизм.
- Легочные нарушения - аспирационная пневмония, торако-диафрагмальная легочная недостаточность.
- Гиперсомния и ночное апноэ.
- Инструментальное обследование - МРТ головного мозга: наличие субкортикальных и перивентрикулярных гиперинтенсивных очагов в головном мозге при проведении МРТ в режиме Т1,Т2 и FLAIR.
- У пациентов с конгенитальной формой МД1 и количеством повторов более 1000 клиническая симптоматика может появляться при рождении или в раннем возрасте. Кроме симптомов, характерных для классической формы МД1, конгенитальная форма МД1 характеризуется также инфантильной гипотонией, дыхательной недостаточностью, серьезным снижением интеллектуальных способностей.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на миотоническую дистрофию 1 типа проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания, а также родственникам и детям больного.
Когда назначается исследование?
- При подозрении на миотоническую дистрофию 1-го типа;
- при дифференциальной диагностике миотонии и мышечной слабости;
- при дифференциальной диагностике миодистрофии;
- при дифференциальной диагностике системных проявлений, ассоциированных с неврологическими симптомами;
- при выявлении характерных изменений при проведении МРТ-исследования головного мозга;
- при раннем выявлении заболевания у родственников;
- при планировании семьи.
Что означают результаты?
Генетическое обследование является основным методом подтверждения диагноза и основано на подсчете числа тройных CTG-повторов с помощью метода фрагментного анализа в гене DMPK. Диагностическая значимость обнаруженного числа CTG-повторов в гене DMPK представлена в таблице:
Количество CTG-повторов
Диагностическая значимость
Менее 36 - нормальные аллели
36-49 - умеренное увеличение
МД1 исключен. Есть риск развития у потомства
50-149 - умеренная экспансия
МД1 подтвержден. Асимптоматичное течение, легкая и классическая формы. Есть повышенный риск развития у потомства
≥ 150 - выраженная экспансия
МД1 подтвержден. Классическая, ювенильная и конгенитальная формы. Есть повышенный риск развития у потомства
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников риска прогрессирования заболевания и назначения лечения рекомендуется получить консультацию специалиста.
Важные замечания
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, психиатр, врач-генетик.
Также рекомендуется
6 Глюкоза в плазме
54 Калий в сыворотке
47 Генетическое обследование на миотоническую дистрофию 2 типа в гене CNBP
Миотонический синдром у детей
Миотонический синдром у детей — это нервно-мышечная патология, для которой характерна затрудненная релаксация мускулатуры после ее напряжения. Встречается при наследственных генетических дефектах либо как проявление неврологических, эндокринных, метаболических, аутоиммунных заболеваний. Синдром характеризуется нарушениями двигательной активности, асимметрией мышц, задержкой психомоторного развития. Для диагностики необходим неврологический осмотр, инструментальные методы (ЭМГ, МРТ) и генетическое обследование. Лечение миотонии симптоматическое с применением физиотерапии, ЛФК и массажа, психолого-педагогической коррекции.
МКБ-10
Общие сведения
Частота встречаемости наследственных форм миотонического синдрома (МС) варьирует от 14 до 23 случаев на 100 тыс. детского населения. В отношении приобретенных вариантов статистических данных нет, поскольку в таких случаях патология не регистрируется как самостоятельная нозологическая единица и входит в структуру других неврологических болезней. Актуальность проблемы в педиатрической практике заключается в прогрессирующем течении заболевания, невозможности назначить этиотропное лечение и значительном ухудшении качества жизни детей при наследственных видах патологии.
Причины
В детском возрасте манифестируют врожденные формы, которые передаются преимущественно по аутосомно-доминантному, реже — аутосомно-рецессивному типу (синдром Шварца-Джампела, в части случаев — болезнь Томпсона). Самая частая форма наследственного МС — миотоническая дистрофия Россолимо-Штейнерта-Баттена-Куршмана, которая составляет до 97% всех генетических миотоний и связана с мутацией гена DMPK на 19-й хромосоме. Реже это заболевание обусловлено генетическими дефектами в 3-й (3q21) и 15-й (15q21-q24) хромосомах.
Вторичным миотоническим синдромом могут сопровождаться:
- родовые травмы с церебральными повреждениями;
- тяжелые формы перинатальной энцефалопатии;
- органические поражения ЦНС (опухоли, последствия перенесенных энцефалитов, ЧМТ);
- врожденные и воспалительные миопатии;
- миоплегии (болезнь Гамсторпа, периодический паралич);
- дефицитные состояния (рахит);
- эндокринные и метаболические расстройства (микседема, гипофункция паращитовидных желез, гиперкалиемия);
- аутоиммунные нарушения (приобретенная нейромиотония);
- длительное лечение клофибратом, калийсберегающими диуретиками, препаратами калия.
Патогенез
В механизме развития заболевания ведущим является нарушение процесса расслабления мышцы в результате метаболического или ионного дисбаланса. При этом изменяются свойства мембран миоцитов, происходит их усиленная деполяризация и повышение возбудимости. При совершении определенного действия с участием мускулатуры миофибриллы долго не могут расслабиться и вернуться в исходное положение, вследствие чего и возникает феномен миотонической задержки.
Лучше всего изучен патогенез дистрофической миотонии 1 типа, связанной с нарушениями на уровне экспрессии нуклеотидного повтора CTG. У детей изменяется концентрация миотонин-протеинкиназы DMPK, которая присутствует в скелетной мускулатуре, миокарде и ЦНС. В редких случаях клинические проявления обусловлены дисфункцией ионных каналов клеточных мембран, которая отмечается при аутоиммунных нарушениях.
Симптомы
Главный признак миотонического синдрома — скованность мышечных групп в конце движения. Это проверяется с помощью симптома «кулака»: после сжатия пальцев ребенок несколько секунд не может разжать кулак, и для этого ему нужно прилагать много усилий. Синдром характеризуется затруднениями жевания, неспособностью быстро открыть глаза после зажмуривания. Для наследственных форм МС у детей типично усиление симптоматики при целенаправленных движениях, в холодное время года.
При врожденных формах миотонического синдрома возможно асимметричное развитие мускулатуры. Если патология проявляется с первых месяцев жизни, родители замечают, что ребенок не способен удерживать голову, слабо сосет грудь, у него хуже сформированы рефлексы. Для грудничков с миотоническим синдромом характерно запоздалое становление моторики. В раннем детстве наблюдается повышенная утомляемость малыша, отсутствие интереса к активным играм, отставание в физическом развитии.
На поздних стадиях заболевания у детей происходит атрофия мышц, поэтому типичные признаки миотонического синдрома ослабевают, а потом исчезают. В клинической картине преобладает выраженная мышечная слабость, особенно в дистальных мышцах конечностей. Это обуславливает изменение походки, приводит к появлению вялых парезов и параличей. Поражение лицевых мышц проявляется постоянно «печальным» выражением лица и бедностью мимики.
Осложнения
Врожденные миотонические синдромы относят к мультисистемным поражениям, поэтому у таких детей зачастую диагностируют сопутствующие сердечно-сосудистые, неврологические и эндокринные нарушения. Особую опасность представляет нарушение иннервации дыхательной мускулатуры, из-за чего у пациентов нарастать дыхательная недостаточность вплоть до асфиксии. Церебральные симптомы, как правило, сочетаются с когнитивными нарушениями.
Лечение наследственных разновидностей миотонического синдрома затруднено, поэтому нередки летальные исходы в молодом возрасте. До 80% смертей вызваны вторичной пневмонией на фоне аспирации пищи и нарушений глотания, жизнеугрожающими аритмиями, которые развиваются вследствие дегенерации проводящей системы сердца. У детей может возникать глубокая умственная отсталость, приводящая к инвалидности.
Диагностика
При физикальном осмотре детский невролог проверяет симптом «кулака», оценивает сохранность рефлексов, мышечную силу. На основании полного физикального осмотра специалисту удается поставить предварительный диагноз. Чтобы подтвердить наличие и выяснить причины миотонического синдрома, назначаются следующие диагностические исследования:
- Электромиография. Патогномоничный признак миотонии на ЭМГ — появление высокоамплитудных разрядов со звуковым феноменом при использовании игольчатых электродов. При прогрессировании болезни присутствуют симптомы миопатии: снижение амплитуды потенциалов, полифазные двигательные единицы.
- Биопсия мышц. На ранних стадиях в биоптате определяются неспецифические миопатические изменения: центрально расположенные ядра, уменьшение длины волокон, признаки денервации — наличие пикнотических ядерных глыбок и маленьких угловых мускульных волокон.
- Магнитно-резонансная томография. По результатам МРТ конечностей врач оценивает степень развития мускулатуры и замещения ее жировой тканью. Учитывая сопутствующее поражение ЦНС, рекомендована МРТ головного мозга для обнаружения структурных изменений белого вещества и желудочков.
- Генетическая диагностика. Исследование генома для выявления типичной мутации — «золотой стандарт» для постановки диагноза врожденных вариантов миотонического синдрома у детей. По показаниям можно проводить тестирование в антенатальном периоде, если в семьях родителей были случаи миотонии.
Лечение миотонического синдрома у детей
Консервативная терапия
Этиопатогенетическое лечение миотонического синдрома пока не разработано, поэтому в детской неврологии ограничиваются симптоматическими средствами. Медикаменты подбираются с учетом конкретной ситуации и выраженности патологических изменений. Для замедления прогрессирования проводится лечение противосудорожными средствами и миорелаксантами, для подавления аутоиммунных реакций назначают глюкокортикоиды.
Решающее значение в терапевтической схеме отводится упражнениям ЛФК, которые укрепляют мышечный корсет, помогают сбалансировать развитие разных групп мышц, что улучшает качество жизни больных детей. Показано физиотерапевтическое лечение в виде электрофореза с кальцием, электростимуляции мышц, бальнеотерапии. Для уменьшения миотонического синдрома полезен массаж.
Реабилитация
Учитывая серьезные двигательные нарушения, лечение дополняется регулярными занятиями в реабилитационных центрах с использованием тренажеров и индивидуальных методик. Нарушения артикуляции у детей корректируются занятиями с логопедом. При задержке умственного развития ребенку может потребоваться помощь олигофренопедагога либо обучение в специализированной школе.
Прогноз и профилактика
Если вовремя начать лечение, при приобретенных формах удается восстановить мышечную активность. Наследственный миотонический синдром отличаются сомнительным прогнозом. Средняя продолжительность жизни пациентов составляет 35 лет при раннем дебюте болезни. Специфические меры профилактики миотонического синдрома не разработаны. Семьям с отягощенным анамнезом требуется медико-генетическое консультирование.
1. Клинический случай дистрофической миотонии 1-го типа/ Н.В. Ноздрюхина, А.А. Струценко, Е.Н. Кабаева// Трудный пациент. — 2019.
2. Миотоническая дистрофия. Современное представление и собственное наблюдение/ Т.И. Стеценко// Современная педиатрия. — 2014.
Миотоническая дистрофия
Это тяжелое наследственное прогрессирующее заболевание, наследуемое по аутосомно-доминантному типу. Миотоническая дистрофия первого типа проявляется преимущественно в возрасте 16-20 лет, второго типа — в 30-40 лет. Мужчины болеют в три раза чаще женщин.
Причины и виды миотонической дистрофии
Генетическая основа заболевания первого типа — повреждение гена миотонинпротеинкиназы (DMPK), локализованного на коротком плече 19 хромосомы, заключающееся в увеличении числа тринуклеотидных повторов CTG. Молекулярно-генетическая причина миотонической дистрофии второго типа заключается в увеличении количества повторов CCTG в интроне 1 гена ZNF9, картируемом на 3 хромосоме.
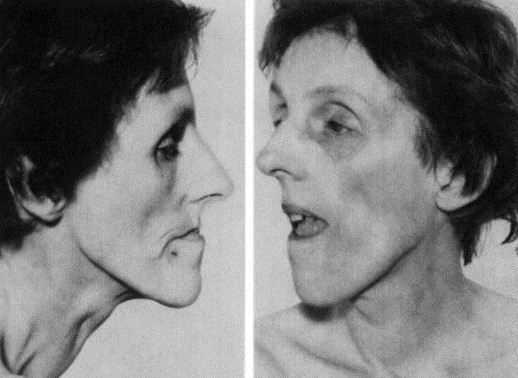
Миотоническая дистрофия второго типа считается более мягкой и проявляется у больных старшей возрастной группы. Эта форма болезни сопровождается полиморфной симптоматикой, которая может включать в себя:
- миотонический феномен в скелетных мышцах;
- мышечную атрофию и парезы;
- кардиомиопатии (нарушение проводимости);
- эндокринные патологии (нарушение секреции гормона роста, инсулинорезистентность, тестикулярную атрофию, раннее лобное облысение у мужчин, бесплодие у женщин);
- катаракту.
Заболевание первого типа характеризуется вовлечением мышц шеи и лица, дистальных отделов конечностей, в то время как при втором типе наблюдается проксимальный паттерн поражения скелетных мышц.
Миотоническая дистрофия проявляется слабостью мышц лица, провисанием челюсти, опущением века, атрофией мышц предплечий и икр. Она вызывает нарушения сердцебиения, апноэ сна, влияет на ЖКТ, обуславливая запоры и другие расстройства пищеварения.
Диагностика миотонической дистрофии
Обследование начинается с изучения истории болезни пациента и семейного анамнеза. Диагностика включает в себя:
- лабораторные анализы (на КФК, ГГТ, ФСГ, тестостерон, IgG);
- генетическое тестирование (поиск мутаций в гене DMPK и ZNF9);
- электромиографию;
- биопсию мышц;
- нейропсихологическое обследование;
- МРТ головного мозга;
- кардиологическое обследование.
Обнаружить миотоническую дистрофию в период внутриутробного развития ребенка позволяет антенатальная диагностика (неинвазивное генетическое тестирование).
Медико-генетический центр «Геномед» проводит молекулярно-генетические исследования для обнаружения этого заболевания: поиск мутаций в генах DMPK и ZNF9, пренатальная ДНК-диагностика.
Лечение и профилактика миотонической дистрофии
Заболевание является неизлечимым, однако комплексный подход к терапии позволяет замедлить его прогрессирование. Лечение включает в себя:
- медикаментозную терапию миотонического компонента;
- физическую терапию (ЛФК);
- имплантацию водителя сердечного ритма;
- удаление хрусталика (при катаракте);
- хирургическое лечение (апноэ сна).
Профилактика повторных эпизодов миотонической дистрофии в семьях с отягощенным анамнезом заключается в проведении предимплантационной и пренатальной ДНК-диагностики.
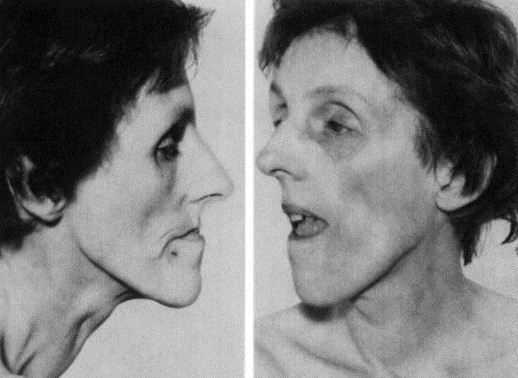
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — наследственное медленно прогрессирующее заболевание, в основе которого лежит дефект миотонин-протеинкиназы, приводящий к развитию миотонии в сочетании с дистрофическими изменениями мышечной ткани. Заболевание проявляется миотоническими спазмами, атрофическими изменениями мышц шеи, лица и дистальных отделов конечностей, снижением интеллекта, аритмиями и эндокринной патологией. Диагностика дистрофической миотонии основывается на клинических данных, результатах генеалогического анализа и исследования ДНК. Лечение симптоматическое, направленное против симптомов миотонии (фенитоин, прокаинамид, хинин, мочегонные) и мышечной дистрофии (анаболические стероиды, АТФ).
Дистрофическая миотония Россолимо-Штейнерта-Куршмана является наследственным заболеванием и передается от родителей к детям по аутосомно-доминантному типу. Классическая форма этого заболевания развивается преимущественно в возрастном периоде от 10 до 20 лет. В более редких случаях встречается врожденная дистрофическая миотония Россолимо-Штейнерта-Куршмана, клинические симптомы которой проявляются сразу же после рождения.
Морфологически при миотонии Россолимо-Штейнерта-Куршмана отмечается сочетание гипертрофических изменений одних мышечных волокон с атрофией других, замещение части мышечных волокон жировой и соединительной тканью. Изучение образцов мышечной ткани под электронным микроскопом показывает деструкцию миофибрилл и изменение размера митохондрий.
Последние исследования генетического набора больных дистрофической миотонией показали, что основу заболевания составляет дефект в гене DMPK, находящемся в 19-й хромосоме и отвечающем за синтез миотонин-протеинкиназы. У больных дистрофической миотонией выявляется значительное увеличение тринуклеотидных CTG-повторов в основной части гена DMPK. При этом именно от количества повторов зависит форма и тяжесть миотонии.
В норме число тринуклеотидных повторов варьирует от 5 до 37. Увеличение повторов до 50-80 приводит к появлению мягкой формы миотонии Россолимо-Штейнерта-Куршмана. Если количество тринуклеотидных повторов находится в промежутке от 100 до 500, развивается поздняя форма заболевания. Врожденные формы дистрофической миотонии возникают при повышении числа CTG-повторов от 500 до 2000. Исследования показали, что увеличение тринуклеотидных повторов происходит в основном в женских гаметах в процессе мейоза. В связи с этим при передаче заболевания от матери у ребенка возникает более тяжелая форма миотонии или ее врожденный вариант.
Симптомы классической формы
В классическом варианте миотония Россолимо-Штейнерта-Куршмана начинает проявляться после первых 5 лет жизни и может манифестировать до 35-летнего возраста. Но наиболее часто клинические проявления заболевания возникают в возрастном диапазоне от 10 до 20 лет. Они представляют собой сочетание типичных симптомов миотонии с признаками миопатии, поражением сердечно-сосудистой системы и ЦНС, эндокринными нарушениями и катарактой.
Из миотонических проявлений для миотонии Россолимо-Штейнерта-Куршмана характерны миотонические спазмы, наиболее выраженные в жевательных мышцах и мышцах-сгибателях кисти. Наблюдаются также механические реакции миотонического типа, выявляемые при ударе неврологическим молоточком. Отличительной особенностью миотонии Россолимо-Штейнерта-Куршмана является наличие атрофических изменений в различных группах мышц. При этом течение заболевания характеризуется постепенным угасанием симптомов миотонии на фоне прогрессирующей мышечной дистрофии.
