Диагностика риска развития миодистрофии Дюшенна. Методы
Добавил пользователь Владимир З. Обновлено: 21.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Мышечная дистрофия: причины появления, симптомы, диагностика и способы лечения.
Определение
Мышечные дистрофии - это большая группа наследственных заболеваний, которые характеризуются прогрессирующей слабостью и дегенерацией скелетных мышц, то есть потерей мышечной массы. К наиболее распространенным миодистрофиям относятся миодистрофия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия), мышечная дистрофия Дюшенна и дистрофия Беккера. Также в эту группу входят дистрофия Эмери-Дрейфуса, миодистрофия Эрба-Рота и другие.

Миодистрофия Дюшенна поражает в основном мальчиков, а распространенность этого заболевания составляет 3,3 на 100 000 населения, 1 из 3500 новорожденных мальчиков страдает данной патологией. Этот вид дистрофии часто выделяют в одну группу с миодистрофией Беккера (частота встречаемости 1 на 20 000 новорожденных). Миодистрофии Дюшенна и Беккера наследуются по Х-сцепленному рецессивному типу. Это означает, что повреждение находится в Х-половой хромосоме и передается от матери к сыну, а дочери являются носительницами и, как правило, сами не болеют.
Миопатия Ландузи-Дежерина (плече-лопаточно-лицевая миопатия) встречается с частотой 0,9-2 на 100 000 населения. Заболевание наследуется по аутосомно-доминантному, аутосомно-рецессивному (самый редкий) или Х-связанному типу. Для аутосомно-доминантного типа наследования достаточно одной копии дефектного гена от одного из родителей.
Частота развития мышечной дистрофии Эмери-Дрейфуса точно не известна, описано 7 генетических форм, но частота установлена лишь для одной из них - Х-сцепленной рецессивной формы, она составляет 1 на 100 000.
Частота встречаемости миодистрофии Эрба-Рота составляет от 1,5 до 2,5 случаев на 100 тыс. населения. Этому типу мышечной дистрофии подвержены и мальчики, и девочки. Наследуется мышечная дистрофия Эрба-Рота аутосомно-рецессивно, то есть патология проявляется, если ребенок получает аномальный ген от каждого из родителей. Каждый родитель может быть носителем дефектного гена, но обычно остается здоровым. Около 30% генных мутаций возникают de novo, то есть не наследуются, а появляются «ниоткуда» и далее могут передаваться потомству.
Причины появления мышечной дистрофии
Причиной развития разных миодистрофий являются патологии в генах - известно порядка 25 генов, ответственных за развитие врожденных миодистрофий.
При мышечной дистрофии Дюшенна вследствие мутации нарушается выработка белка дистрофина, который обеспечивает прочность, стабильность и функциональность мышечных волокон, и его нехватка приводит к повреждению мембран мышечных клеток (миоцитов).
Классификация заболеваний
Мышечные дистрофии могут классифицироваться в зависимости от того, какой белок подвергся мутации. Кроме того, их подразделяют по типу наследования: аутосомно-доминантные, аутосомно-рецессивные, Х-сцепленные.
Симптомы мышечной дистрофии
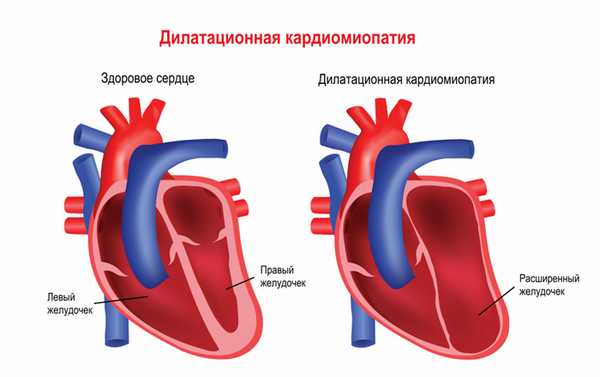
Мышечная дистрофия Дюшенна обычно манифестирует в возрасте 2-3 лет. Патологические процессы сначала происходят в мышцах ног, дети могут ходить на пальцах, вразвалку, отмечается избыточное выгибание позвоночника вперед - лордоз. Детям становится сложно бегать, прыгать, подниматься по лестнице, вставать с пола. Для разных типов миодистрофий характерным является симптом Говерса - вследствие слабости мышц бедер и тазового пояса больному, чтобы подняться из положения на корточках, приходится опираться руками об пол, затем подниматься, опираясь руками об колени. Мышечная слабость прогрессирует, у детей развивается сколиоз и сгибательные контрактуры - когда ребенок не может полностью разогнуть конечность. Дети часто падают, поэтому велик риск переломов рук или ног. Отдельные мышечные группы могут замещаться жировой или фиброзной тканью, в результате чего появляется псевдогипертрофия мышц, особенно заметная на лодыжках. Если страдает миокард (сердечная мышца), то существует предрасположенность к развитию нарушений ритма и проводимости сердца, а также дилатационной кардимиопатии (состояния, когда камеры сердца увеличены, а стенки истончены), приводящей к сердечной недостаточности.
В 20-30% случаев при мышечной дистрофии Дюшенна появляются нарушения интеллекта и памяти.
К 12 годам большинство детей вынуждено пользоваться инвалидной коляской. В возрасте 15-20 лет пациентам уже требуется респираторная поддержка, умирают больные миодистрофией Дюшенна от дыхательных или кардиальных осложнений в возрасте 12-25 лет.
Миодистрофия Беккера дебютирует в 10-20 лет и медленно прогрессирует, способность к самостоятельной ходьбе сохраняется в течение 15-20 лет от начала заболевания. Симптоматика схожа с миодистрофией Дюшенна, слабость распространяется на мышцы бедер, таза, плеч, пациенты ходят на носочках или вразвалку, также наблюдается гипертрофия мышц голеней.
Развитие заболевания очень индивидуально, некоторым пациентам требуется инвалидное кресло к 30 годам, некоторые длительное время обходятся тростью.
У пациентов с миодистрофией Беккера также отмечается поражение сердечной мышцы с развитием сердечной недостаточности.
Первые признаки миодистрофии Ландузи-Дежерина проявляются в основном в возрасте 10-20 лет. Сначала атрофия и мышечная слабость наблюдаются в плечевом поясе с поражением мышц лопаток и плеч, потом распространяются на лицо с характерной асимметричностью. Начальными проявлениями являются затруднение подъема рук над головой, выступающие «крыловидные» лопатки и сколиоз. При прогрессировании заболевания страдают лицевые мышцы, при этом пациент не может крепко зажмурить глаза и сжать губы. Позже мимика становится скудной, а речь неразборчивой. Характерными симптомами являются поперечная улыбка («улыбка Джоконды»), вывороченные губы («губы тапира»), «полированный» лоб. Иногда атрофия распространяется на мышцы ног. Другими клиническими признаками миодистрофии Ландузи-Дежерина могут быть аномалии сосудов сетчатки глаза, отек и отслойка сетчатки, снижение слуха.
Для миодистрофии Эмери-Дрейфуса характерны контрактуры локтевых и голеностопных суставов, возникающие в раннем детстве (укорочение ахилловых сухожилий приводит к тому, что ребенок не может опуститься на пятки), тугоподвижность позвоночника, медленно прогрессирующая слабость лопаточно-плечевых и тазово-перонеальных мышц (мышц бедра и голени), а также выраженная кардиомиопатия с нарушениями ритма и проводимости. Тяжесть заболеваний сердца часто определяет прогноз течения болезни вследствие высокой вероятности внезапной сердечной смерти или развития прогрессирующей сердечной недостаточности.
Миодистрофия Эрба-Рота сопровождается слабостью мышц поясничной области и конечностей. Первые признаки появляются в возрасте 10-20 лет: трудности при беге, быстрой ходьбе, прыжках, характерен симптом Говерса. Со временем начинает меняться осанка, походка, снижается тонус мышц плечевого пояса. С прогрессированием заболевания больной может полностью потерять способность ходить. Тотальная гипотрофия мышц туловища приводит к тому, что у пациента начинают выступать лопатки, талия становится очень тонкой, усиливается поясничный лордоз. Характерен симптом свободных надплечий — при попытке приподнять больного, удерживая его подмышки, плечи пациента свободно движутся вверх и голова будто бы «проваливается» между ними.
Диагностика мышечной дистрофии
Предварительный диагноз врач может установить уже при осмотре, наблюдая за попытками ребенка побежать, прыгнуть, подняться по ступенькам, встать с пола.
Для подтверждения диагноза проводятся:
- Анализ крови на сывороточную креатинкиназу, АСТ, АЛТ.
Прогрессирующая мышечная дистрофия Дюшенна
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Причины
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Миодистрофия Дюшенна: особенности болезни

Редкие генетические болезни — это несчастливый лотерейный билет, который может выпасть каждому человеку. Многие из этих недугов приводят к инвалидизации и даже преждевременной гибели пациента. Довольно редким заболеванием является прогрессирующая миодистрофия Дюшенна: 1 случай на 4000 человек. Дети с подобной патологией требуют совершенно особенного подхода со стороны врачей и родителей: им требуется не только медицинская, но и психологическая реабилитация.
Миодистрофия Дюшенна: определение болезни

Прогрессирующая миодистрофия Дюшенна — это наследственная патология, поражающая мышечную систему. Сначала страдают мышечные группы нижних конечностей и тазового пояса, но постепенно возникает обездвиженность. Развитие болезни связано с мутацией в 21-ом локусе плеча в Х-хромосоме, которая кодирует белок дистрофин. Так как представители мужского пола имеют генотип ХУ, именно они в подавляющем большинстве становятся обладателями миодистрофии. Женщины же могут являться бессимптомными носительницами мутации (около 70% случаев мышечной дистрофии Дюшенна).
Дистрофин представляет собой белок, который входит в состав клеточной мембраны мышечных клеток и обеспечивает ее устойчивость к растяжению и целостность. При нарушении синтеза дистрофина нарушается структура мышечных клеток, они разрушаются и замещаются соединительной или жировой тканью. В среднем симптомы заболевания начинают появляться в первые 4-7 лет жизни ребёнка, однако известны случаи возникновения первых признаков недуга в подростковом возрасте.
Основные симптомы заболевания
В некоторых случаях внимательные родители могут отметить отставание ребёнка в моторном развитии. Малыш позже начинает держать головку, переворачиваться, самостоятельно сидеть или стоять. При тяжелой форме недуга хождение возможно только с поддержкой взрослых. Такие дети отличаются от сверстников большей неуклюжестью: они часто падают и имеют шаткую походку.
Первые симптомы мышечной слабости чаще всего обнаруживаются на 4-5 году жизни. Изначально малыш быстро утомляется при ходьбе на большие расстояния или при подъемах по лестнице. С течением времени походка приобретает «утиный» характер: мелкие шаги с сильным прогибом корпуса. Чтобы подняться с пола, ребёнку приходиться опираться руками о собственные коленки (признак Говерса).
Атрофические изменения мускулатуры начинаются с мышечных групп бедра и тазового пояса. В результате этого формируются «осиная» талия и крыловидные выпирающие лопатки. Постепенно у маленьких пациентов исчезают мышечные рефлексы, а затем формируются мышечные контрактуры. Другие типичные симптомы при дистрофии Дюшенна — это нарушения костно-суставной системы:
- искривление позвоночника;
- деформации грудины и грудной клетки;
- плоскостопие.
Методы диагностики миодистрофии Дюшенна

Первичный диагноз болезни устанавливается на основании данных анамнеза, неврологического осмотра. Для его подтверждения требуется проведение лабораторных и инструментальных исследований:
- увеличение креатинфосфокиназы в биохимическом анализе крови (свидетельствует о повреждении мышечных клеток);
- данные электронейромиографии свидетельствуют о замедлении проведения импульса;
- биопсия мышечных волокон (определяется гибель миоцитов и замещение их соединительной тканью);
- генетическое исследование (составляется генеалогическое древо пациентов или проводится ДНК-диагностика).
Лечение миодистрофии Дюшенна
В настоящее время не разработано специфического лечения болезни. Вся терапия направлена в основном на поддержание оптимального состояния пациента, его реабилитацию и адаптацию к жизни в современном обществе. Из медикаментозных препаратов используются глюкокортикостероиды (преднизолон, Дефлазакорт). Они препятствуют ухудшению силы и снижению двигательной активности при миодистрофии. Цель стероидной терапии — отсрочить момент использования дополнительных поддерживающих методов.
При нарушении функции дыхательной мускулатуры пациенту требуется искусственная легочная вентиляция в период сна, которая осуществляется с помощью маски. При прогрессировании болезни приходится прибегнуть к трахеостомии.
Реабилитация больных
Пациентам с миодистрофией обязательно посещение лечебной физкультуры, массажа и физиотерапии. Эти методики позволяют улучшить кровообращение в конечностях и препятствуют появлению пролежней. При прогрессировании заболевания требуется применение специальных ортопедических приспособлений (корсетов, ходунков, костылей) и инвалидных колясок, которые помогают ребёнку передвигаться.
Крайне важным аспектом является психологическая реабилитация. Родителям и ребёнку рекомендуется регулярно посещать не только детского невролога и логопеда, но и психолога. Групповые занятия позволят малышу частично адаптироваться в обществе. Обучение детей с миодистрофией Дюшенна проводится как в специализированных школах, так и в домашних условиях.
Прогнозы и профилактика
Из всех известных в настоящее время форм мышечной дистрофии патология Дюшенна имеет самое неблагоприятное течение. Раннее возникновение недуга приводит к полной инвалидизации пациентов, достигших 15-летнего возраста. Средняя продолжительность жизни таких людей не превышает 25-30 лет, однако за рубежом известны случаи, когда больные доживали и до 50 лет. Летальный исход в 90% случаев связан с нарушением функции дыхательной мускулатуры и присоединением вторичных инфекций.
Специфическая профилактика направлена на своевременное выявление женщин, которые являются носительницами аномального белка, а также на предупреждение появления на свет больного малыша. Для этого в период планирования беременности проводится консультация генетика и пренатальная ДНК-диагностика.
Миодистрофия Дюшенна
Миодистрофия Дюшенна - это тяжелое генетически обусловленное заболевание, манифестирующее уже в первые пять лет жизни ребенка, проявляющееся прогрессирующей мышечной слабостью. Данная патология сопровождается достаточно быстрым усугублением симптоматики и при отсутствии лечения в короткие сроки приводит к полной обездвиженности пациента. Прогноз при этой болезни неблагоприятный. Как правило, люди с данным диагнозом не доживают до двадцати пяти лет.
Свое название миодистрофия Дюшенна получила по фамилии французского невролога, впервые описавшего ее в 1853 году. Данное заболевание встречается среди представителей мужского пола. В среднем уровень ее распространенности составляет около одного случая на четыре тысячи новорожденных мальчиков. Кроме этого, известны единичные случаи, когда патология диагностировалась и у девочек.
Симптомы миодистрофии Дюшенна достаточно специфичны. Как правило, болезнь манифестирует в возрасте от одного года до пяти лет.
Чаще всего уже на первом году жизни можно заметить некоторое отставание ребенка в моторном развитии. Такие дети позже начинают сидеть, самостоятельно вставать и ходить. При начале ходьбы отмечается большая, нежели у сверстников, неуклюжесть, неустойчивость походки.
Примерно на третьем или четвертом году жизни возникает мышечная слабость. На первых порах она проявляется быстрой утомляемостью во время ходьбы на большие расстояния или при подъеме по лестнице. Спустя некоторое время возникает характерная для данного заболевания «переваливающаяся» походка, напоминающая ходьбу утки. У ребенка возникают трудности при беге, приседании, преодолении препятствий.
Первоначально дистрофические изменения захватывают мышцы нижних конечностей. Однако патологический процесс крайне быстро распространяется на мышцы спины, плечевого пояса, верхних конечностей. В результате мышечной атрофии талия ребенка становится узкой, лопатки располагаются под заметным углом к поверхности ребер.
Характерным симптомом является увеличением в объеме голеней (ложная гипертрофия икроножных мышц). Сухожильные рефлексы выпадают, сухожилья укорачиваются, пассивные движения в конечностях ограничиваются.
В результате прогрессирования данной болезни присоединяются различные изменения со стороны костно-суставной системы. Они могут быть представлены нарушением конфигурации позвоночного столба в виде его искривления в передне-задней или боковой плоскости, выпячиванием или западением грудины, деформацией стоп.
Как правило, возникают расстройства со стороны сердечно-сосудистой системы. В 2016 году ученые из Самарского государственного медицинского университета опубликовали результаты работы, в которой было установлено, что для миодистрофии Дюшенна характерны кардиологические изменения в виде нарушения ритма и проводимости сердца, нарушения процессов реполяризации в миокарде, что может быть расценено как ранние проявления кардиомиопатии.
Примерно у половины пациентов наблюдаются нейроэндокринные нарушения, представленные, например, неправильной работой гипоталамо-гипофизарной системы. У трети больных детей выявляется отставание в интеллектуальном развитии. Кроме этого, могут присоединяться расстройства аутистического спектра, проблемы с памятью и так далее.
Примерно к семилетнему возрасту у ребенка обнаруживаются выраженные двигательные ограничения. В среднем уже к двенадцати годам пациент утрачивает возможность ходить, а к пятнадцатилетнему возрасту - полностью двигаться.
В результате того, что дистрофические процессы распространяются и на дыхательную мускулатуру, возникают проблемы с процессом дыхания, в легкие начинает поступать меньшее количество кислорода.
Причины
Причины миодистрофии Дюшенна кроются в генетических нарушениях. В основе развития данного заболевания лежит мутация в гене, находящемся в Х-хромосоме 23 пары, а именно в двадцать первом локусе ее короткого плеча. Этот ген кодирует структурный белок дистрофин. При отсутствии данного белка клеточная мембрана мышечной клетки повреждается, сами мышечные клетки разрушаются, замещаются жировым и соединительнотканным компонентами. Все перечисленное приводит к постепенно усугубляющемуся снижению сократительной способности мышц, утрате их тонуса и силы, атрофическим изменениям в мышечных волокнах.
Тип наследования при этой болезни - Х-сцепленный. Примерно в семидесяти процентах случаев болезнь передается от матери, являющейся носительницей дефектного гена. Остальные тридцать процентов - это новые мутации в женских половых клетках.
Методы диагностики
Диагностика миодистрофии Дюшенна начинается со сбора жалоб, анамнеза, неврологического осмотра.
При объективном обследовании выявляются:
- изменения в походке по типу «утиной»;
- псевдогипертрофия икроножных мышц;
- снижение мышечного тонуса и силы;
- снижение или утрата сухожильных рефлексов: на начальных стадиях - выпадение коленных при сохранении ахилловых и рефлексов с верхних конечностей, на поздних стадиях - арефлексия верхних и нижних конечностей;
- изменения со стороны костно-суставной системы и ряд других отклонений.
Из лабораторных исследований ведущая роль отводится биохимическому анализу крови. В 2020 году ученые из Казанского государственного медицинского университета опубликовали результаты работы, в которой был сделан вывод о том, что для миодистрофии Дюшенна характерны повышение активности аланинаминотрансферазы, аспартатаминотрансферазы, креатинфосфокиназы, её МВ-изоформы (специфического маркера некроза миокарда).
Из инструментальных исследований проводятся:
Выявляются уменьшение величины средней длительности потенциалов двигательных единиц, снижение амплитуды отдельных потенциалов двигательных единиц, снижение амплитуды М-ответа.
- Ультразвуковое обследование мышечных волокон.
Обнаруживаются участки замещения мышечной ткани жировым и соединительнотканным компонентами;
Характерны различные изменения со стороны функциональной активности сердечно-сосудистой системы.
Дополнительно проводится ДНК диагностика, позволяющая выявить мутации в гене дистрофина. Однако стоит заметить, что если мутация не обнаружена, однако присутствуют клинические и лабораторные признаки миодистрофии Дюшенна, это не является поводом для исключения диагноза. В таких ситуациях назначается биопсия мышц с последующим направлением полученного материала на морфологическое и иммунохимическое исследование.
Лечение
Лечение миодистрофии Дюшенна носит симптоматический характер. Этиотропная терапия на сегодняшний день не разработана.
Замедлить прогрессирование патологических изменений в мышцах можно с помощью глюкокортикостероидов. Дополнительно показаны витамины, в особенности группы В, препараты кальция, анаболические стероиды, ингибиторы холинэстеразы для облегчения нервно-мышечной передачи.
Для того чтобы продлить двигательную активность ребенка, назначаются лечебная гимнастика, массаж, различные физиотерапевтические процедуры. В 2014 году ученые из Первого Московского государственного медицинского университета им. И.М.Сеченова опубликовали результаты работы, в которой было установлено, что лечебная гимнастика, пассивные и активные растяжки не могут привести к излечению прогрессирующей миодистрофии Дюшенна, но способствуют улучшению качества жизни пациентов, страдающих от этого заболевания, уменьшению частоты развития осложнений.
Осложнения
Осложнения миодистрофии Дюшенна могут быть следующие:
- остеопороз;
- искривление позвоночника;
- частые респираторные инфекции;
- развитие дыхательной недостаточности;
- нарушения со стороны сердца и ряж других.
Профилактика
Профилактика миодистрофии Дюшенна сводится к медико-генетическому консультированию пар на этапе планирования беременности, пренатальной ДНК-диагностике.
Какие вопросы следует задать врачу
Что такое миодистрофия Дюшенна?
Какими симптомами проявляется данное заболевание?
Почему развивается миодистрофия Дюшенна?
Как ее диагностировать?
Можно ли вылечить миодистрофию Дюшенна?
Как предупредить ее развитие?
Советы пациенту
Парам, планирующим завести ребенка, настоятельно рекомендуется обращаться за консультацией к врачу-генетику. В ходе генетического обследования удается выявить риски развития у будущего малыша не только миодистрофии Дюшенна, но и ряда других генетических заболеваний.
Прогрессирующая мышечная дистрофия Дюшенна/Беккера (ПМДД), делеции и дупликации гена DMD
Синонимы: Анализ крови на делеции и дупликации гена DMD при прогрессирующей мышечной дистрофии Дюшенна/Беккера.
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD); Deletions and duplications in DMD gene; Duchenne and Becker muscular dystrophies (DMD/BMD), deletions/duplications in DMD gene.
Краткое описание исследования «Прогрессирующая мышечная дистрофия Дюшенна/Беккера (ПМДД), делеции и дупликации гена DMD»
Нейромышечное заболевание, обусловленное мутацией в гене дистрофина и приводящее к прогрессирующей дегенерации мышечных волокон.
Основная функция дистрофина заключается в обеспечении устойчивости и эластичности мышечного волокна при последующих мышечных сокращениях. При отсутствии дистрофина вследствие мутации мембрана разрушается, в ней появляются участки некроза, что приводит к вымыванию содержимого саркоплазмы в кровяное русло. Происходит постепенная гибель мышечных волокон и замещение их соединительнотканными структурами, которые увеличивают плотность и объем мышц, вызывая феномен псевдогипертрофии. Заболевание встречается в двух клинических формах, являющихся аллельными генетическими вариантами.
Заболевание проявляется в возрасте 1-5 лет, быстро прогрессирует и приводит к летальному исходу до 25-летнего возраста. Для большинства больных характерна задержка темпов раннего моторного развития. При начале самостоятельной ходьбы, в возрасте старше 14 месяцев, отмечаются частые падения, спотыкания, моторная неловкость, быстрая утомляемость. Постепенно походка становится переваливающейся, возникают затруднения при подъеме по лестнице и из положения на корточках, когда больные вынуждены использовать вспомогательные приемы Говерса («взбирание по самому себе»). На ранних стадиях заболевания обнаруживаются псевдогипертрофии мышц, возникающие за счет разрастания соединительной и жировой ткани на месте гибнущих мышечных волокон. Наиболее часто они локализуются в икроножных, дельтовидных, четырехглавых и трехглавых мышцах и создают ложное впечатление атлетического телосложения больного. По мере прогрессирования заболевания псевдогипертрофии мышц трансформируются в их гипотрофии. Распространение патологического процесса имеет восходящий характер. Первыми поражаются мышцы тазового пояса и проксимальных отделов нижних конечностей, затем мышцы плечевого пояса, спины и проксимальных отделов верхних конечностей. Уже на ранних стадиях болезни снижаются или угасают коленные рефлексы. Ахиллов рефлекс, а также сухожильные рефлексы с рук могут длительное время оставаться сохранными. По мере развития патологического процесса в мышцах возникают вторичные деформации позвоночника (усиление лордоза и кифоза, сколиоз), грудной клетки (по типу седловидной и килевидной) и стоп, а также ретракции сухожилий с развитием контрактур в суставах. Характерным признаком является кардиомиопатия, которая проявляется симптомами гипертрофии левого желудочка и аритмией. У 25-30% больных диагностируется олигофрения в степени дебильности. Пациенты сохраняют способность к самостоятельной ходьбе до 10-12-летнего возраста, после чего пользуются инвалидной коляской. Смерть больных наступает от сердечной недостаточности или от интеркуррентных инфекций.
Наиболее часто заболевание возникает в возрастном интервале от 10 до 20 лет с появления слабости и утомляемости мышц тазового пояса и ног. Ранними симптомами у значительного числа больных бывают болезненные мышечные крампи. Клинические проявления сходны с таковыми при ПМДД, однако имеют значительно меньшую степень выраженности. Характерной особенностью ПМДБ является вовлечение в патологический процесс миокарда. Гипертрофическая или дилатационная кардиомиопатия диагностируется у 50-60% больных. В 40-50% случаев выявляются гипогенитализм и атрофия яичек. Интеллект, как правило, не страдает. Заболевание прогрессирует достаточно медленно и в большинстве случаев приводит к инвалидизации больного не ранее 40-летнего возраста.
Описаны клинические проявления ПМДД у женщин, которые являются носительницами мутации в гене дистрофина в гетерозиготном состоянии. Клинические признаки могут появиться в различные возрастные периоды, но чаще провоцируются гормональными перестройками в организме женщины (начало менструаций, беременность, климакс). Появление клинических симптомов может быть обусловлено двумя причинами: 1) наличие полной или мозаичной форм синдрома Шерешевского-Тернера; 2) феноменом несбалансированной лайонизации. На электромиограмме выявляются признаки первично-мышечного поражения в виде усиления интерференции и снижения амплитуды М-ответа. Высокую диагностическую значимость имеет определение активности фермента креатинфосфокиназы в плазме крови больного. Этот показатель у больных ПМДД в 50-100 раз превышает норму и может быть выявлен до возникновения выраженных клинических признаков. Для диагностики и дифференциальной диагностики ПМДД/ПМДБ используются иммуногистохимические методы анализа дистрофина в биоптате мышечного волокна. При использовании антисывороток на различные районы дистрофина при ПМДД иммунореактивных форм белка, как правило, не выявляется. У больных с ПМДБ наблюдается прерывистое окрашивание мышц при иммунохимическом анализе, что свидетельствует об относительной сохранности отдельных структур цитоскелета. Специфического морфологического дефекта не существует. В биоптате мышц больных выявляются изменения, характерные для группы прогрессирующих мышечных дистрофий в целом.
Мышечная дистрофия Дюшенна (МДД): 1:2500-4000 новорожденных мальчиков. Частота МДБ (Беккера) составляет 1 на 20000 мальчиков.
Х-сцепленный рецессивный, т. е. им страдают почти исключительно мальчики, женщины же с поврежденным геном в одной из Х-хромосом являются носительницами МДД. Но в редких случаях миодистрофией Дюшенна могут болеть и девочки. Причинами этого могут быть преимущественная инактивация Х-хромосомы с нормальным аллелем у гетерозиготных носительниц мутантного гена дистрофина, Х-аутосомная транслокация, затрагивающая этот ген, гемизиготность по мутантному аллелю и наличие фенокопий (заболеваний, связанных с нарушением других белков, входящих в дистрофин-гликопротеиновый комплекс). Приблизительно в 2/3 случаев сын получает хромосому с повреждением от матери-носительницы, в остальных случаях заболевание возникает в результате мутации de novo в половых клетках матери или отца, либо в предшественниках этих клеток. Приблизительно 30% всех случаев заболевания связаны с возникновением свежих мутаций в гене дистрофина, а остальные 70% обусловлены носительством матерью пробанда патологической мутации в одной из Х хромосом. Считается, что 6-7% всех спорадических случаев заболевания являются следствием гонадного мозаицизма - существования в яичниках женщины нескольких генераций ооцитов с нормальными и мутантными аллелями гена дистрофина.
DMD (DYSTROPHIN) - ген дистрофина, находится в Х-хромосоме в регионе Хр21.2-р21.1, состоит из 79 экзонов. У 60%-70% больных выявляют крупные делеции, захватывающие один или несколько экзонов гена и локализованные в двух «горячих» регионах - в области 5' конца (экзоны 6-19) и 3' конца (экзоны 40-43). У 5% больных обнаруживаются дупликации, в остальных случаях - точковые мутации. Различия в тяжести клинических проявлений при двух аллельных вариантах заболевания связывают с различиями в характере мутации в гене дистрофина. При мышечной дистрофии Дюшенна мутации в гене дистрофина приводят к сдвигу рамки считывания и преждевременной терминации трансляции, при этом синтез белка прекращается. При мышечной дистрофии Беккера структурные перестройки гена не приводят к сдвигу рамки считывания, ДНК-полимераза может «перескакивать» делетированные экзоны, что приводит к синтезу внутренне усеченного белка, который может до некоторой степени выполнять свои функции.
С какой целью выполняют анализ «Прогрессирующая мышечная дистрофия Дюшенна/Беккера (ПМДД), делеции и дупликации гена DMD»
Генетическое исследование направлено на поиск мутаций в гене DMD (поиск делеции и дупликации) с целью диагностики мышечной дистрофии Дюшенна/Беккера - нейромышечного заболевания, обусловленного мутацией в гене дистрофина и приводящего к прогрессирующей дегенерации мышечных волокон.
Читайте также:
- Связки и мениски коленного сустава. Функции связочного аппарата колена
- Уход после артроскопической операции по поводу кальцифицирующего тендинита плечевого сустава
- Доступ и ход операции панкреатикодуоденэктомии по Уипплу
- Путь звуковой волны. Афферентная иннервация улитки.
- Введение в применение и кинетику лекарственных препаратов