Диагностика синдрома Кернса-Сейра по КТ, МРТ головного мозга
Добавил пользователь Дмитрий К. Обновлено: 22.01.2026
Синдро́м Ке́рнса — Се́йра (англ. Kearns-Sayre syndrome , сокращённо KSS) — митохондриальная миопатия с типичным началом до 20-летнего возраста. KSS является более серьезным синдромным вариантом хронической прогрессирующей внешней офтальмоплегии (сокращенно CPEO), синдром, который характеризуется изолированным поражением мышц, контролирующих движения век (поднимающая верхнее веко, круговая мышца глаза) и контролирующих движения глаз (экстраокулярных мышц). Это приводит к птозу и офтальмоплегии соответственно. KSS включает в себя триаду уже описанных: CPEO, двустороннюю пигментную ретинопатию и блокаду сердца. Другие области участия может включать в себя мозжечковую атаксию, проксимальную мышечную слабость, глухоту, сахарный диабет, дефицит гормона роста, гипопаратиреоз или другие эндокринные нарушения. В обоих этих заболеваниях, вовлечение мышц может начаться односторонним, но всегда развивается в двусторонний дефицит, и, конечно является прогрессирующим.
Содержание
История
Это триада CPEO, двусторонняя пигментная ретинопатия и блокада сердца была впервые описана в докладе о случае у двух пациентов в 1958 году доктором медицинских наук Томасом П. Кернс, (англ. Thomas P. Kearns ) и доктором медицинских наук Джорджем Помероя Сейр (англ. George Pomeroy Sayre ). Второй случай был опубликован в 1960 году Ягером и соавторами, которые сообщили об этих симптомах у 13-летнего мальчика.Предыдущие случаи внезапной смерти пациентов с CPEO были опубликованы, как от сердечной аритмии. Другие случаи отмечали особую пигментацию сетчатки, но ни одна из этих публикаций не была документирована как три патологии, возникающие вместе в качестве генетического синдрома. Кернс опубликовал определяющий случай в 1965 году, описывающий 9 несвязанных случаев этой триады. В 1988, была впервые выявлена связь между KSS и крупномасштабными удалениями мышечной митохондриальной ДНК (сокращенно мтДНК) После этого открытия, многочисленные делеции в митохондриальной ДНК были связаны с развитием КСС.
Этиология
Признаки и симптомы
Лица с KSS предстают первоначально со сходными симптомами с типичной CPEO. Начало в первой и второй декадах жизни.
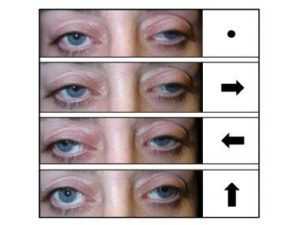
Первым симптомом этого заболевания является односторонний птоз, или проблемы при открытии век, который постепенно прогрессирует и приводит к двустороннему птозу. Когда птоз усиливается, пострадавший обычно запрокидывает шею, поднимая подбородок в попытке предотвратить окклюзию зрительной оси опустившимися веками. Наряду с коварным развитием птоза, движения глаз в конечном итоге становятся ограниченными, в результате чего, лицо больше полагается на поворот головы из стороны в сторону или вверх и вниз для просмотра объектов в периферическом поле зрения.
KSS приводит к пигментации сетчатки, прежде всего, в задней части глазного дна. Наблюдается диффузная депигментация пигментного эпителия сетчатки с наибольшим эффектом в жёлтом пятне. Этим KSS отличается от пигментного ретинита, где пигментируется периферия. Вид сетчатки при KSS аналогичен тому, которое наблюдалось при миотонической дистрофии типа 1 (сокращенно DM1). Умеренная куриная слепота может наблюдаться у пациентов с KSS. Потеря остроты зрения, как правило, мягкая и происходит только у 40-50 % больных.
Нарушения сердечной проводимости
Это чаще всего происходит после образования птоза и офтальмоплегии.Атриовентрикулярная блокада (сокращенно «AV») является наиболее распространенным дефицитом сердечной проводимости. Это часто прогрессирует до третьей степени желудочковой блокады, которая является полным блокированием проводимости от предсердия в желудочек. Симптомы сердечной блокады включают обморок, непереносимость физических нагрузок и брадикардию
Церебральная фолатная недостаточность
У пациентов с синдромом Кернса — Сейра очень часто обнаруживается церебральная фолатная недостаточность - синдром, при котором уровни 5-MTHF в спинномозговой жидкости снижены, несмотря на нормальные уровни фолиевой кислоты и 5-MTHF в плазме крови. Назначение фолиниевой кислоты может в некоторых случаях облегчить симптомы недостаточности и даже скорректировать наблюдаемые на снимках мозга отклонения, особенно если терапия была начата на ранних стадиях заболевания. Предполагаемая причина церебральной фолатной недостаточности у пациентов с синдрмом Кернса-Сейра - дисфункция сосудистого сплетения, нарушающая поступление фолатов в спинномозговую жидкость.
Согласно описанию заболевания, представленному Кернс в 1965 году, а также описаниям в более поздних публикациях, некоторые симптомы возникают не у всех пациентов. Среди этих симптомов - слабость мышц лица, глотки, туловища и мышц конечностей, потеря слуха, небольшой рост, электроэнцефалографические изменения, мозжечковая атаксия и повышенный уровень белка в спинномозговой жидкости.
Генетика
KSS является результатом делеций в митохондриальной ДНК (мтДНК), которые вызывают определённый фенотип. мтДНК передается исключительно от яйцеклетки матери. Митохондриальная ДНК состоит из 37 генов, найденных в одной кольцевой хромосоме размерностью 16569 спаренных оснований в длину. Среди них 13 генов кодируют белки дыхательной цепи переноса электронов(сокращенно «ЭTЦ»), 22 кодируют транспортировку РНК (тРНК) и два кодируют ряд больших и малых субъединиц, которые образуют рибосомные РНК (рРНК). 13 белков, участвующие в ЭТЦ в митохондриях, необходимы для окислительного фосфорилирования. Мутации в этих белках приводит к нарушениям производства энергии в митохондриях. Этот дефицит клеточной энергии быстрее всего проявляется в тканях, которые сильно зависят от аэробного метаболизма, таких как мозг, скелетные и сердечные мышцы, органы чувств и почки. Это лишь один фактор, участвующий в представлении митохондриальных заболеваний.
Есть и другие факторы, влияющие на проявление митохондриальной болезни, кроме величины и расположения мутации. Митохондрии дублируются посредством деления клеток во время беременности и в течение всей жизни. Поскольку мутация митохондриальной болезни чаще всего встречается на ранних сроках беременности при этих заболеваниях, только митохондрии в мутированной линии являются дефектными. Это приводит к неравномерному распределению дисфункциональных митохондрий в каждой клетке, и в различных тканях тела. Это называется гетероплазмия, которая характерна для митохондриальных заболеваний, включая KSS. Распределение мутантных мтДНК в каждой клетке, ткани и органе, зависит от того, когда и где произошла мутация. Это может объяснить, почему два пациента с одинаковыми мутациями в мтДНК могут представлять совершенно разные фенотипы и, в свою очередь различные синдромы, Публикация в 1992 году Fischel-Ghodsian и др. определила удаление одного и того же 4977-б.п. в мтДНК у двух пациентов с двумя совершенно различными заболеваниями. Один из пациентов имел характерный KSS, а другой пациент — совсем другое заболевание, известное как синдром Пирсона. Усложняет дело то обстоятельство, что в некоторых случаях синдром Пирсона, как было выявлено, может прогрессировать внутри KSS позднее в жизни. Более поздние исследования привели к выводу, что дублирование мтДНК может также играть важную роль в определении присутствия фенотипа. Дублирование мтДНК, кажется, характерно для всех случаев KSS и синдрома Пирсона, в то время как они отсутствуют в CPEO.
Удаления мтДНК в KSS различаются по размеру (1.3-8kb), а также положению в митохондриальном геноме. Наиболее распространенным удалением является 4.9kb и простирается от позиции 8469 до позиции 13147 в геноме. Это удаление присутствует примерно у ⅓ людей с KSS.
Диагностика
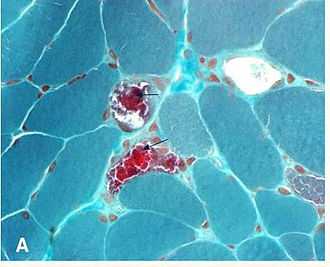
Нейроофтальмологи, как правило, участвуют в диагностике и лечении KSS. Человек должен быть заподозрен в КСС на основе клинических данных экспертизы. Подозрение на миопатию должно быть усилено для пациентов с отсутствием офтальмоплегии и определенного набора параличей черепных нервов (паралич глазодвигательного нерва, паралич блокового нерва, паралич отводящего нерва). Первоначально исследования изображения часто проводится, чтобы исключить более общие патологии. Диагноз может быть подтвержден мышечной биопсией, которая может быть дополнена PCR для определения мутаций мтДНК. Биопсия: Это не обязательно биопсия глазной мышцы, для демонстрации гистопатологические аномалий. Сечение мышечных волокон, трихромное окрашение пятна Гомори рассматривается с помощью световой микроскопии. В мышечных волокнах, с более высоким содержанием мутированных митохондрий, существует более высокая концентрация митохондрий. Это придаёт этим волокнам темно-красный цвет, в результате чего общий вид биопсии описывается как «рваные красные волокна». Аномалии могут быть также продемонстрированы в образцах биопсии мышц с использованием других гистохимических исследований, таких как пятен митохондриальных ферментов, с помощью электронной микроскопии, биохимических анализов мышечной ткани (то есть активность ферментной цепи переноса электронов) и с помощью анализа мышечной митохондриальной ДНК.
Лабораторные исследования
Уровень молочной кислоты и пировиноградной кислоты крови, как правило, повышается в результате увеличения анаэробного метаболизма и уменьшением соотношения АТФ/АДФ. При исследовании СМЖ обнаруживается повышенный уровень белка, как правило, > 100 мг/дл, а также повышенный уровень молочной кислоты.
Ведение и лечение
Генетика, симптомы, диагностика и лечение синдрома Кернса-Сейра
Болезни, обусловленные мутациями в митохондриальной ДНК, проявляются не при рождении, а во второй, реже - в первой декаде жизни.
В ряду генетических заболеваний числится Синдром Кернса-Сейра (Kearns-Sayre syndrome, KSS) — митохондриальная миопатия, поражающая глаза и мышцы пациентов. Неоднозначная динамика симптомов, их не одновременное проявление, а также отказ от биопсии и детальных генетических исследований осложняет точную постановку диагноза.
История появления диагноза
В 60-е годы доктора медицины Томас Кернс и Джозеф Сейр обратили внимание на три симптома, встречающиеся одновременно (нарушения зрения, пигментацию сетчатки глаза, нарушения проводимости нервных импульсов в сердце), описали несколько клинических случаев протекания болезни. Только в конце 80-х годов, после проведения исследований в области изменений в митохондриальных структурах стало понятно, как связанны между собой различные нарушения здоровья у одного пациента.
Синдром получил название по именам специалистов, впервые описавших характерную клиническую картину.
Дальнейшее изучение KSS показало, что симптоматика синдрома может быть более вариабельной (мышечная недостаточность в конечностях, слабоумие, нарушения в других сенсорных системах, общая задержка развития, низкий рост), но речь все равно идет о системном заболевании из-за нарушений в ДНК митохондрий.

Патогенез и общий характер симптоматики
Среди зафиксированных случаев (всего около 300 детально описанных), только в нескольких можно говорить о семейной наследственности (у родителей зафиксированы те же симптомы, что и у детей). Остальные — мутации на ранних сроках беременности, подлежащие диагностированию после 4 года жизни ребенка.
Симптомы при синдроме Кернса Сейра развиваются по нарастающей:
- уменьшение двигательной активности;
- значительное ухудшение четкости зрения;
- птоз одного или двух век;
- минимизация движений глазным яблоком до полного их отсутствия;
- брадикардия, аритмия;
- нарушения слуха, координации движений;
- проблемы с глотанием пищи и слюны (реже);
- атрофия мышц конечностей;
- прекращение работы одного из желудочков сердца и смерть.
Этиология болезни такова: «испорченный» геном митохондрий продолжает копироваться в искаженной форме при каждом делении клетки. Поскольку среди его главных функций - нормальный энергетический обмен, то начинают страдать те органы, где затраты энергии максимальны (нервные волокна, сенсорная система, мышцы). При отсутствии симптоматического лечения митохондриальной миопатии наступает смерть (средние показатели — до 24-36 лет).
Примечательно, что параллельно с «испорченными» клетками развиваются и работают те, у которых митохондрии в норме.
Генетический аспект
Митохондриальные проблемы передаются по материнской линии при значительном количестве измененных генов в органелле. На практике это означает, что у здоровой по виду женщины-носителя могут родиться и здоровые, и больные дети.
Наука пока не выявила точной закономерности развития синдрома. Понятно, что важным является первый триместр беременности, когда происходит активное деление клеток и закладка систем органов. Какой именно фактор влияет на более активное деление дефектных клеток, чем здоровых, пока не выявлено.
Синдром Кернса-Сейра встречается одинаково часто у мужчин и женщин разных рас.
Характерный клинический случай
Синдром Кернса-Сейра может иметь разные первоначальные проявления и дальнейшую клиническую картину. К примеру, у 25-летнего мужчины с выраженным птозом обеих век, атрофией мышц конечностей и спины, отсутствием движений глазными яблоками, пониженным зрением, болезнь проявилась в младшем школьном возрасте.
При физических нагрузках (бег) появлялись сильные головокружения, рвота, боль в животе. В 12 лет специалисты заметили птоз левого века, через 5 лет такая же участь постигла правое веко. При этом отмечалось двоение в глазах.
На фоне симптоматического лечения опущение уменьшилось, но препараты помогали все меньше, что сочли последствием привыкания. Для предотвращения полной слепоты рекомендовали укорочение век. С 23-летнего возраста начала развиваться тугоухость, прогрессирует общая мышечная слабость.
Нарушений в интеллектуальном развитии, работе сердца не обнаружено. Постоянное наблюдение у кардиолога и невропатолога дает возможность стабилизировать состояние, снизить интенсивность прогрессирования болезни.
Симптомы, характерные для синдрома Кернса-Сейра
Митохондриальная миопатия может развиваться и другим путем, когда при раннем выявлении болезни прослеживается быстрое ухудшение состояния без отзыва организма на лечение. Например, родители 5-летнего мальчика обратились к врачам с жалобами на одновременный птоз обеих век, нарушения движений глазными яблоками. При дальнейшем наблюдении выявили также нарушения сердечного ритма, психофизическое отставание в развитии, торможение полового созревания. За 6 лет наблюдений синдром прогрессировал до:
- координационных нарушений;
- затрудненного жевания и постоянного поперхивания;
- атрофии мимических мышц;
- атриовентрикулярной блокады;
- наджелудочковых экстрасистол.
Главным достижением лечения в данном случае стала временная нормализация сердечного ритма.
Динамика синдрома индивидуальна, зависит от количественных изменений в митохондриях, систематически принимаемых препаратов, а также от сопутствующих болезней.
Диагностический подход
Точное диагностирование заболевания возможно только при проведении биопсии мышц на предмет выявления патологии. Косвенными показателями болезни являются:
- совокупность симптомов и их динамика;
- завышенные уровни пировиноградной и молочной кислот в крови пациента;
- отсутствие отклика на прозерин, калимин;
- данные электрокардиографии;
- исследования на возможные нарушения в работе эндокринной системы (выявлены у 3% пациентов, существенно усугубляют протекание болезни).
Выявление болезни обычно занимает длительный промежуток времени ввиду медленного нарастания симптомов, неготовности родителей к проведению биопсии и детального генетического анализа.
Возможности современной медицины
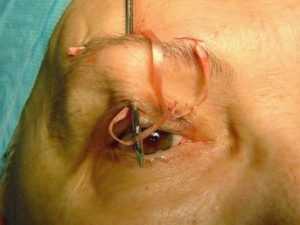
Оперативное подрезание век
Синдром Кернса Сейра не поддается полному лечению на данном этапе развития медицины. Предполагается, что возможно купирование всех симптомов при стимуляции к делению здоровых клеток, постепенному замещению ими больных волокон.
Механизм подобной операции не разработан.
Экспериментальные операции показали, что спутниковые клетки могут способствовать регенерации мышечных волокон.
Пациент может рассчитывать на:
- вживление кардиостимулятора для предотвращения остановки сердца;
- введение коферментов для понижения уровня пировиноградной и молочной кислот (наблюдается улучшение подвижности глаза);
- укорочение век (временно улучшает зрение);
- использование слухового аппарата;
- нормализацию уровня гормонов.
Все названые меры снижают дискомфорт в быту, но не предотвращают полностью дальнейшее развитие мышечной слабости.
Последствия и прогноз
Митохондриальная миопатия имеет не оптимистический прогноз: прогресс мышечной слабости до остановки сердца или дыхания. При надлежащем лечении возможно существенное увеличение продолжительности жизни, но с признанной инвалидностью (2-3 группа).
В любом случае, речь идет о пожизненном наблюдении у кардиолога и невропатолога с постоянной корректировкой препаратов, их доз в зависимости от показателей анализов крови.
Профилактических методов нет, ввиду незначительной изученности пусковых механизмов.
Действенным методом уменьшения негативного влияния синдрома на качество жизни станет своевременное обращение к врачу, прохождение всего комплекса обследований и начало симптоматического лечения.
Перед запланированной беременностью родителям стоит пройти тест на определение возможных генетических отклонений у общего ребенка.
XIV Международная студенческая научная конференция Студенческий научный форум - 2022

ДЕФЕКТЫ мтДНК: KSS-СИНДРОМ КЕРНСА-СЕЙРА, СИНДРОМ ПИРСОНА, СИНДРОМЫ MELAS, MERRF, LHON, NARP, ПРОГРЕССИРУЮЩАЯ НАРУЖНАЯ ОФТАЛЬМОПЛЕГИЯ
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке "Файлы работы" в формате PDF
Понятие «митохондриальная патология» сформировалось в медицине к концу двадцатого века благодаря выявленным в клинической генетике мутациям генов, ответственным за синтез митохондриальных белков. Митохондриальные болезни — наследственные заболевания, связанные с нарушениями работы органелл эукариотической клетки — митохондрий. В митохондриях происходит синтез 95% клеточной энергии (в виде молекул аденозинтрифосфорной кислоты или АТФ), которая расходуется на поддержание жизнедеятельности организма. Если структура митохондрий, в особенности митохондриальной ДНК (мтДНК), нарушается, в клетке вырабатывается меньше энергии, что приводит к нарушению функции клетки, а затем это нарушение распространяется на ткани, органы, системы органов и организм. При митохондриальных заболеваниях страдают больше всего клетки головного мозга, скелетной мускулатуры, сердца, печени, почек, эндокринной и дыхательной систем.
Патологии, вызванные дефектами мтДНК, встречаются с каждым десятилетием всё чаще. Однако эффективных методов лечения данных заболеваний до сих пор не выявлено. За последние годы появилось много научных работ, посвященных нарушениям энергетического обмена в митохондриях, экспериментальных работ по созданию эффективной терапии митохондриальной патологии, что убеждает в актуальности теоретического исследования дефектов мтДНК [2,5].
KSS -синдром Кернса-Сейра, синдром Пирсона, MELAS , MERRF , LHON , NARP , прогрессирующая наружная офтальмоплегия относятся к редким наследственным заболеваниям, возникающим вследствие точечных мутаций участков мтДНК. Синдром Пирсона, MELAS , MERRF , LHON , NARP , прогрессирующая наружная офтальмоплегия передаются только по материнской линии, так как отцовские митохондрии не попадают в яйцеклетку при зачатии.
Синдром Кернса - Сейра (сокращённо KSS) возникает вследствие делеции участка мтДНК. Для заболевания характерна триада симптомов: наружная офтальмоплегия (слабость глазодвигательных мышц с опущением век), пигментная ретинопатия (разрушение сетчатки) и нарушения сердечной проводимости. Распространенность синдрома составляет 1-3 случая на 100 000 человек. В большинстве случаев синдром Кернса - Сейра приобретенный. Это означает, что мутации не передаются от родителей, а возникают после зачатия. Делеции мтДНК при данной патологии приводят к нарушению процесса окислительного фосфорилирования, в результате чего страдают энергозависимые органы. Начало заболевания приходится на первое - второе десятилетие жизни.Для постановки диагноза следует обратиться к генетику, неврологу, кардиологу, офтальмологу, ЛОРу, эндокринологу, гастрологу. Проводят обследование зрения, ЭКГ, УЗИ сердца, анализы крови и спинномозговой жидкости, биопсию мышц, КТ или МРТ головного мозга, генетическое тестирование [7].
Синдром Пирсона возникает вследствие дупликации или делеции мтДНК. Из-за дефектов мтДНК возникают нарушения кроветворения - сидеробластная анемия в течение первых месяцев жизни пациента, недостаточность функций поджелудочной железы - хронический понос, частые срыгивания, интенсивные колики, периодические приступы рвоты. Частота встречаемости заболевания ‒ 1:5000-1:10000 новорожденных. Патология связана с мутацией в 3p21 ген LAMB2. Этот ген ответственен за кодирование β2-цепь ламинина. Эта субъединица ламинина является важным компонентом клубочковых и базальных мембран сетчатки и нервно-мышечных соединений. Ламинины регулируют пролиферацию и дифференциацию клеток, прилегающих к базальной мембране. Симптомы, характерные для данного синдрома, начинают беспокоить в течение первых 3 месяцев жизни. Для верификации диагноза синдрома Пирсона назначаются следующие лабораторно-инструментальные исследования: гематологические анализы, биохимическое исследование, биопсия костного мозга, молекулярно-генетические исследования [1].
В настоящее время известно не менее 23 точечных мутаций и одна делеция в генах MTTL1, MTTQ, MTTH, MTTK, MTTS1, MTND1, MTND5, MTND6, MTTS2, которые вызывают синдром MELAS. Наиболее распространены у пациентов мутации в гене переноса РНК — MTTL1. В 80 % всех случаев удается обнаружить мутацию 3243A>G. Она приводит к нарушению митохондриальной трансляции и синтеза белка, в том числе субъединиц комплексов митохондриальной электрон-транспортной цепи. В результате нарушается выделение митохондриальной энергии. Отсутствие достаточного количества энергии провоцирует дисфункцию органов, в клетках которых находятся мутированные митохондрии, и стимулирует пролиферацию митохондрий в гладких мышцах и эндотелиальных клетках мелких кровеносных сосудов, что приводит к их поражению. Синдром MELAS характеризуется мигренью и мышечной слабостью, инсультоподобными приступами, инфарктами головного мозга. Из-за накопления молочной кислоты наблюдается рвота, спазмы, затруднение дыхания, нарушение координации, потеря слуха. Частота встречаемости заболевания - от 1:15 000 до 1:20 000 человек. Возраст дебюта синдрома MELAS 5-20 лет. В биохимическом анализе крови определяется высокий уровень молочной кислоты, глюкозы и гликированного гемоглобина, уменьшение концентрации кальция. При анализе крови на гормоны обнаруживают снижение содержания соматотропного и паратиреоидного гормонов. В общем анализе мочи часто выявляется протеинурия. Так же производится: МРТ головного мозга, МР-спектроскопия, ЭЭГ, ЭНМГ, биопсия мышц, исследование ДНК [8].
Возникновение синдрома MERRF вызвано изменениями нуклеотидной последовательности мтДНК. Наиболее часто встречается замена аденина на гуанин в положении 8344 транспортной РНК (тРНК) лизина или замена тимина на цитозин в положении 8356. Мутации затрагивают ген лизиновой тРНК, что приводит к нарушению митохондриального синтеза обогащенных лизином белков вследствие преждевременного обрыва трансляции и развитию выраженного дефекта дыхательной цепи митохондрий с нарушением окислительного фосфорилирования. Ранними признаками синдрома являются снижение толерантности к физической нагрузке, развитие болей в икроножных мышцах, снижение памяти и внимания. Типичные клинические проявления развернутой стадии заболевания - миопатия, миоклонус эпилепсия в сочетании с мозжечковой атаксией, нейросенсорная тугоухость.По разным эпидемиологическим данным частота встречаемости синдрома MERRF составляет от 0,25:100000 до 1,5:100000 населения. Начало заболевания приходится на второе десятилетие жизни. Пациентов с синдромом MERRF курируют врачи-неврологи. Больные детского возраста находятся под совместным наблюдением детских невропатологов и педиатров. При осмотре обращается внимание на общее снижение мышечного тонуса, ослабление сухожильных и наличие патологических рефлексов (Бабинского, Оппенгейма), невыполнение координационных тестов - позы Ромберга, пяточно-коленной, пальценосовой проб. В биохимическом анализе крови отмечается увеличение концентрации лактата и пирувата. В ликворе выявляется высокое содержание белка. Дополнительное обследование включает: ЭЭГ томография, ДНК-анализ, ЭНМГ. При окраске гистологических срезов по методу Гомори более чем в 5% мышечных волокон обнаруживается наличие «рваных красных волокон» [4].
В основе развития синдрома LHON (атрофия зрительного нерва Лебера) лежит мутация в мтДНК. В 95% случаев у пациентов с LHON выявляется одна из трех мажорных мутаций в митохондриальной ДНК: m.3460G>A, m.11778G>A или m.14484T>C. Данные мутации изменяют структуру генов, кодирующих белки первого комплекса дыхательной цепи митохондрий. Эти гены кодируют мембранную часть белка НАДН-дегидрогеназы, участвующего в процессах окислительного фосфорилирования. Окислительное фосфорилирование использует серию из четырех крупных мультиферментных комплексов, которые встроены во внутреннюю мембрану митохондрий для преобразования кислорода и моносахаридов в энергию. Мутации в любом из генов нарушают этот процесс, вызывая синдром LHON. В результате сочетанного воздействия генетического дефекта в мтДНК, приводящего к определенному биохимическому дефекту, факторов внешней среды и модифицирующих эндогенных факторов при синдроме LHON происходит избирательная дегенерация ганглиозных клеток сетчатки и их аксонов, образующих зрительные нервы. В большинстве случаев при рождении ребенка патология не определяется - при осмотре глазного дна изменения наблюдаются лишь в нескольких процентах случаев. По мере роста ребёнка родители могут замечать, что он не задерживает взгляд на окружающих предметах, может болезненно реагировать на свет (фотофобия), часто тереть глаза и указывать на них пальцем (симптом Франческетти). Обнаруживается нистагм, который возникает еще в течение первых 2-3 месяцев жизни, замедленная реакция зрачка на свет или ее полное отсутствие. Обычно к десяти годам большинство больных с амаврозом Лебера полностью слепнут. Данный синдром встречается с частотой 3,2 на 100000 населения. Начало заболевания приходится на второе-третье десятилетие жизни.
В современной офтальмологии диагностика амавроза Лебера производится на основании осмотра глазного дна, мониторинга динамики изменений в нем, данных электроретинографии. Дифференциальную диагностику производят с различными формами пигментной абиотрофии сетчатки (при ней сохраняется нормальная или немного сниженная амплитуда волн на электроретинограмме) и некоторыми типами атрофии зрительных нервов [9].
У больных с синдромом NARP обнаруживается гетероплазмическая мутация T=> G в положении 8993 (ген АТФазы 6 - субъединицы V комплекса дыхательной цепи). При данном синдроме синтез АТФ нарушается из-за недостаточности V комплекса дыхательной цепи митохондрий. Ведущими клиническими симптомами заболевания являются мышечная слабость, атаксия, пигментная дегенерация сетчатки по типу "соль с перцем" или макулодистрофия. Другими частыми симптомами болезни являются полиневропатия, эпилептические приступы и умственная отсталость. Частота встречаемости синдрома NARP составляет 1 на 100 000 населения. Как правило, симптомы, характерные для данного синдрома, начинают появляться в раннем возрасте. По данным лабораторных исследований нередко обнаруживают лактат-ацидоз, однако его может и не быть. При морфологическом исследовании мышц иногда встречается феномен «рваных» красных волокон. ДНК диагностика для выявления наиболее частых мутаций мтДНК, характерных для данного синдрома. Так же производится: ЭНМГ, электроретинография, МРТ головного мозга [ 6 ] .
При мутациях в ядерных генах POLG, TWNK, RRM2B и TK2 происходит нарушение синтеза мтДНК и развивается прогрессирующая наружная офтальмоплегия. В результате в мтДНК мышечных клеток отсутствуют сегменты, размер которых варьирует в пределах 2-10 тысяч нуклеотидов. Также хроническая прогрессирующая наружная офтальмоплегия может быть следствием мутаций в генах самой мтДНК. Эти гены отвечают за синтез трансмиттеров, которые помогают собирать аминокислоты в белки. При мутациях в мтДНК нарушается сборка белков, которые участвуют в окислительном фосфорилировании. Первыми симптомами данного заболевания является птоз, двоение в глазах, так как офтальмоплегия, как правило, симметрична. В результате слабости круговой мышцы глаза, пациенты могут страдать от кератопатии (повреждение роговицы). Лицевые мышцы могут быть также вовлечены, приводя к атрофии мимических мышц, делающих лицо тощим и невыразительным с возникновением проблем с жеванием. Исследование показало, что у 40 % пациентов с митохондриальными заболеваниями наблюдается прогрессирующая наружная офтальмоплегия. Данное заболевание может начаться в любом возрасте и прогрессировать в течение 5-15 лет [10].
К настоящему времени специфического лечения синдромов, вызванных дефектами мтДНК, не разработано. В большинстве случаев проводится симптоматическая и полиативная терапия. Идёт активный процесс изучения патологий, связанных с дефектами мтДНК. На стадии разработки находятся специфические методы лечения митохондриальных заболеваний, проводятся экспериментальные работы по экстракорпоральному (in vitro) оплодотворению с использованием переноса ядра оплодотворённой яйцеклетки в безъядерную цитоплазму другой яйцеклетки с нормально функционирующими митохондриями (замена ядра).
Таким образом, при митохондриальной патологии в патологический процесс вовлекается большое число органов и систем. Среди клинических проявлений наибольшее значение имеют миопатия, нарушение движения глаз (птоз, офтальмоплегия), кардиомиопатия, инсультоподобные приступы, пигментный ретинит, атаксия, задержка физического развития и умственная отсталость. Появление этих признаков требует проведения углублённого обследования с использованием лабораторных методов диагностики [2,3].
Каган М. Ю. Врожденная патология β2 ламинина (синдром Пирсона): клинические и генетические аспекты // Педиатрическая фармакология, 2010. 114-117 с.
Леонтьева И.В., Е.А. Николаева Е.А. Митохондриальные кардиомиопатии: статья. - ОСП «Научно-исследовательский клинический институт педиатрии им. Ю.Е. Вельтищева» ГБОУ ВПО РНИМУ им. Н.И. Пирогова, Москва, 2016.
Митохондриальная патология у детей/ В.М. Студеникин, О.В. Глоба// Лечащий врач. — 2016. — №1.
El-Hattab A.W., Adesina A.M., Jones J., Scaglia F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015 Sep- Oct;116(1-2):4-12. [PMID: 26095523]
Hudson G., Carelli V., Horvath R., Zeviani M., Smeets H.J., Chinnery P.F. X-Inactivation patterns in females harboring mtDNA mutations that cause Leber hereditary optic neuropathy // Molecular Vision: journal. — 2007. — vol. 13. — P. 2339—2343.
Yu Wai Man CY; and others. Assessment of visual function in chronic progressive external // Eye : journal. — 2006. — Vol. 20, no. 5. — P. 564—568.
Синдром Кернса-Сейра
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Синдром Кернса-Сейра - это заболевание впервые описано в 1958 г. Большинство случаев обусловлено крупными делециями мтДНК протяжённостью 2-10 тыс. п.н. Наиболее рапространена делеция протяжённостью 4977 п.н. Крайне редко встречаются дупликации или точковые мутации.
Код по МКБ-10
Причины синдрома Кернса-Сейра
Большинство случаев синдрома Кернса-Сейра - спорадические, что можно объяснить высокой скоростью мутирования митохондриального генома. Предполагают, что делеции наиболее часто возникают в митохондриях соматических клеток в период раннего эмбрионального развития. Почти в 50% случаев больные имеют наряду с этой мутацией дупликацию D-петли, унаследованную от матери. Аномально слившиеся в результате делеции гены могут транскрибироваться, но не способны к трансляции и, следовательно, развивается дефицит кодируемых белков.
Симптомы синдрома Кернса-Сейра
Заболевание манифестирует в возрасте 4-20 лет и включает триаду симптомов:
- офтальмоплегию с птозом верхнего века и ограничением движений глазных яблок;
- прогрессирующую слабость мышц проксимальных отделов конечностей;
- пигментную дегенерацию сетчатки.
По мере прогрессирования синдрома Кернса-Сейра присоединяются другие симптомы: поражения сердца (нарушение ритма, атриовентрикулярная блокада, расширение полости желудочков), органа слуха (нейросенсорная глухота), органа зрения (атрофия зрительного нерва), снижается интеллект. Больные умирают от сердечно-сосудистой недостаточности спустя 10-20 лет после начала заболевания. При лабораторном исследовании выявляют: лактат-ацидоз и повышение 3-гидроксибутирата в крови; при морфологическом исследовании биоптатов мышечной ткани обнаруживают феномен RRF («рваные» мышечные волокна).
Диагностика синдрома Кернса-Сейра
Диагноз уточняют при молекулярно-генетическом исследовании и выявлении крупной делеции в мтДНК. Однако при анализе полученных данных необходимо принимать во внимание существование гетероплазмии, в клетках периферической крови содержится лишь около 5% мутантной ДНК. Большую информацию можно получить при молекулярно-генетическом анализе биоптатов мышц, в которых содержится до 70% мутантной ДНК митохондрий.
Что показывает МРТ мозга?
Головной мозг является самым сложным органом, он насчитывает целых 5 отделов, поэтому так важно знать, что показывает МРТ мозга. Речь идет о самой современной и информативной методике обследования.
В чем заключаются главные преимущества магнитно-резонансной томографии?
МРТ исследованием головного мозга не стоит пренебрегать, если врач выписывает направление на прохождение такой диагностики. Данная процедура очень важна для выявления серьезных отклонений в ее функционировании на ранних стадиях.
Это неинвазивная методика обследования. В ходе прохождения такой диагностики пациент не ощущает дискомфорта, за исключением того, что ему приходится находиться внутри томографа. При этом аппараты бывают закрытыми и полузакрытыми. Вторую разновидность используют для обследования людей, испытывающих страх перед замкнутым пространством.
К положительным свойствам данной процедуры относят ее безболезненность. При ее проведении нет необходимости выполнять хирургическое вмешательство. Она позволяет обследовать все отделы головы человека.
Делать МРТ можно пациентам разного возраста. Диагностика дает возможность обнаруживать патологии, когда они только зарождаются. Своевременная постановка диагноза очень важна, когда речь идет о здоровье.
МРТ снимки головного мозга четко показывают, есть патологические изменения в его отделах или нет. При их выявлении врач назначает эффективное лечение. Чем раньше начинается терапия, тем больше шансов, что она окажется успешной.
Среди других преимуществ МРТ головного мозга можно выделить:
- Возможность обследования всех частей головы, невзирая на наличие костных структур.
- Отсутствие токсичного воздействия.
- Получение четкой «картины» сосудов и мягких тканей, находящихся на разных уровнях, даже в том случае, если контрастное вещество при проведении процедуры не используется.
Решение о том, как именно выполнять МРТ головного мозга с контрастом или без, принимает лечащий врач, исходя из имеющихся показаний.
Для чего проводится процедура?
В список показаний, при наличии которых нужно выполнить исследование головного мозга, входят:
- головные боли;
- черепно-мозговые травмы;
- проблемы с памятью;
- ишемические инфаркты;
- вегетососудистая дистония.
МРТ дает возможность оценивать степень активности коры головного мозга. Благодаря этой процедуре удается оценить, насколько сильно пострадал этот орган в результате травмы. С ее помощью выявляют болезни Паркинсона и Альцгеймера, доброкачественные и злокачественные новообразования.
Магнитно-резонансную томографию назначают при шизофрении и прочих расстройствах психики, чтобы оценить, какое воздействие они оказали на структуру мозга.
При наличии нарушений в целостности миелиновой оболочки коры МРТ их выявит. Процедура помогает диагностировать кисты, карциномы, кровоизлияния в головной мозг, обнаруживать воспалительные и инфекционные процессы. Ее часто назначают при аутизме, рассеянном склерозе. Перфузия головного мозга дает возможность оценивать кровоток на различных участках. Перед столь основательной диагностикой рекомендуется сдать анализы. Их результаты укажут на область головы, подлежащую более детальному обследованию. К примеру, если у пациента наблюдается повышенный уровень пролактина - это может указывать на проблемы в работе мозжечка.
МРТ картина головного мозга позволяет оценить состояние не только мозжечка, но и других отделов, которые отвечают за память и мыслительный процесс. Тщательно обследовать затылочные доли, отвечающие за визуальное восприятие предметов, оценить плотность серого и белого вещества.
Рентгеновское облучение при проведении МРТ не используется и это большой плюс. Результаты процедуры не приходится долго ждать, их расшифровка занимает минимальное количество времени. Данная процедура отличается высокой диагностической информативностью. В последние годы она стала незаменимой в выявлении патологий головного мозга.
Особенности процедуры МРТ, принцип действия
Исследуемые ткани подвергаются воздействию электромагнитных волн в пределах магнитного поля, в которых содержится различное количество атомов водорода. Они формируют определенное количество энергии, которую регистрирует и обрабатывает специальный прибор. Речь идет о томографе. Такая обработка позволяет получать изображения различных отделов головного мозга. Развитие патологических явлений характеризуется структурными трансформациями в тканях органа. В результате меняется содержание атомов водорода в пораженной области. Это приводит к изменению получаемых в ходе обследования сигналов.
МРТ головы визуализирует различные мозговые отделы, дает их характеристику. Главной целью такой диагностики является обнаружение пораженных участков в мозге, определения их формы, размеров, характера и точной локализации.
При проведении подобной диагностики невозможно пропустить опухоль, она определяет ткань происхождения новообразования, отличается атрофические участки от постравматических. Позволяет определять характер инсультов, выявлять инфекционные поражения, отеки мозга, смещение его структур, оценивать состояние желудочков.
МРТ здорового мозга не выявляет отклонений. При наличии подозрений на наличие опухоли в голове, нужно в обязательном порядке провести данную процедуру. Она выявляет не только первичные новообразования, но и метастазы, поражения гипофиза.
Даже при небольшой травме головы рекомендуется сделать МРТ. Процедура позволяет исключить абсцесс головного мозга, менингоэнцефалит. Ее проводят при наличии симптоматики повышенного внутричерепного давления.
Врачи назначают магнитно-резонансную томографию при часто повторяющихся головных болях, сбоях моторики. Такое обследование показано людям, без причины падающим в обмороки. Тем, у кого имеются нарушения поведения и когнитивных функций.
МРТ незаменимо как в неврологической сфере, так и в эндокринологии, психиатрии, других областях медицины.
Магнитно-резонансную и компьютерную томографию часто делают после операций, чтобы оценить результаты проведенного хирургического вмешательства. Методика незаменима в динамическом наблюдении. С ее помощью обследуют не только головной мозг, но и другие органы - поджелудочную железу, желчный пузырь.
Противопоказания к магнитно-резонансной томографии
Противопоказаний к проведению магнитно-резонансной томографии немного, но они есть. К ним относят:
- Наличие имплантата в организме (в том числе кардиостимулятора).
- Чрезмерная подвижность пациента, например, если он пребывает в состоянии алкогольного опьянения. При проведении МРТ обследуемый должен лежать без движения.
- Инородные предметы в теле (в ходе обследования они могут сместиться и повредить сосуды).
К относительным противопоказаниям относят болезни, детский возраст до 7 лет, металлические протезы суставов за пределами действия электромагнитных волн, болезни крови (в этом случае нельзя применять контраст), выраженную клаустрофобию (у пациента в закрытом пространстве может резко ухудшиться состояние).
Как подготовиться к обследованию?
К МРТ головного мозга не нужно долго и основательно готовиться. За пару часов до диагностики желательно воздержаться от приема пищи. Не стоит применять перед процедурой декоративную косметику. За пределами кабинета, в котором проводится диагностика, необходимо оставить телефон, банковские карты и часы.
Врач обязательно расскажет пациенту о ходе обследования. Если тот страдает боязнью замкнутого пространства, пропишет успокаивающие препараты.
При необходимости будет выполнена процедура с контрастированием, позволяет получать более точную картину. После введения контрастного вещества может появиться головная боль и тошнота. Перед применением контраста необходимо провести тест на чувствительность.
Как проходит процедура?
Перед диагностикой аппаратом МРТ пациента просят подписать согласие. После этого ему предлагается надеть халат (можно обследоваться и в своей одежде, главное, чтобы на ней не было металлических элементов) лечь спиной на специальную платформу. Голову обследуемого фиксируют, чтобы обеспечить ее неподвижность.
Платформа, с лежащей на ней пациентом, заезжает в «трубу» томографа. При работе оборудование издает шум, поэтому рекомендуется пользоваться берушами.
В кабинете для МРТ предусмотрена система двухсторонней голосовой связи. Врач контролирует ход процедуры из соседнего кабинета.
По времени обследование занимает примерно полчаса. При использовании контраста около 50 минут. Контраст вводят одномоментно или дозатором.
Результаты
Расшифровкой результатов магнитно-резонансной томографии занимается врач-рентгенолог, обладающий соответствующей квалификацией. Он анализирует полученные результаты и исходя из этого составляет заключение. Пациент получает итоги сканирования на ДВД-диске, после чего передает их врачу.
В нашем медицинском центре проведение МРТ головного мозга и органов брюшной полости выполняется с использованием современного оборудования. Мы задействуем лучшие томографы, гарантирующие высокую точность результатов обследования.
Читайте также: