Диагностика прогрессирующего надъядерного паралича по КТ, МРТ, ПЭТ головного мозга
Добавил пользователь Евгений Кузнецов Обновлено: 01.02.2026
Отсутствие или минимальный ответ на препараты L-ДОФА
Явный ответ на L-ДОФА
Нейровизуализационные изменения в постановке диагноза ПНП
Тактика ведения пациентов с ПНП основывается на принципе мультидисциплинарной бригады, в которую вовлечены как неврологи, так и врачи смежных специальностей: логопеды, реабилитологи, физиотерапевты, клинические фармакологи и т.д. Медикаментозное лечение при ПНП является симптоматическим. При двигательных нарушениях, связанных с экстрапирамидными нарушениями, применяют препараты леводопы в сочетании с ингибитором дофа-декарбоксилазы (например, карбидопа), которые эффективны при паркинсоническом фенотипе ПНП (ПНП-П). В остальных случаях эффективность леводопы низкая. Однако, учитывая ограниченные терапевтические возможности, леводопу применяют в дозах до 1000 мг/сут. Ученые из университета Оттавы C. Barclay и A. Lang [26] изучили документы и видеозаписи всех пациентов с ПНП с 1983 по 1993 г., учитывая дистонические симптомы. Из 83 случаев у 38 зафиксированы дистонические проявления: 20 (24%) пациентов страдали блефароспазмом (один случай спровоцирован леводопой), 22 (27%) — дистонией в конечностях (один случай был вызван противоэпилептической терапией, второй — приемом леводопы), 14 (17%) — аксиальной дистонией, один — оромандибулярной дистонией, вызванной приемом леводопы, а у 2 — наблюдались краниальные дистонии. В своем наблюдении авторы также описывают 6 пациентов, у которых дистония в конечностях проявилась одним из первых симптомов заболевания, что затруднило диагностику корково-базальной ганглионарной дегенерации. Таким образом, при возникновении у пациентов с ПНП дистонии дозировку дофаминергических препаратов следует уменьшить или отменить, чтобы исключить возможность побочного действия препарата. Считается, что этиология леводопа-индуцированной дистонии по механизму развития схожа с таковой при паркинсонизме, сочетание же ее с оромандибулярной дистонией связано с особенностью многоуровневого поражения нигростриальной и стриопалидарной систем и процессов нейропластичности в них при ПНП. В лечении фокальной дистонии при ПНП, включая апраксию открытия века, можно использовать инъекции ботулинического токсина [27]. Описан опыт лечения пациентов с ПНП с использованием глубокой стимуляции головного мозга (ГСГМ). В рандомизированном контролируемом исследовании с односторонней глубокой стимуляцией ножкомостового ядра покрышки головного мозга приняли участие 8 больных с ПНП Ричардсона (ПНП-Р) и отметили улучшение постуральных функций, однако при сравнении результатов через 6 и 12 мес в режиме стимуляции ВКЛ и ВЫКЛ метод не показал эффективности [28]. ГСГМ в настоящее время не рекомендуется в лечении ПНП [27].
Согласно опубликованным исследованиям, в лечении когнитивных нарушений у пациентов с ПНП применяли ингибиторы холинэстеразы, которые улучшали когнитивные функции, однако было замечено, что у данной группы пациентов быстрее прогрессировали двигательные нарушения [28]. Необходимо отметить, что пациенты с ПНП могут страдать психоэмоциональными нарушениями, например депрессией, которая также требует применения специфической терапии, однако в литературе не было найдено конкретных рекомендаций по применению антидепрессантов при ПНП. В связи с чем можно индивидуально рассмотреть назначение антидепрессантов и рекомендовать занятие с психотерапевтом как альтернативный метод помощи пациенту.
Согласно исследованиям, физиотерапевтические реабилитационные методики уже доказали свою эффективность в лечении БП, у пациентов, страдающих ПНП, это еще предстоит выяснить, однако уже сегодня есть наблюдения, в которых отмечают положительный эффект комплексного реабилитационного лечения пациентов с ПНП [29]. В одной из работ, в которой исследовалась эффективность логопедической терапии по протоколу LSVT Ли Сильвермана у пациентов с БП и ПНП, было высказано предположение о потенциальной пользе логопедических методов лечения. По результатам исследования, увеличение максимальной продолжительности фонации и громкости голоса при чтении были одинаковыми в обеих группах, однако улучшение качества голоса и артикуляции были более значимыми в группе пациентов с БП по сравнению с группой ПНП [30]. Во избежание аспирации во время приема пищи необходимо составить специальную диету для пациента, а при развитии дисфагии рассмотреть вопрос наложения гастростомы, учитывая когнитивные нарушения.
Пациенты с ПНП страдают нарушениями координации и ходьбы, в связи с чем обучение методикам по поддержанию равновесия, а также «обучению падений» может минимизировать вероятность получения травмы. Для безопасного передвижения необходимо использование средств опоры: тростей, ходунков, при выраженных нарушениях ходьбы — инвалидной коляски.
Прогрессирование заболевания при ПНП обычно происходит довольно быстро [31]. Большинство пациентов становятся уход-зависимыми в течение 3—4 лет после дебюта заболевания, значительно снижается качество их жизни, в связи с чем важно вовремя организовать паллиативную помощь. Летальный исход в среднем наступает через 6—9 лет после постановки диагноза, однако продолжительность жизни при ПНП с акинезией и застываниями составляет около 11 лет [32—35].
В систематическом обзоре и метаанализе 2017 г. предиктором более короткой выживаемости был фенотип ПНП-Р по сравнению с фенотипом ПНП-П, при котором наблюдалось раннее начало постуральных нарушений с частыми падениями и нарушением когнитивных функций [36]. Ранний дебют дисфагии, которая наблюдается в фенотипах как ПНП-Р, так и ПНП-П, также был предиктором более короткой выживаемости.
Клинический случай
Пациентка И., 79 лет, поступила на лечение в неврологическое отделение клиники Башкирского государственного медицинского университета (БГМУ) по направлению участкового невролога. При поступлении жалобы на частые падения преимущественно назад. Пациентка не может опустить взгляд вниз, чтобы прочесть записи, посмотреть в тарелку при приеме пищи, в связи с чем во время еды часто проливает жидкую пищу, замедленность движений, снижение памяти на недавние события, нарушение концентрации внимания, дополнительные движения рта с прикусом внутренних поверхностей щек, боли в шее, бессонница, которую пациентка объясняет сложностью найти удобное положение в кровати. Со слов родственников, около 3 мес они наблюдают, что пациентка часто заходит в туалетную комнату — до 20 раз в день, но при этом может не совершать в ней каких-либо действий. Пациентка объясняет свой поступок как «захотела в туалет, но приду еще раз позже». При этом нарушений функций тазовых органов не отмечается. Считает, что заболела год назад (в 78 лет), когда впервые стала отмечать нарушение координации в виде шаткости при ходьбе, неустойчивости и падений, далее присоединилась скованность движений. Оценка двигательных функций по унифицированной шкале оценки болезни Паркинсона составила 58 баллов. Пациентке был выставлен диагноз «болезнь Паркинсона» и назначен противопаркинсонический препарат (леводопа+карбидопа 250/25 мг) по 1 таблетке 3 раза в сутки, однако эффект от препарата ощутила только в 1-й месяц приема, в виде некоторого уменьшения брадикинезии, снижения тонуса в конечностях, улучшения походки. В ходе наблюдения и лечения участковый невролог увеличил дозу препарата до 6 приемов в сутки, но значимого эффекта более не отмечалось. В 2019 г. у пациентки участились падения назад и вперед, появились дополнительные движения рта с прикусом щек. Родственники отметили изменение мимики — «суровое» выражение лица, нарушения памяти, поведенческие расстройства, бессонницу. После проведения неврологического осмотра пациентка была направлена на стационарное обследование и лечение в клинику БГМУ.
Наследственный анамнез проследить не удалось, так как пациентка росла в детском доме, однако у нее есть родная сестра, у которой не зафиксировано неврологических нарушений. Объективное исследование: пациентка нормального питания (индекс массы тела 21,1 кг/м 2 ), рост 154 см, пропорциональное соотношение частей тела.
Неврологический статус: паралич взора, выражение лица пациентки с фиксированным взглядом вперед, приподнятыми бровями и нахмуренным лбом, положительный вестибулярный глазной рефлекс, положительные рефлексы орального автоматизма (Маринеску—Радовичи, хоботковый рефлекс), речь замедленная, «рычащая» дизартрия, мышечный тонус повышен по пластическому типу с двух сторон, преимущественно проксимально, олигобрадикинезия в конечностях. Умеренная пирамидная недостаточность с гиперрефлексией. Координаторные пробы выполняет с промахиванием с двух сторон. В пробе Ромберга неустойчива. Походка: брадибазия, постуральная неустойчивость с частыми падениями назад и вперед. Оромандибулярный гиперкинез. Ортостатическая проба отрицательная. Для исследования когнитивной сферы использованы нейропсихологические тесты: краткая шкала психического статуса — 24 балла, батарея тестов на лобную дисфункцию —7 баллов, Монреальская когнитивная шкала — 22 балла. При проведении нейропсихологического исследования выявлена умеренная степень деменции лобного типа.
Медперсоналом и родственниками зафиксировано повторяющееся поведение (пациентка так же, как и дома, ходила в туалетную комнату до 20 раз в день).
Проведенные стандартные клинико-лабораторные исследования соответствовали референтным значениям с учетом возраста пациентки.
МРТ-исследование проведено на магнитно-резонансном томографе «Siemens» с напряженностью магнитного поля 1,5 Тл, с получением стандартных Т1- и Т2-взвешенных изображений (Т1-ВИ, Т2-ВИ) в фронтальной, аксиальной и сагиттальной плоскостях, с последующей оценкой визуальных изменений головного мозга (см. рисунок). Выявлены признаки атрофии среднего мозга с симптомами колибри в сагиттальной (см. рисунок, а) и Микки Мауса в аксиальной плоскостях (см. рисунок, б).
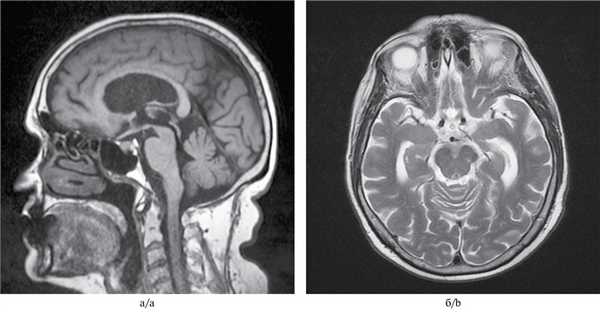
МРТ головного мозга 79-летней пациентки с ПНП.
а — Т1-взвешенное изображение, сагиттальная плоскость. Атрофия среднего мозга, сохранение объема моста и атрофия крыши среднего мозга. Симптом клюва колибри; б — Т2-взвешенное изображение на аксиальном срезе на уровне среднего мозга. Атрофия среднего мозга с формированием симптома Микки Мауса.
Учитывая прогрессирование заболевания в течение года, с постуральными нарушениями, акинетико-ригидным синдромом, параличом взора, дизартрией, умеренным когнитивным нарушением, стереотипными поведенческими нарушениями, данные МРТ головного мозга (атрофия среднего мозга), а также недавно возникший оромандибулярный гиперкинез на фоне приема противопаркинсонического препарата, выставлен диагноз «ПНП с леводопа-индуцированной дискинезией в виде оромандибулярного гиперкинеза».
Пациентке был отменен противопаркинсонический препарат, в результате чего дискинезия уменьшилась, через 2 нед и более не наблюдалась.
Пациентка дала информированное согласие на публикацию полученных результатов ее обследований и демонстрацию данных в печати.
Таким образом, ПНП является гетерогенным заболеванием с разнообразными клиническими фенотипами, диагностические критерии которых представлены Международным обществом по болезни Паркинсона и двигательным расстройствам в 2017 г. и являются основным руководством для практического врача. Однако атипичные проявления ПНП могут усложнить диагностику данного заболевания. Представленный клинический случай интересен сочетанием ПНП с леводопа-индуцированной дискинезией. К сожалению, на сегодняшний день нет патогенетического лечения ПНП, однако создание безопасной среды, персонифицированный подход и выбор оптимальной тактики ведения пациента в составе мультидисциплинарной бригады способствуют поддержанию качества жизни и его безопасности.
Прогрессирующий надъядерный паралич ( Прогрессирующая надъядерная офтальмоплегия , Синдром Стила-Ричардсона-Ольшевского )
Прогрессирующий надъядерный паралич — это дегенеративное церебральное заболевание с преимущественным поражением среднего мозга, ядерно-корковых путей, подкорковых образований. Составляющими клинической картины выступают акинетико-ригидная форма паркинсонизма, атаксия, офтальмоплегия, когнитивное снижение, псевдобульбарный синдром. Диагностика осуществляется по клиническим данным, результатам церебральной МРТ и цереброваскулярных исследований. В терапии препаратами выбора являются леводопа, мемантин, антидепрессанты из группы ингибиторов обратного захвата серотонина.
МКБ-10

Общие сведения
Прогрессирующий надъядерный паралич (ПНП) — дегенеративное поражение головного мозга неясной этиологии. Наряду с болезнью Альцгеймера, мультисистемной атрофией, кортикобазальной дегенерацией, болезнью Пика, ПНП относится к таупатиям, характеризующимся образованием включений тау-протеина в нейронах и глиальных клетках. Прогрессирующий надъядерный паралич впервые был подробно описан в 1963-64 годах канадскими неврологами Стилом и Ричардсоном в соавторстве с патоморфологом Ольшевским, в честь которых носит название синдром Стила-Ричардсона-Ольшевского. Распространённость заболевания согласно различным информационным источникам варьирует в пределах 1,4-6,4 случая на 100 тыс. населения. Манифестация клинической симптоматики приходится на возрастной период от 55 до 70 лет, с возрастом вероятность развития заболевания увеличивается. Лица мужского пола в большей степени подвержены болезни по сравнению с женщинами.

Причины ПНП
Этиофакторы, запускающие дегенеративные процессы определённой церебральной локализации, остаются неизвестными. Большинство случаев болезни имеют спорадический характер. Отдельные семейные варианты с предположительным аутосомно-доминантным наследованием были выявлены после 1995 года. Молекулярно-генетические исследования показали, что некоторые формы ПНП обусловлены дефектами кодирующего тау-белок гена, локализованного в локусе 17q21.31. Наиболее вероятным представляется мультифакторный механизм возникновения патологии, реализующийся на фоне генетической предрасположенности.
Патогенез
Ведущим патогенетическим механизмом считается дисметаболизм церебральных внутриклеточных белков, сопровождающийся избирательной агрегацией отдельных белков (тау-протеина, убиквитина) в определённых группах мозговых клеток. Патологические включения нарушают жизнедеятельность нейронов, запускают процесс деградации и запрограммированной гибели (апоптоза). Дегенеративные изменения носят селективный характер, распространяются преимущественно на средний мозг, зубчатые мозжечковые ядра и подкорковые структуры: черную субстанцию, бледный шар, таламус, ретикулярную формацию, субталамическое ядро. В меньшей степени поражается кора префронтальных и височных зон.
Патоморфологическая картина ПНП представлена наличием нейрофибриллярных клубочков, глиальных включений, нитевидных белковых образований в нейронах указанных церебральных структур. Макроскопически определяется атрофия среднего мозга с существенным уменьшением его сагиттального размера. Поражение среднего мозга обуславливает надъядерный паралич глазодвигательной мускулатуры, дегенерация кортико-бульбарных трактов — псевдобульбарные проявления. Нейрохимические исследования выявляют пониженную концентрацию дофамина в стриатуме, лежащую в основе паркинсонического симптомокомплекса.
Симптомы ПНП
Прогрессирующий надъядерный паралич характеризуется неспецифичным клиническим дебютом. Симптоматика этого периода представлена непривычной утомляемостью, сниженной работоспособностью, цефалгиями, головокружением, пониженным настроением, сужением круга интересов, нарушениями сна, включающими бессонницу ночью и гиперсомнию днём. В последующем присоединяются симптомы акинетико-ригидного паркинсонизма. Постуральный тремор у большинства пациентов отсутствует. Мышечная ригидность выражена преимущественно в аксиальной мускулатуре — мышцах, идущих вдоль шейного отдела позвоночника, соединяющих его с черепом. Больные жалуются на скованность в шее, спине. Повышение тонуса в задних мышцах шеи приводит к типичному «горделивому» положению головы пациента. Характерна паркинсоническая атаксия, обусловленная расстройством координации положения туловища и нижних конечностей относительно центра тяжести. Затруднения в поддержании равновесия в процессе ходьбы приводят к частым падениям назад.
Отличительной особенностью ПНП выступает офтальмоплегия, возникающая в среднем спустя 2-3 года от дебюта заболевания. На фоне замедленного движения глазных яблок происходит паралич взора в вертикальной плоскости, пациент не может опустить глаза вниз. Из-за редкого моргания больной ощущает дискомфорт, жжение в глазах. Возможны расплывчатость зрения, расстройство конвергенции, блефароспазм. Прогрессирующий надъядерный офтальмопарез сопровождается ограничением взора вниз и вверх, со временем может приводить к глазодвигательным нарушениям в горизонтальной плоскости. При развитии полной офтальмоплегии формируется ретракция верхних век, что придаёт лицу удивлённое выражение.
В клинической картине ПНП относительно рано возникают псевдобульбарные проявления: дизартрия, дисфагия, насильственный плач или смех. Происходят изменения личностно-эмоциональной сферы, больные становятся замкнутыми, апатичными, демотивированными, безразличными. Когнитивные нарушения в большинстве случаев присоединяются в разгаре болезни, в 10-30% случаев — на стадии дебюта. Характерно интеллектуальное снижение, расстройства абстрактного мышления и памяти, зрительно-пространственная апраксия, элементы агнозии. Деменция наблюдается у 60% пациентов с 3-летним стажем заболевания.
Осложнения
В начальном периоде падения больного без возможности скоординировать свои движения приводят к ушибам и переломам. Спустя несколько лет прогрессирующий олигобрадикинетический синдром приковывает пациентов к постели. При отсутствии должного ухода обездвиженность опасна развитием контрактур суставов, пролежней, застойной пневмонии. Прогрессирующий псевдобульбарный паралич обуславливает попёрхивание пищей с риском асфиксии, аспирационной пневмонии. Ночные апноэ могут стать причиной внезапной смерти во сне. Серьёзным осложнением является присоединение интеркуррентных инфекций (пневмонии, цистита, пиелонефрита), поскольку на фоне сниженного иммунитета существует высокий риск развития сепсиса.
Диагностика
Вероятными ранними критериями ПНП являются начало после 40-летнего возраста, прогрессирующий характер, парез горизонтального взора, выраженная постуральная неустойчивость с эпизодами падений. Постановка достоверного диагноза возможна при наличии гистологически подтверждённых патогномоничных для ПНП изменений в тканях мозга. Перечень необходимых диагностических исследований включает:
- Осмотр невролога. В неврологическом статусе ведущим синдромом является симметричная олигобрадикинезия. Наблюдается гипомимия, ретроколлис (патологическая установка шеи), парез вертикального взора, симптомы орального автоматизма, повышение сухожильных рефлексов. Выражена постуральная неустойчивость.
- Нейропсихологическое тестирование. Проводится психиатром, нейропсихологом с использованием специальных тестов, заданий (шкалы MMSE, MоCА, теста рисования часов). Требуется для оценки наличия и степени выраженности когнитивного снижения. Надъядерный паралич проявляется замедленным мышлением, быстрой истощаемостью, умеренной выраженностью интеллектуальных нарушений.
- МРТ головного мозга. Выявляет расширение III желудочка, атрофические изменения среднего мозга, базальных ганглиев, премоторных зон лобной коры и височных областей. Позволяет исключить внутримозговую опухоль, энцефалит, рассеянный склероз, инсульт.
- Оценку церебральной гемодинамики. Данные о кровоснабжении мозга могут быть получены путём дуплексного сканирования, УЗДГ, МРТ сосудов. Необходимы для исключения дисциркуляторной энцефалопатии, сосудистого паркинсонизма, сосудистой деменции.
Дифференциальная диагностика осуществляется с болезнью Паркинсона, вторичным паркинсонизмом травматической, инфекционной, токсической, сосудистой этиологии, деменциями альцгеймеровского типа, поздней формой нейроакантоцитоза. От классической болезни Паркинсона надъядерный паралич отличается симметричностью паркинсонизма с момента его появления, быстрым развитием когнитивных расстройств, офтальмоплегией, ретроколлисом, выраженной атаксией, малым эффектом дофаминергической терапии. Достоверно дифференцировать прогрессирующий надъядерный паралич от прочих таупатий можно по особенностям патоморфологических изменений.
Лечение ПНП
Эффективная терапия, способная остановить прогрессирующий дегенеративный процесс, пока не найдена. Осуществляется симптоматическое лечение, направленное на облегчение состояния пациента. Проведенные фармакотерапевтические исследования не сопровождались плацебо-контролем, слабо доказывают эффективность медикаментозной терапии. В лечении когнитивных нарушений возможно применение мемантина, ингибиторов ацетилхолинэстеразы, для коррекции психоэмоциональной сферы — антидепрессантов с психоактивирующим действием (флуоксетина, пароксетина).
Большинство неврологов считают необходимым назначение стартовой дофаминергической терапии. У половины больных наблюдается определённое облегчение состояния на фоне приёма препаратов леводопы, однако данный эффект длится не более двух лет. Противопаркинсонические фармпрепараты прочих групп (ингибиторов МАО, агонистов дофаминовых рецепторов, ингибиторов КОМТ) не показали своей эффективности.
Прогноз и профилактика
При надъядерном параличе наблюдается безостановочное прогрессирование симптоматики. Проводимая терапия не оказывает существенного эффекта на течение болезни. Продолжительность жизни пациентов колеблется в пределах 5-15 лет. Летальный исход обусловлен интеркуррентными инфекциями, затяжным апноэ сна, аспирационной пневмонией. В связи с отсутствием ясного понимания этиологии и патогенеза нозологии разработка профилактических мероприятий не представляется возможной, исследования заболевания и методов его лечения продолжаются.
1. Клинические особенности надъядерного паралича/ Валикова Т.А., Алифирова В.М., Пугаченко Н.В., Цыренжапова Р.Б., Бичик А.Б.// Бюллетень сибирской медицины - 2009. - №3 (2).
2. Cлучай прогрессирующего надъядерного паралича с кортикобазальным синдромом/ Федотова Е.Ю., Чечеткин А.О., Иванова-Смоленская И.А., Иллариошкин С.Н.// Атмосфера. Нервные болезни. - 2009 - №2.
3. Трудности диагностики прогрессирующего надъядерного паралича/ Ситкали И.В., Раздорская В.В.// Бюллетень медицинских интернет-конференций. - 2015 - Т.5, №4.
4. Трудности дифференциальной диагностики прогрессирующего надъядерного паралича и болезни Паркинсона/ Магжанов Р.В., Давлетова А.И., Ибатуллин Р.А., Туник В.Ф., Идрисова Р.Ф., Бахтиярова К.З.// Анналы клинической и экспериментальной неврологии. - 2016.
МРТ при болезни Паркинсона

Ранняя диагностика болезни Паркинсона на данный момент остается сложной задачей для медицинского сообщества и требует комплексного подхода - данных истории болезни, экспертизы врача-невролога и результатов исследования МРТ головного мозга. С ростом продолжительности жизни и неблагоприятной экологической обстановкой все больше жителей Санкт-Петербурга имеют шансы дожить до диагноза Паркинсон. Этот недуг становиться одной из причин роста инвалидности населения в области неврологических заболеваний. Именно поэтому очень важно выявить эту патологию вовремя на ранней стадии развития. Ведь к более адекватному ответу на лечение приводит лишь своевременно начатая терапия. Механизма появления и развития этой патологи до конца еще не открыт. Считается, что это заболевание отчасти зависит от наследственности, а отчасти может быть вызвано триггерными факторами, например, экологической обстановкой.
Методы диагностики болезни Паркинсона
Диагностика этого недуга инструментальными методами при помощи КТ или МРТ головы, а также ПЭТ - это сложная задача, поскольку прямых и точных признаков болезни Паркинсона пока не выявлено. Если выбирать из современных методов диагностики, то сейчас набирает силу метод позитронно-эмиссионной томографии, на котором при подозрении на болезнь Паркинсона исследование проводится с применением флуородопа. Недостатком данного исследования является его низкая доступность даже в таких крупных городах, как Санкт-Петербург. К тому же это очень дорогостоящая диагностика. Цена исследования методом позитронно-эмиссионной томографии составляет 30 000 руб.
Основной диагностический недостаток ПЭТ заключается в том, что в ходе сканирования используются радиоактивные препараты, и не всегда получается анатомически точно локализовать найденный патологический очаг. Поэтому в медицинских центрах СПб наиболее распространенной формой аппаратной диагностики при болезни Паркинсона является МРТ головного мозга, которая более доступна широкому кругу пациентов.
Что показывает МРТ при болезни Паркинсона
В диагностике болезни Паркинсона очень важен правильный сбор анамнеза, особенно информации о том, как развивалось заболевание, какие симптомы появились первично. Врачу необходимо установить, есть ли у пациента такие важные признаки, как:
- олигобрадикинезия;
- ригидность мышц;
- постуральная неустойчивость;
- развитие депрессии на фоне изменений состояния пациента.
Перед неврологом стоит задача определить и другие симптомы, которые могут говорить о каких-то других заболеваниях. Для этого врачи часто назначают сделать МРТ головного мозга при подозрении на болезнь Паркинсона. Она позволяет оценить состояние черной субстанции вещества головного мозга и провести дифференциальную диагностику для исключения следующих патологий:
- демиелинизация;
- новообразования;
- возрастные дегенеративные изменения головного мозга.


МРТ головного мозга при Паркинсоне
МРТ головного мозга при Паркинсоне следует сделать на высокопольном аппарате с напряженностью магнитного поля 1.5 Тесла и выше. Такое обследование, как правило, проходит без контраста и позволяет визуализировать очаги поражения черной субстанции мозга. Особой подготовки данное сканирование не требует, однако характерный для больных Паркинсоном тремор может создать некоторые диагностические сложности. Высокопольный томограф очень чувствителен к любому движению. Оно может смазать изображения на получаемых в ходе процедуры снимках. Длительность МРТ головы обычно составляет 15-20 минут, и пациент все это время должен лежать неподвижно. Иногда больным Паркинсона на поздних стадиях заболевания такая неподвижность дается с трудом.
ЗАПИСЬ НА ПРИЕМ
Болезнь Паркинсона МРТ признаки
Для дифференциальной диагностики болезни Паркинсона в медицинских центрах СПб используют протокол - МРТ головного мозга в SWI-режиме (в режиме изображений, взвешенных по магнитной восприимчивости). Такая МРТ показывает не только признаки вторичного паркинсонизма, но и визуализирует типичные для этой болезни изменения черной субстанции (отсутствие нигросомы-1) при первичной форме заболевания. Изображение не поврежденных тканей мозга в середине черной субстанции напоминает хвост ласточки. А вот у больных болезнью Паркинсона данное разделение на две доли исчезает.
Как сделать МРТ головного мозга при Паркинсоне
Выявление болезни Паркинсона на МРТ головного мозга требует обследования на высокопольном или сверхвысокопольном томографе по специальной импульсной последовательности. Такое исследование проводит не каждая МРТ клиника, оборудованная томографическим аппаратом. Уточнить адреса и цены диагностических центров, где можно осуществить данную разновидность томографии головного мозга, можно с помощью Бесплатной службы записи на диагностику в СПб. Наши сотрудники будут рады подобрать Вам правильный медицинский центр. Звоните, мы знаем все о диагностике в СПб!
Диагностика прогрессирующего надъядерного паралича по КТ, МРТ, ПЭТ головного мозга
ФГБОУ ДПО «Российская медицинская академия непрерывного профессионального образования» Минздрава России
ФГБУ ГНЦ «Федеральный медико-биологический центр им. А.И. Бурназяна» ФМБА России
ФГБУ «Государственный научный центр Российской Федерации — Федеральный медицинский биофизический центр им. А.И. Бурназяна» ФМБА России
Прогрессирующий надъядерный паралич
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2021;121(5): 111‑119
Прогрессирующий надъядерный паралич (ПНП) является прогрессирующим нейродегенеративным заболеванием, которое характеризуется началом в возрасте старше 50 лет, синдромом паркинсонизма с ранним развитием постуральной неустойчивости, отсутствием или преходящей реакцией на препараты леводопы, а также нейропсихологическими нарушениями, дисфагией, дизартрией и глазодвигательными расстройствами. В обзоре приводится анализ современных данных по этиологии, клинической картине, дифференциальной диагностике заболевания, приведены критерии диагноза ПНП. Описана морфологическая картина и нейровизуализационные особенности головного мозга, а также современные представления о перспективах лечения. Значительный клинический полиморфизм, а также сходство с другими нейродегенеративными заболеваниями, проявляющимися синдромом паркинсонизма, усложняет диагностику ПНП. Установка точного диагноза позволяет определить прогноз и дальнейшую тактику ведения пациентов.
Дата принятия в печать:
В 1964 г. К. Ричардсон, Д. Ольшевский и Д. Стил впервые описали клинические и морфологические особенности 8 пациентов с прогрессирующим надъядерным параличом (ПНП) [1]. Особенностью клинической картины являлись паралич вертикального взора, быстро прогрессирующая постуральная неустойчивость, псевдобульбарный синдром и акинетико-ригидный синдром, преимущественно в аксиальной мускулатуре, деменция подкорково-лобного типа. В литературе уже имелись описания 51 подобного случая, из них 22 — с данными аутопсий, однако именно их классическая работа очень точно и полно отразила особенности заболевания и по-прежнему является интересной для специалистов.
ПНП относится к группе гетерогенных заболеваний, отличающихся от идиопатической болезни Паркинсона (БП) клиническими признаками, низкой реакцией или отсутствием терапевтического эффекта леводопы, а также неблагоприятным прогнозом.
В 2005 г. D. Willims и соавт. [2] проанализировали 103 случая подтвержденного ПНП, разделив их на 2 фенотипа. Классический синдром, описанный авторами в 1964 г., был назван синдромом Ричардсона, второй фенотип получил название ПНП-паркинсонизм. За последнее десятилетие были описаны также и другие варианты ПНП, названные атипичными.
Частота ПНП, по данным различных исследований, составляет 5—18 человек на 100 тыс. населения, но истинная распространенность этого заболевания до конца не известна [3, 4]. Ежегодная заболеваемость варьирует от 0,3 до 1,1 случая на 100 тыс. населения, с увеличением до 5,3 случаев у лиц старше 50 лет. Мужчины и женщины страдают ПНП примерно одинаково, однако при синдроме Ричардсона соотношение мужчин и женщин составляет 1:1,8 [5]. Средний возраст начала заболевания составляет 55—70 лет. Дебют заболевания начинается без явных предвестников, в последующем наблюдается вариабельное прогрессирование симптомов. Прогноз ПНП неблагоприятный, средняя продолжительность жизни после дебюта составляет около 7 лет [6]. Наиболее частая причина смерти — аспирационная пневмония.
Основными клиническими признаками ПНП являются:
1. Паркинсонизм, характеризующийся двусторонней и симметричной брадикинезией с преобладанием аксиальной ригидности и отсутствием реакции на препараты леводопы.
2. Псевдобульбарный синдром (дизартрия, дисфония, дисфагия, насильственный плач или смех).
3. Глазодвигательные нарушения: парез вертикального взора, который может отсутствовать в 50% случаев и редко является первым симптомом ПНП.
4. Лобный синдром: брадифрения, дефицит познавательных и исполнительных функций, палилалия, эхолалия, хватательный симптом, снижение вербальной беглости.
У больных ПНП достаточно рано появляются нейроофтальмологические нарушения, предшествующие параличу вертикального взора, нарушение способности подавления вестибулоокулярного рефлекса, отсутствие быстрой фазы оптокинетического нистагма, нестабильность фиксации взгляда, скачкообразные подергивания глазных яблок (дисфиксационные саккады), затуманивание зрения, неуверенность при инициализации движения глаз по команде, замедление и гипометрия саккад, снижение скорости и амплитуды произвольных движений глазных яблок, ограничение или отсутствие конвергенции глазных яблок, снижение частоты мигания, апраксия открывания и закрывания век, блефароспазм [7—9].
Когнитивные нарушения при ПНП проявляются замедленностью мышления, нарушением исполнительных функций — 80% [10], лобной или фронто-темпоральной деменцией — 30% [2], апраксией — 36% [10], апатией — 91% [6], депрессией — 50% [11].
Немоторные симптомы встречаются при ПНП не реже, чем при БП. Так, по данным сравнительного исследования F. Radicati и соавт., кардиоваскулярные симптомы при ПНП встречались в 50% случаев, тогда как при БП в 47%, нарушения сна и усталость отмечались в 92% случаев (при БП — в 89%), нарушения настроения и апатия — в 88% (при БП — в 69%), нарушение восприятия, галлюцинации — в 36% (при БП — в 27%), гастроинтестинальные симптомы — в 86% (при БП — в 71%), урологические — в 92% (при БП — в 72%)[12].
Клинические критерии диагностики ПНП были разработаны в 1996 г. (рис. 1) [7]. Согласно критериям, возможный диагноз ПНП требует либо паралича вертикального взора (вверх или вниз), либо замедления вертикальных саккад в сочетании с выраженной постуральной неустойчивостью и частыми падениями, развивающимися на первом году. Вероятный диагноз ПНП требует сочетания паралича вертикального взора и выраженной постуральной неустойчивости. Достоверный диагноз ПНП требует наличия клинически возможного или вероятного ПНП и гистологических изменений, типичных для ПНП.
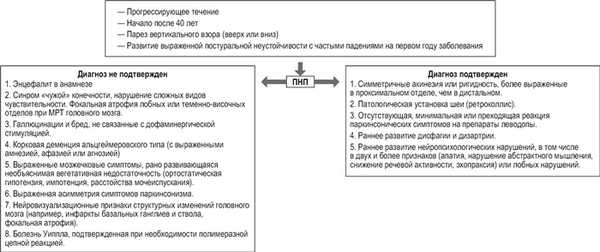
Рис. 1. Клинические критерии диагностики ПНП.
В 2017 г. были разработаны новые клинические критерии ПНП, основанные на оценке уровня достоверности окуломоторной дисфункции, постуральной нестабильности, акинезии и когнитивной дисфункции. Однако эти критерии несколько сложны для использования в повседневной практике и могут с успехом применяться при проведении научно-исследовательских работ [13]. Эти клинические критерии при сопоставлении с данными при аутопсии мозга имеют высокую специфичность: от 95 до 100% для возможного и от 80 до 93% — для вероятного диагноза ПНП [14—16].
Следует отметить, что в обычной практике диагноз устанавливается спустя 3—4 г. после появления первых симптомов заболевания.
В последние годы описано несколько различных клинических фенотипов ПНП [17, 18].
Классический фенотип ПНП (синдром Ричардсона)
Наиболее характерный признак классического ПНП — паралич вертикального взора, особенно при взгляде вниз (парез взора вверх менее специфичен и возможен у пожилых больных с паркинсонизмом любой этиологии, в том числе и при БП). Однако в большинстве случаев паралич взора вниз развивается лишь спустя 2—3 года от начала болезни. До развития паралича взора у пациентов выявляются более легкие глазодвигательные нарушения: замедление и гипометрия вертикальных саккадических движений и нарушение плавности вертикальных следящих движений, а также нарушение подавления вестибулоокулярного рефлекса и отсутствие или замедление быстрой фазы оптико-кинетического нистагма в вертикальной плоскости [2, 19]. Характерной чертой ПНП является исключительная редкость мигания, иногда больные испытывают затруднения при произвольном открывании, реже закрывании глаз (апраксия открывания и закрывания глаз).
Помимо глазодвигательных нарушений для классического варианта ПНП характерны симметричный синдром паркинсонизма, преимущественного аксиальных отделов, отсутствие классического тремора покоя, раннее развитие постуральных нарушений с частыми падениями. На ранней стадии у таких больных не отмечается затруднений инициации ходьбы, уменьшение длины шага и площади опоры, как при БП. Затруднения больного скорее связаны с тем, что он не может правильно скоординировать движения туловища и нижних конечностей таким образом, чтобы не происходило резких смещений центра тяжести тела относительно площади его опоры. Голова и туловище больного нередко бывают отклонены кзади. Больной может неожиданно упасть на спину, даже не предпринимая попыток удержать равновесие. При ПНП больные чаще падают назад, хотя падения могут происходить в любую сторону [20, 21].
Кроме того, у пациентов с синдромом Ричардсона быстро развиваются грубые дизартрия и дисфагия, отмечается преобладание повышенного тонуса мышц — разгибателей спины с формированием ретроколлиса, умеренная пирамидная недостаточность с гиперрефлексией и патологическими стопными рефлексами, развивается деменция с выраженными чертами лобной психики, речевыми персеверациями с повторением слов, слогов и целых фраз [22].
Фенотип ПНП-паркинсонизм
Для этого фенотипа характерно более благоприятное течение [17]. У таких пациентов часто присутствуют асимметричный тремор, брадикинезия, ригидность, умеренный эффект леводопы, при этом отмечается более медленный темп прогрессирования заболевания, чем при классическом фенотипе Ричардсона, что на ранних этапах клинически схоже с БП, и дифференциальная диагностика затруднена [23]. Однако присоединение в последующем лекарственных дискинезий, вегетативных нарушений, зрительных галлюцинаций более характерно для БП, чем для ПНП [18].
Фенотип ПНП-акинезия с застываниями при ходьбе
Для этого фенотипа характерны прогрессирующие застывания при ходьбе, изолированные расстройства ходьбы в течение нескольких лет, лишь затем присоединяются другие типичные проявления ПНП. В течение первых 5 лет для этого фенотипа не характерны тремор, ригидность, деменция, ограничение вертикального взора [17].
Фенотип ПНП-кортикобазальный синдром
При этом редком фенотипе отмечаются прогрессирующая асимметричная ригидность конечностей, апраксия, миоклонии, синдром «чужой» конечности, дистония и низкий эффект препаратов леводопы. Прижизненная дифференциальная диагностика невозможна, по рекомендации Международного общества расстройств движений, следует рассматривать этот фенотип как возможный диагноз ПНП [13].
Фенотип ПНП с нарушениями речи
Этот фенотип является клиническим вариантом первичной прогрессирующей афазии, характеризуется нарушением грамматизма речи, ее прерывистостью с искажением слов (речевая апраксия), лишь в последующем присоединяются другие моторные симптомы ПНП [24].
Фенотип ПНП с лобными симптомами (поведенческий вариант)
Для этого редкого фенотипа характерны симптомы поведенческого варианта лобно-височной деменции в течение нескольких лет до появления моторных симптомов ПНП. В клинической картине доминируют личностные расстройства, асоциальное поведение, когнитивные нарушения [25].
Фенотип ПНП с мозжечковой атаксией
Фенотип характеризуется церебеллярной атаксией в качестве первых симптомов заболевания, лишь затем присоединяются характерные симптомы ПНП. В первые годы болезни необходимо проводить дифференциальную диагностику с мультисистемной атрофией, однако для этого фенотипа не характерны выраженные вегетативные нарушения [26].
Клинические варианты ПНП, их примерная частота и соответствующие им патоморфологические нарушения представлены в табл. 1 [27—34]. Пациенты с различными фенотипами, кроме классического фенотипа ПНП (синдром Ричардсона), составляют 76% [28]. Продолжительность жизни при различных фенотипах ПНП значительно отличается.
Таблица 1. Клинические варианты ПНП [27—34]
Примерная частота встречаемости от всех случаев ПНП, %
Средняя продолжительность жизни (годы)
Ранняя постуральная нестабильность, падения, супрануклеарный паралич, аксиальная ригидность, дизартрия, дисфагия, прогрессирующая деменция
Зубчатое ядро, стриатум, бледный шар, средний мозг, верхняя ножка мозжечка
Тремор, ригидность, брадикинезия, наличие ответа на леводопу, позднее развитие когнитивных нарушений
Черная субстанция, субталамическое ядро
Дистония, апраксия, речевая апраксия, корковые расстройства чувствительности
Лобная и теменная кора
ПНП-акинезия с застываниями
Ранние нарушения ходьбы, застывания, микрография, нарушения речи, гипофония, течение заболевания — до 11—15 лет
Моторная кора, мост, мозжечок
ПНП-поведенческий вариант лобно-височной деменции
Когнитивные и поведенческие расстройства, поздний паркинсонизм
ПНП-боковой амиотрофический склероз
Слабость бульбарных мышц, мышц конечностей, спастичность
Лобная кора, кортикоспинальный тракт
Процент смертности в течение 5 лет наблюдения за больными с разными фенотипами ПНП составил: при ПНП-Ричардсона — 29,2%, при ПНП-постуральных нарушениях — 16,7%, при ПНП-паркинсонизме — 5,3%, при ПНП-кортико-базальном синдроме — 28,6%, при ПНП-фронтотемпоральной деменции — 33,3% [28].
Таким образом, фенотипический спектр ПНП значительно более широкий и вариабельный, чем было представлено ранее в отдельных исследованиях. Слишком жесткие клинические критерии, определяющие фенотипы ПНП, могут не отражать всей их вариабельности. Более целесообразно использовать в клинической практике преимущественные варианты ПНП (в течение первых 2 лет заболевания), что может помочь в ранней диагностике и определении прогноза течения заболевания у этих пациентов.
Для определения степени тяжести ПНП и темпа прогрессирования заболевания в научно-клинических исследованиях применяется рейтинговая шкала, которая содержит 6 разделов. Она состоит из вопросов сбора анамнеза, оценки психического состояния, оценки функции бульбарных нервов, оценки надъядерных механизмов движения глаз, оценки функций конечностей, оценки походки/отклонений положения тела от срединной линии [34].
Морфологические изменения при ПНП
ПНП относится к группе таупатий — нейродегенеративных заболеваний, характеризующихся отложением в клетках головного мозга патологически фосфорилированного тау-протеина — низкомолекулярного белка, который является компонентом нейронального цитоскелета. Функцией тау-белка является полимеризация тубулина в процессе сборки микротрубочек, которые являются транспортной системой клетки [35].
При ПНП тау-протеин отделяется от микротрубочек, аномально фосфорилируется, накапливается в цитоплазме и становится токсичным для клеточных мембран, превращаясь в нейрофибриллярные клубочки, которые и определяются под микроскопом. В результате этого процесса нарушается транспорт по микротрубочкам, нарушаются контакты между нейронами и происходит гибель клеток [36].
Ген человеческого тау-белка расположен на хромосоме 17q21 и содержит 16 экзонов — участков гена ДНК, несущих генетическую информацию о структуре белка. В человеческом мозге найдено 6 различных изоформ тау-протеина, генерирующихся поочередным сплайсингом (склеиванием) экзонов 2, 3 и 10. Они разделяются на две группы, отличающиеся по наличию 3 или 4 повторяющихся доменов (3R и 4R), связанных с микротрубочками. В здоровом мозге тау-протеин формируется с 3 повторами, тогда как при ПНП имеется соотношение 3:1 в пользу белка с 4 повторами. Достижения в области нейронаук в течение последних десятилетий привели к открытию того, что анормальный гиперфосфорилированный 4R -изоформный микротубулоассоциированный тау-протеин (hp4R-MAPT) накапливается в нейронах и глиальных клетках коры, подкорковых ядрах, стволе и мозжечке [37].
Отличительный морфологический признак ПНП — появление в астроцитах hp4R-MAPT («астроциты с хохолком») в стриатуме и лобной коре (поля 6 и 8 по Бродману) [38]. При ПНП гибель нейронов, глиоз и накопление тау-протеина происходит в нейронах и глиальных клетках лобной коры, черной субстанции, стриатума, бледного шара, субталамического ядра, ядер ствола мозга (ростральное интерстициальное ядро медиального продольного пучка, интерстициальное ядро Кахала, ядро Даркшевича, ядро Эдингера-Вестфаля, верхние бугорки четверохолмия, педункулопонтинное ядро, ядра шва, голубое пятно) [39]. Т-позитивные пучковидные астроциты являются характерным морфологическим признаком ПНП [40].
Следует отметить, что гетерогенность форм ПНП коррелирует и с морфологическими изменениями в головном мозге [39]. Более выраженное отложение тау-протеина в коре мозга отмечается при фенотипах ПНП-кортикобазальный синдром, ПНП-лобно-височная деменция. При фенотипах ПНП-паркинсонизм и ПНП-акинезия с застываниями преобладает патология субталамического ядра и черной субстанции. Клинико-морфологические корреляции при ПНП представлены в табл. 2 [39—41].
Таблица 2. Клинико-морфологические корреляции при ПНП [39—41]
Нейродегенеративные заболевания и их диагностика
МРТ головного мозга. Демонстрация атрофии и нормы при цветовой обработке.
Под термином “нейродегенеративные заболевания” (НДЗ) определяется большая группа заболеваний преимущественно позднего возраста, для которых характерна медленно прогрессирующая гибель определенных групп нервных клеток и одновременно - постепенно нарастающая атрофия соответствующих отделов головного и/или спинного мозга. В основе развития этих заболеваний лежит нарушение метаболизма и изменение конформации клеточных белков с их последующим накоплением и агрегацией в определенных группах нейронов. При НДЗ страдают преимущественно нейроны и глиальные клетки базальных ганглиев и стволовых структур, вырабатывающие ацетилхолин, дофамин, серотонин.
Классификация делит НДЗ на 2 большие группы - спорадические и ирритативные.
- Спорадические НДЗ:
- Прогрессирующий надъядерный паралич (болезнь Стила —Ричардсона — Ольшевского).
- Мультисистемная атрофия.
- Деменция с тельцами Леви.
- Паркинсоническая деменция (синдром Гуам).
- Кортикобазальная дегенерация.
- Болезнь Альцгеймера.
- Ирритативные НДЗ:
- Болезнь Гентингтона.
- Болезнь Галлервордена—Шпатца.
- Болезнь Вильсона—Коновалова.
- Болезнь Фара.
- Болезнь Бессена — Корнцвейга.
Болезнь Альцгеймера - прогрессирующее нейродегенеративное заболевание, характеризующееся постепенным развитием деменции. Происхождение заболевания точно неизвестна. Биохимические изменения состоят в снижении активности холин-ацетил-трансферазы коры головного мозга и гиппокампов. Патологические проявления заключаются в образовании специфических амилоидных бляшек, нейрофибриллярных тяжей и реактивном глиозе. Развивается атрофия, захватывающая преимущественно кору вокруг Сильвиевых щелей и гиппокампы, с вторичным расширением желудочков, особенно височных рогов
Заболевание впервые описано Alois Alzheimer в 1907 году. Процесс напоминает естественное старение, но резко ускоренное. Начинается с нарушений памяти, затем потерянность, невозможность повседневного самообслуживания, повторяющиеся вопросы. Позже присоединяются глубокие нарушения психики, речи, потеря веса, судороги.
Частота составляет 0,51% для лиц в возрасте 70-74 лет с возрастным прогрессирующим увеличением частоты. Клинические проявления состоят в нарушении памяти, депрессии, поведенческих нарушениях и галлюцинациях. На поздних стадиях к психическим расстройствам добавляется экстрапирамидная симптоматика. Заболевание занимает 4 место по смертности. Диагноз ставится на основании клинического и нейрофизиологического обследования, а также нейровизуализации. Типичные проявления на КТ состоят в диффузной атрофии (особенно,височных долей), вторичном расширении борозд и желудочков. Чувствительность КТ (без измерения объемов) в сравнении с нормальной возрастной группой около 80%, специфичность около 70%. Измерение объемов гиппокампов при выполнении МРТ с тонкими срезами повышает точность до 85%.
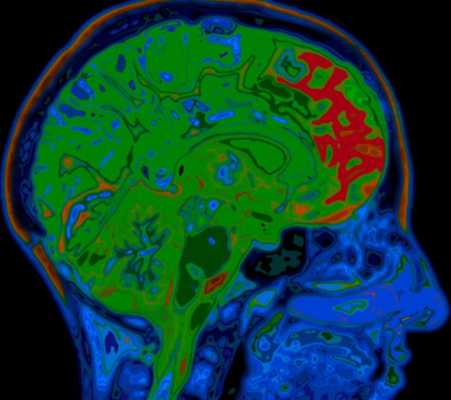
МРТ головного мозга. Т2-взвешенная сагиттальная МРТ. Болезнь Пика. Цветовая обработка изображения.
МРТ головного мозга служит метода выбора оценки структурных изменений. Атрофические изменения выражены во всём медиобазальном отделе височной доли. Чувствительность и специфичность МРТ при начальной деменции около 80%. Измерение объемов гиппокампов и амигдалы повышает точность до до 85%.
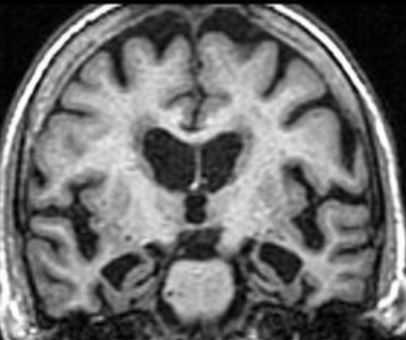
МРТ головного мозга. Т1-взвешенная корональная МРТ. Диффузная атрофия при болезни Альцгеймера.
Дифференциальную диагностику при МРТ головного мозга надо проводить с болезнью Паркинсона, мультиинфарктной деменцией и лобнотеменной деменцией (болезнь Пика).
В этих же зонах отмечается гипоперфузия и снижение активации при фМРТ. Кроме МРТ при исследовании болезни Альцгеймера важное значение имеет ПЭТ с [ 18 F] флюоро-2-деоксиглюказой (FDG). Гипометаболизм хорошо коррелирует с тяжестью заболевания и предсказывает его развитие.
Синдромы Паркинсона включают группу заболеваний, близких по клинике к болезни Паркинсона. К синдромам Паркинсона относится быстро прогрессирующая деменция с тельцами Леви . При МРТ головного мозга низкий сигнал наблюдается не только от компактной части черного вещества, но и от скорлупы, которая становится даже темнее бледного шара. При оливопонтоцеребеллярной атрофии на сагиттальных МРТ головного мозга видно уменьшение объема моста и мозжечка. При прогрессирующем надъядерном параличе обнаруживается атрофия пластины четверохолмия. Описаны характерные симптомы при МРТ - «пингвина», «Микки Мауса» и другие, смысл которых заключается в описании признаков атрофии.
При деменции, связанной с болезнью Паркинсона при МРТ головного мозга отмечается снижение толщины коры в проекции парагиппокампальной части левой средней затылочно-височной извилины и уменьшение объема левого нижнего продольного пучка. Уменьшение толщины коры левой парагиппокампальной зоны связано с высоким риском депрессии. Отмечено, что дневная сонливость коррелирует с уменьшением толщины коры фузиформной зоны, определяемое при МРТ.
Для прослеживания динамики и прогнозирования также прибегают к различным измерениям при МРТ головного мозга:
- коэффициент средний мозг-мост в норме 0,24, а при прогрессирующем надъядерном параличе становится меньше 0,12.
- Индекс паркинсонизма - отношение ширины верхней ножки мозжечка в корональной плоскости к площади среднего мозга в средней сагиттальной плоскости умноженной на отношение ширины средней ножки мозжечка к ширине верхней ножки мозжечка - больше 13,55 свидетельствует в пользу паркинсонических синдромов. При МРТ выявляется атрофия хвостатых ядер с вторичным расширением передних рогов; атрофия скорлупы и коры лобных долей. Отношение ширины передних рогов к расстоянию между хвостатыми ядрами (по их краям), измеряемое в поперечной плоскости уменьшается с 2,2-2,6 до значений близких к 1,0. Другой коэффициент - расстояние между хвостатыми ядрами (между их головками к ширине черепа по внутренним пластинкам) - увеличивается (в норме 0,09-0,12).При МРТ головного мозга выявляется диффузная атрофия мозга, расширение периваскулярных пространств Вирхова- Робена и лейкоараиоз. Последний является следствием стеноза и окклюзии глубоких вен мозга. На Т2-зависимых МРТ изображениях лейкоараиоз выглядит как небольшие очаги гиперинтенсивности. В целом эти признаки неспецифические и отражают старение мозга. При МРТ головного мозга на томограммах обоих типов взвешенности обнаруживается повышенный сигнал от моста и покрышки мозжечка. Типично изменение сигнала от периферии моста. В отличии от опухолей при МРТ нет отека и масс-эффекта. Самые ранние проявления обнаруживаются на диффузионное-взвешенных МРТ головного мозга, примерно через 24 часа от начала тетрапареза.
Прогрессирующий надъядерный паралич проявляется в виде нарушения взора вверх, экстрапирамидной симптоматики и умственных нарушениях. Заболевание развивается у лиц около 60 лет. Этиология неизвестна, почти все случаи спорадические. Частота 1-1,5 случаев на 100 тыс. населения. Заболевание характеризуется патологическим скопление в головном мозге тау-протеина. При МРТ головного мозга наблюдается диффузная атрофия, причем на сагиттальных Т1-взвешенных МРТ отмечается характерный симптом “пингвина”. Атрофические изменения моста и среднего мозга приводят к расширению водопровода и III желудочка, контур которых напоминает очертания пингвина.
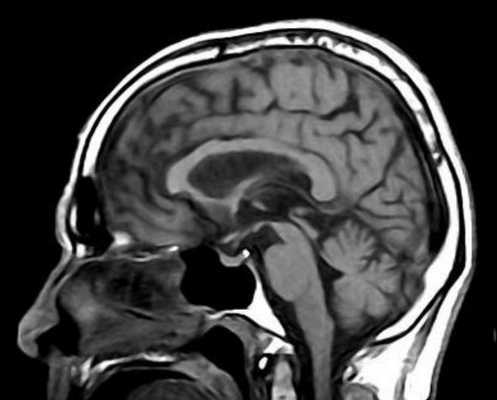
МРТ головного мозга. Т12-взвешенная сагиттальная МРТ. Прогрессирующий надъядерный паралич. Симптом “пингвина”.
Центральный понтинный миелиноз (осмотическая деменция) представляет собой приобретенное метаболическое расстройство. Чаще встречается у алкоголиков. Гипонатрийемия приводит к демиелинизации, видимой при МРТ. Центральный понтинный миелиноз часто сопровождается экстрацентральным, когда есть поражение выше ствола. Клинические проявления сводятся к заторможенности (вплоть до летаргии), спастическому тетрапарезу и поражению нижних черепных нервов.
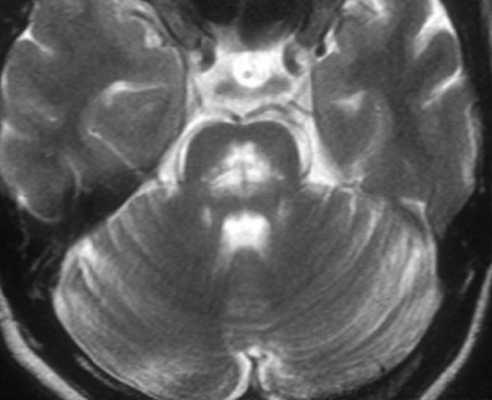
МРТ головного мозга. Т2-взвешенная аксиальная МРТ. Центральный понтинный миелиноз,
Болезнь Бинсвангера (субкортикальная атеросклеротическая энцефалопатия, деменция мелких сосудов). Это состояние, связанное с множественными инфарктами мелких ветвей, что при МРТ головного мозга видно как лакунарные ОНМК. Заболевание постепенно прогрессирует. Вариантом болезни Бинсвангера можно считать наследуюмую семейную артериопатическую лейкоэнцефалопатию.
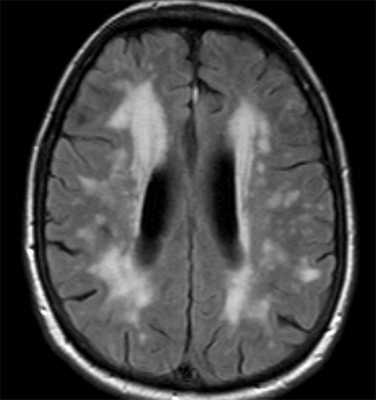
МРТ головного мозга. Т2-взвешенная МРТ типа FLAIR. Болезнь Бинсвангера.
Болезнь Гентингтона относится к наследуемым заболеваниям, проявляется в среднем возрасте и быстро прогрессирует. В клинической картине преобладают хореоатетоз и деменция.
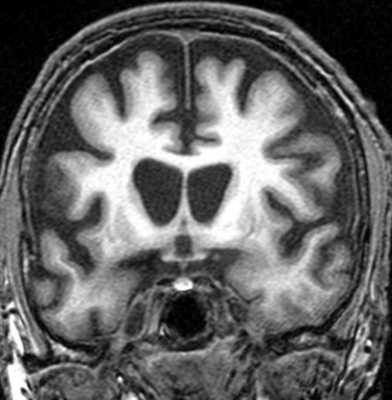
МРТ головного мозга. Т1-взвешенная корональная МРТ. Болезнь Гентингтона.
Болезнь Фара очень редкое наследственное заболевание, проявляющееся в кальцификации базальных ганглиев и зубчатого ядра. На Т2-зависимых МРТ ядра резко гипоинтенсивны, что соответствует кальцинатам, хорошо видимым на КТ. Нередко в области зрительных бугров обнаруживаются мелкие гиперинтенсивные очажки.
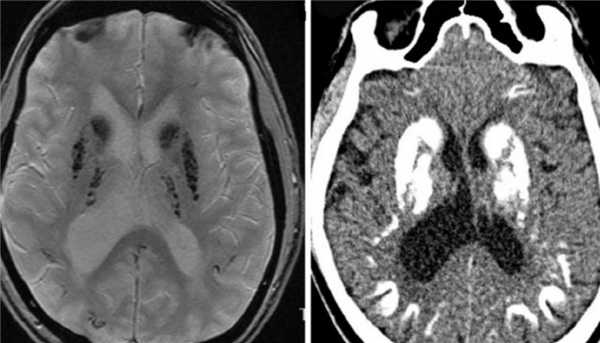
МРТ и КТ головного мозга в аксиальной плоскости. Болезнь Фара.
Об МРТ в СПб при нейродегенераторных заболеваниях можно также читать на странице нашего другого сайта. Исследование мы выполняем в закрытом аппарате, но можно и на открытом МРТ. МРТ СПб позволяет выбирать место МРТ, но в этом случае мы рекомендуем Вам обследоваться у нас.
Читайте также:
- Алкилирующие средства в онкогинекологии
- PC-вирус и бронхиальная астма. Интерфероны при вирусных инфекциях.
- Рентгенограмма, КТ при метастазе в стенку тонкой кишки и лимфоме тонкой кишки
- Пример гипотермии при алкоголизме. Влияние гипотермии на абстинентный синдром
- Сдавление головного мозга. Острые травматические гематомы.