Чаще всего при миотонии Россолимо-Штейнерта-Куршмана поражаются мышцы дистальных отделов конечностей, мимическая мускулатура, грудино-ключично-сосцевидные и височные мышцы. Поражение мимических мышц проявляется характерным маскообразным печальным выражением лица больных дистрофической миотонией. Атрофические изменения мышц глотки и гортани приводят к развитию миопатического пареза гортани с нарушением голоса и затруднением глотания. Миопатические изменения могут возникать в дыхательной мускулатуре. Наряду с миотоническими спазмами они приводят к ухудшению легочной вентиляции, появлению приступов апноэ во сне, возникновению застойной или аспирационной пневмонии.
Нарушения сердечно-сосудистой системы наблюдаются примерно в половине случаев дистрофической миотонии. К ним относятся аритмии, связанные с нарушением проводимости, и гипертрофия левого желудочка. Наиболее распространена блокада ножек пучка Гиса. Из признаков поражения ЦНС чаще всего наблюдается гиперсомния и снижение интеллектуальных способностей, доходящее до легкой степени дебильности.
Эндокринные расстройства при миотонии Россолимо-Штейнерта-Куршмана затрагивают в основном половую сферу. У мужчин они проявляются снижением либидо, крипторхизмом, импотенцией, гипогонадизмом, у женщин — гирсутизмом, нарушениями менструального цикла (олигоменореей, дисменореей) и ранним климаксом. Типичным является изменение структуры волос в сочетании с алопецией. У мужчин отмечается выпадение волос на висках и в области лба, у женщин — диффузное или очаговое облысение.
Симптомы врожденной формы
Первые признаки врожденной формы миотонии Россолимо-Штейнерта-Куршмана могут проявляться еще в период внутриутробного развития. Как правило, они выражаются в значительном снижении двигательной активности плода, которое диагностируется акушером-гинекологом по данным акушерского УЗИ в III триместре беременности.
После рождения ребенка преобладают симптомы миопатии. Отмечается диффузная гипотония мышц, более выраженная в мимической, жевательной и глазодвигательной мускулатуре, а также в мышечных группах дистальных отделов конечностей. Характерны затруднения вскармливания и дыхательные расстройства. Миотоническая симптоматика начинает проявляться несколько позже. Врожденная дистрофическая миотония сопровождается задержкой моторного развития и олигофренией. Типично быстрое прогрессирование симптомов заболевания, часто приводящее к смертельному исходу еще в раннем детстве.
Типичное сочетание миотонии с признаками дистрофических изменений мышечной ткани, умственной отсталостью, нарушениями со стороны сердечно-сосудистой и эндокринной систем позволяет неврологу предположить миотонию Россолимо-Штейнерта-Куршмана. Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе наследования заболевания, и данные ДНК-анализа. Дополнительно проводится электромиография, электронейрография, исследования половых гормонов, ЭКГ. К диагностике пациентов с миотонией Россолимо-Штейнерта-Куршмана могут дополнительно привлекаться генетики, кардиологи, эндокринологи, гинекологи, андрологи.
При диагностике дистрофической миотонии ее необходимо дифференцировать ее от других видов миотонии. Так, наличие мышечных атрофий позволяет отличить миотонию Россолимо-Штейнерта-Куршмана от миотонии Томсена, для которой типична мышечная гипертрофия. От миотонии Беккера заболевание отличается ранним поражением мышц лица и доминантным типом наследования. Кроме того, следует проводить дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршман с миопатиями, БАС и амиотрофией Шарко-Мари-Тута.
Лечение миотонии Россолимо-Штейнерта-Куршмана
Радикальной терапии миотонии Россолимо-Штейнерта-Куршмана пока не существует. Пациентам, имеющим это заболевание, показана диета со сниженным содержанием калия. Им также следует избегать переохлаждения, которое провоцирует миотонические спазмы. Уменьшению миотонических проявлений способствует прием хинина, прокаинамида, фенитоина в сочетании с ацетазоламидом. Показаны анаболические стероиды ( нандролона деканоат, метиландростендиол, метандростенол), небольшие дозы АТФ, витамины группы В.
Читайте также: