Дисгенезия мозолистого тела. Голова и шея при врожденной патологии
Добавил пользователь Владимир З. Обновлено: 21.01.2026
Аномалии развития головного мозга — это результат происходящих во внутриутробном периоде нарушений формирования отдельных церебральных структур или головного мозга в целом. Зачастую имеют неспецифическую клиническую симптоматику: преимущественно эпилептический синдром, задержку психического и умственного развития. Тяжесть клиники напрямую коррелирует со степенью поражения головного мозга. Диагностируются антенатально при проведении акушерского УЗИ, после рождения — при помощи ЭЭГ, нейросонографии и МРТ головного мозга. Лечение симптоматическое: противоэпилептическое, дегидратационное, метаболическое, психокоррегирующее.
МКБ-10
Общие сведения
Причины
Наиболее весомой причиной сбоев внутриутробного развития является влияние на организм беременной и на плод, различных вредоносных факторов, обладающих тератогенным действием. Возникновение аномалии в результате моногенного наследования встречается лишь в 1% случаев. Наиболее влиятельной причиной пороков головного мозга считается экзогенный фактор. Тератогенным эффектом обладают многие активные химические соединения, радиоактивное загрязнение, отдельные биологические факторы. Немаловажное значение здесь имеет проблема загрязнения среды обитания людей, обуславливающая поступление в организм беременной токсических химических веществ.
Различные эмбриотоксические воздействия могут быть связаны с образом жизни самой беременной: например, с курением, алкоголизмом, наркоманией. Дисметаболические нарушения у беременной, такие как сахарный диабет, гипертиреоз и пр., могут также стать причиной церебральных аномалий плода. Тератогенным действием обладают и многие медикаменты, которые может принимать женщина в ранние сроки беременность, не подозревая о происходящих в ее организме процессах. Мощный тератогенный эффект оказывают инфекции, перенесенные беременной, или внутриутробные инфекции плода. Наиболее опасны цитомегалия, листериоз, краснуха, токсоплазмоз.
Патогенез
Дифференцировка нейробластов (зародышевых нервных клеток) приводит к образованию нейронов, формирующих серое вещество, и глиальных клеток, составляющих белое вещество. Серое вещество отвечает за высшие процессы нервной деятельности. В белом веществе проходят различные проводящие пути, связывающие церебральные структуры в единый функционирующий механизм. Рожденный в срок новорожденный имеет такое же число нейронов, как и взрослый человек. Но развитие его мозга продолжается, особенно интенсивно в первые 3 мес. жизни. Происходит увеличение глиальных клеток, разветвление нейрональных отростков и их миелинизация.
Сбои могут произойти на различных этапах формирования головного мозга. Если они возникают в первые 6 мес. беременности, то способны приводить к снижению числа сформированных нейронов, различным нарушениям в дифференцировке, гипоплазии различных отделов мозга. В более поздние сроки может возникать поражение и гибель нормально сформировавшегося церебрального вещества.
Виды аномалий мозга
Анэнцефалия — отсутствие головного мозга и акрания (отсутствие костей черепа). Место головного мозга занято соединительнотканными разрастаниями и кистозными полостями. Может быть покрыто кожей или обнажено. Патология несовместима с жизнью.
Энцефалоцеле — пролабирование церебральных тканей и оболочек через дефект костей черепа, обусловленный его незаращением. Как правило, формируется по средней линии, но бывает и асимметричным. Небольшое энцефалоцеле может имитировать кефалогематому. В таких случаях определить диагноз помогает рентгенография черепа. Прогноз зависит от размеров и содержимого энцефалоцеле. При небольших размерах выпячивания и наличии в его полости эктопированной нервной ткани эффективно хирургическое удаление энцефалоцеле.
Микроцефалия — уменьшение объема и массы головного мозга, обусловленное задержкой его развития. Встречается с частотой 1 случай на 5 тыс. новорожденных. Сопровождается уменьшенной окружностью головы и диспропорциональным соотношением лицевого/мозгового черепа с преобладанием первого. На долю микроцефалии приходится около 11% всех случаев олигофрении. При выраженной микроцефалии возможна идиотия. Зачастую наблюдается не только ЗПР, но и отставание в физическом развитии.
Макроцефалия — увеличение объема головного мозга и его массы. Гораздо менее распространена, чем микроцефалия. Макроцефалия обычно сочетается с нарушениями архитектоники мозга, очаговой гетеротопией белого вещества. Основное клиническое проявление — умственная отсталость. Может наблюдаться судорожный синдром. Встречается частичная макроцефалия с увеличением лишь одного из полушарий. Как правило, она сопровождается асимметрией мозгового отдела черепа.
Кистозная церебральная дисплазия — характеризуется множественными кистозными полостями головного мозга, обычно соединенными с желудочковой системой. Кисты могут иметь различный размер. Иногда локализуются только в одном полушарии. Множественные кисты головного мозга проявляются эпилепсией, устойчивой к антиконвульсантной терапии. Единичные кисты в зависимости от размера могут иметь субклиническое течение или сопровождаться внутричерепной гипертензией; зачастую отмечается их постепенное рассасывание.
Голопрозэнцефалия — отсутствие разделения полушарий, в результате чего они представлены единой полусферой. Боковые желудочки сформированы в единую полость. Сопровождается грубыми дисплазиями лицевого черепа и соматическими пороками. Отмечается мертворождение или гибель в первые сутки.
Агирия (гладкий мозг, лиссэнцефалия) — отставание развития извилин и тяжелое нарушение архитектоники коры. Клинически проявляется выраженным расстройством психического и моторного развития, парезами и различными формами судорог (в т. ч. синдромом Веста и синдромом Леннокса-Гасто). Обычно заканчивается летальным исходом на первом году жизни.
Пахигирия — укрупнение основных извилин при отсутствии третичных и вторичных. Сопровождается укорочением и выпрямлением борозд, нарушением архитектоники церебральной коры.
Микрополигирия — поверхность коры мозга представлена множеством мелких извилин. Кора имеет до 4-х слоев, тогда как в норме кора насчитывает 6 слоев. Может быть локальной или диффузной. Последняя, полимикрогирия, характеризуется плегией мимических, жевательных и глоточных мышц, эпилепсией с дебютом на 1-ом году жизни, олигофренией.
Гипоплазия/аплазия мозолистого тела. Часто встречается в виде синдрома Айкарди, описанного только у девочек. Характерны миоклонические пароксизмы и сгибательные спазмы, врожденные офтальмические пороки (колобомы, эктазия склеры, микрофтальм), множественные хориоретинальные дистрофические очаги, обнаруживаемые при офтальмоскопии.
Фокальная корковая дисплазия (ФКД) — наличие в коре головного мозга патологических участков с гигантскими нейронами и аномальными астроцитами. Излюбленное расположение — височные и лобные зоны мозга. Отличительной особенностью эпиприступов при ФКД является наличие кратковременных сложных пароксизмов с быстрой генерализацией, сопровождающихся в своей начальной фазе демонстративными двигательными феноменами в виде жестов, топтания на одном месте и т. п.
Гетеротопии — скопления нейронов, на этапе нейронной миграции задержавшихся на пути своего следования к коре. Гетеротопионы могут быть единичными и множественными, иметь узловую и ленточную форму. Их главное отличие от туберозного склероза — отсутствие способности накапливать контраст. Эти аномалии развития головного мозга проявляются эписиндромом и олигофренией, выраженность которых прямо коррелирует с числом и размером гетеротопионов. При одиночной гетеротопии эпиприступы, как правило, дебютируют после 10-летнего возраста.
Диагностика
Тяжелые аномалии развития головного мозга зачастую могут быть диагностированы при визуальном осмотре. В остальных случаях заподозрить церебральную аномалию позволяет ЗПР, гипотония мышц в неонатальном периоде, возникновение судорожного синдрома у детей первого года жизни. Исключить травматический или гипоксический характер поражения головного мозга можно при отсутствии в анамнезе данных о родовой травме новорожденного, гипоксии плода или асфиксии новорожденного. Пренатальная диагностика пороков развития плода осуществляется путем скринингового УЗИ при беременности. УЗИ в I триместре беременности позволяет предупредить рождение ребенка с тяжелой церебральной аномалией.
Одним из методов выявления пороков головного мозга у грудничков является нейросонография через родничок. Намного более точные данные у детей любого возраста и у взрослых получают при помощи МРТ головного мозга. МРТ позволяет определить характер и локализацию аномалии, размеры кист, гетеротопий и других аномальных участков, провести дифференциальную диагностику с гипоксическими, травматическими, опухолевыми, инфекционными поражениями мозга. Диагностика судорожного синдрома и подбор антиконвульсантной терапии осуществляется при помощи ЭЭГ, а также пролонгированного ЭЭГ-видеомониторинга. При наличии семейных случаев церебральных аномалий может быть полезна консультация генетика с проведением генеалогического исследования и ДНК-анализа. С целью выявления сочетанных аномалий проводится обследование соматических органов: УЗИ сердца, УЗИ брюшной полости, рентгенография органов грудной полости, УЗИ почек и пр.
Лечение аномалий мозга
Терапия пороков развития головного мозга преимущественно симптоматическая, осуществляется детским неврологом, неонатологом, педиатром, эпилептологом. При наличии судорожного синдрома проводится антиконвульсантная терапия (карбамазепин, леветирацетам, вальпроаты, нитразепам, ламотриджин и др.). Поскольку эпилепсия у детей, сопровождающая аномалии развития головного мозга, обычно резистентна к противосудорожной монотерапии, назначают комбинацию из 2 препаратов (например, леветирацетам с ламотриджином). При гидроцефалии осуществляют дегидратационную терапию, по показаниям прибегают к шунтирующим операциям. С целью улучшения метаболизма нормально функционирующих мозговых тканей, в какой-то степени компенсирующих имеющийся врожденный дефект, возможно проведение курсового нейрометаболического лечения с назначением глицина, витаминов гр. В и пр. Ноотропные препараты используются в лечении только при отсутствии эписиндрома.
При умеренных и относительно легких церебральных аномалиях рекомендована нейропсихологическая коррекция, занятия ребенка с психологом, комплексное психологическое сопровождение ребенка, детская арт-терапия, обучение детей старшего возраста в специализированных школах. Указанные методики помогают привить навыки самообслуживания, уменьшить степень выраженности олигофрении и по возможности социально адаптировать детей с церебральными пороками.
Прогноз и профилактика
Прогноз во многом определяется тяжестью церебральной аномалии. Неблагоприятным симптомом выступает ранее начало эпилепсии и ее резистентность к осуществляемой терапии. Осложняет прогноз наличие сочетанной врожденной соматической патологии. Эффективной мерой профилактики служит исключение эмбриотоксических и тератогенных влияний на женщину в период беременности. При планировании беременности будущим родителям следует избавиться от вредных привычек, пройти генетическое консультирование, обследование на наличие хронических инфекций.
Агенезия мозолистого тела
Агенезия мозолистого тела — это врожденное отсутствие мозолистого тела либо его части. Аномалия обусловлена генетическими нарушениями, сосудистыми мальформациями, тератогенными факторами. Основные признаки заболевания: двигательные расстройства, задержка психоречевого развития, судорожные приступы. При негрубом (частичном) варианте патологии возможно малосимптомное течение. Для диагностики состояния назначается церебральные КТ или МРТ, нейросонография у новорожденных, генетические исследования. Лечение симптоматическое: медикаментозная коррекция осложнений, реабилитационные программы.
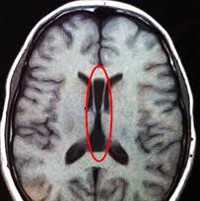
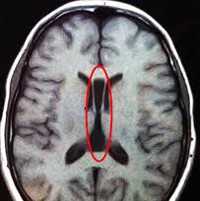
Агенезия мозолистого тела (АМТ) — один из наиболее частых пороков нервной системы. Распространенность болезни в популяции составляет от 0,05% до 7% среди новорожденных, причем в группе детей с замедленным становлением психики агенезия встречается у 2,3%. Калифорнийская программа по изучению врожденных пороков предоставляет другие данные по частоте агенезии — 1,4 на 10000 живых новорожденных. Впервые состояние было описано в 1812 году в ходе аутопсии, проведенной немецким анатомом И. Рэйлем, и названо «природной моделью рассеченного мозга».
Точные этиологические факторы заболевания не установлены. В современной неврологии преобладает мультифакториальная теория, согласно которой для формирования врожденного порока ЦНС требуется комбинация неблагоприятных экзогенных и эндогенных причин. Ученые выделяют несколько наиболее вероятных предпосылок развития агенезии:
- Генетические аномалии. Повреждения мозолистого тела отмечаются при различных наследственных синдромах: Миллер-Дикера, Рубинштейна-Тауби, Доннаи-Кугана. Состояние входит в состав не менее 7 аутосомно-доминантных, 23 аутосомно-рецессивных, 12 Х-сцепленных врожденных заболеваний.
- Сосудистые нарушения. Причиной недоразвития мозолистого тела могут выступать артериовенозные мальформации или аневризмы, которые характеризуются отсутствием нормальной капиллярной сети. При этом возникает феномен обкрадывания, клетки МТ не получают должного количества кислорода, питательных веществ.
- Токсические влияния. Болезнь связана с действием тератогенных химических факторов: лекарственных препаратов, солей тяжелых металлов, пестицидов и бытовой химии. Негативное влияние на формирование ЦНС плода оказывает вдыхание табачного дыма (активное или пассивное курение) или прием беременной алкоголя во время гестации.
- Внутриутробные инфекции. Аномалии формирования неврологических структур, в том числе агенезия мозолистого тела, встречаются при проникновении возбудителей в организм плода на 2-3 месяце беременности. Нейротропные свойства демонстрируют герпетические инфекции, токсоплазмоз, цитомегаловирус.
Основным фактором риска выступает недоношенность. У новорожденных, родившихся до 27-недельного срока гестации МТ истончено в задних отделах, между 28 и 30 неделями — только в области валика. У рожденных после 30 недели в неонатальном периоде изменения не обнаруживаются, хотя при нейропсихологическом исследовании у школьников зачастую выявляется дефицит межполушарной передачи познавательной информации.
Агенезия возникает при нарушении дифференциации нервной трубки в период со 2 до 5 месяца внутриутробного развития. При полном отсутствии МТ третий мозговой желудочек остается открытым, не формируются столбы свода мозга, отсутствуют прозрачные перегородки. В 60% случаев при АМТ передней комиссуры нет вообще. В 10% она увеличена и берет на себя часть функций мозолистого тела у новорожденных, а также на следующих этапах постнатального периода.
Характерным анатомическим изменением является колпоцефалия, при которой расширены задние отделы боковых церебральных желудочков. Состояние не относится к истинной гидроцефалии новорожденных, а обусловлено уменьшением кортикальных ассоциативных путей. Еще один типичный признак порока — пучки Пробста, представляющие собой неправильно ориентированные аксоны, расположенные параллельно межполушарной щели.
Классификация
В практической неврологии состояние подразделяют на тотальное, когда орган полностью отсутствует, и частичное (парциальное), при котором визуализационные методы не обнаруживают отдельные участки МТ. Это имеет решающее значение для тяжести клинической картины, возможных осложнений. В соответствии с патогенетическими особенностями формирования врожденных пороков, выделяют следующие 3 формы болезни:
- Агенезия. Закладка эмбрионального зачатка МТ отсутствует полностью.
- Аплазия. Эмбриональный зачаток мозолистого тела есть, но не развивается.
- Гипоплазия. МТ недостаточно развито из-за нарушений на одном из этапов эмбриогенеза: размеры и масса органа уменьшены, его функциональная активность снижена.
Симптомы
Клиническая картина агенезии мозолистого тела широко варьирует от практически бессимптомных форм (при гипоплазии) до критических нервно-психических расстройств при его грубом недоразвитии, сопровождающемся другими врожденными пороками ЦНС. У новорожденных признаки патологии могут вовсе отсутствовать и проявляться по мере взросления младенца задержкой психомоторного развития.
Двигательные нарушения определяются у 35-40% пациентов. Они проявляются мышечной гипотонией или дистонией, гипер- или гипорефлексией, нарушением глотательного и сосательного рефлексов. Дети позже начинают держать голову, испытывают затруднения при обучении сидению, ползанию, ходьбе. Могут отмечаться координационные нарушения, неуклюжая походка. Из пароксизмальных расстройств у новорожденных и детей первого года жизни преобладают судороги.
Мозолистое тело поддерживает связь между церебральными зонами, формирует межполушарную организацию высших психических процессов. При его агенезии либо гипоплазии у детей выявляются когнитивные расстройства. У новорожденных пациентов и в раннем детстве наблюдается задержка речи, снижение динамического компонента игровой деятельности. В дошкольном и школьном возрасте возникают проблемы с концентрацией внимания, расстройства памяти, при тотальной АМТ снижен коэффициент интеллекта.
Осложнения
Около 65% случаев заболевания сопровождаются сопутствующими врожденными патологиями, среди которых преобладают мальформации кортикального развития (22,8%), межполушарные кисты (14,3%), голопрозэнцефалия (14,3%). К более редким сопутствующим аномалиям относят кисты и гипоплазию мозжечка, синдром Арнольда-Киари. До 20% новорожденных, кроме структур ЦНС, имеют пороки нескольких внутренних органов.
У 75% больных с тотальным поражением наблюдается симптоматическая эпилепсия височно-лобной локализации, в 66% случаев выражены когнитивные нарушения. У 16% пациентов формируются расстройства аутистического спектра. Изредка встречаются патологии органа зрения в виде хориоретинальных лакунарных очагов, сочетанной аномалии зрительных нервов.
В качестве первичного метода обследования в пренатальном периоде проводится акушерское УЗИ. У новорожденных для скрининговой диагностики используется нейросонография, однако этот метод не всегда показывает хорошую информативность, особенно при парциальной агенезии. Для верификации диагноза назначаются следующие методы исследования:
- КТ головного мозга. При компьютерной томографии определяются широко расставленные передние рога, высокое стояние третьего желудочка, параллельный ход медиальных стенок боковых желудочков. КТ производится в рамках постнатальной диагностики.
- МРТ головного мозга. Для максимально точной визуализации степени агенезии или гипоплазии мозолистого тела новорожденным выполняется магнитно-резонансная томография в трех плоскостях. По показаниям МРТ может рекомендоваться беременным женщинам для исключения несовместимых с жизнью сочетанных пороков ЦНС.
- Нейропсихологическое обследование. Для изучения когнитивных функций у детей применяется шкала интеллекта Векслера (WISC-Revised), адаптированное чтение и правописание (Schonnel Graded Reading and Spelling Tests), оценка вербальной беглости, тест контролируемых устных ассоциаций (Controlled Oral Word Association Test).
- Генетический анализ. Для подтверждения или исключения наследственных заболеваний, сопровождающихся агенезией мозолистого тела, показаны кариотипирование, секвенирование генома, проводимое как новорожденным, так и детям другого возраста. Исследования также проводят в антенатальном периоде для принятия решения о сохранении или прерывании беременности.
Лечение агенезии мозолистого тела
Специфическая терапия отсутствует. Медикаментозное лечение назначается неонатологом или педиатром индивидуально с учетом ведущих патологических синдромов: у новорожденных, детей раннего возраста используются антиконвульсанты, нейрометаболические препараты, дегидратационная терапия. Основу медицинской помощи составляет комплексная реабилитация, которая включает следующие составляющие:
- Нейрологопедические программы. Занятия с детским логопедом проводятся для становления речевой функции, ликвидации проявлений дизартрии, улучшения артикуляции.
- Дефектологические программы. Помощь коррекционных педагогов требуется детям с интеллектуальными нарушениями, которые не могут проходить обучение в обычных классах.
- Нейроакустические программы. Формирование и гармонизация высших психических функций производятся с помощью звуковой терапии, музыкотерапии.
Прогноз определяется видом врожденной аномалии мозолистого тела, наличием сопутствующих пороков развития ЦНС. Благоприятный исход наблюдается при частичной гипоплазии МТ, а в случае комбинированных церебральных пороков у новорожденных могут быть жизнеугрожающие осложнения. Профилактические меры включают медико-генетическое консультирование, исключение тератогенных влияний в гестационном периоде.
1. Эпилептические проявления когнитивные и аутистические расстройства у пациентов с агенезией мозолистого тела: результаты нейропсихологического тестирования/ О.А. Милованова, О.А. Комиссарова, Т.Ю. Тараканова, С.В. Бугрий// Эпилепсия и пароксизмальные состояния. — 2018. — №4.
2. Влияние особенностей строения мозолистого тела и доминирующего полушария на протекание психических процессов в юношеском возрасте/ У.С. Чернышова, Т.Ю. Хабарова, Д.А. Соколов// Центральный научный вестник. — 2016.
4. Молекулярная эмбриология: на пути каталогизации генов врожденных пороков развития головного мозга/ В.П. Пишак, М.А. Ризничук// Международный журнал педиатрии, акушерства и гинекологии. — 2014. — №5.
Дисгенезии головного мозга
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
Распространенными неврологическими проблемами для детского возраста являются патологии, появление которых основано на неправильном внутриутробном развитии головного мозга. Подобные нарушения называют «дисгенезии головного мозга» речь идет о мультифакторых состояниях, зачастую не имеющих выраженной клинической картины. В большинстве случаев родители обращаются за помощью к врачу с жалобами на сложности с обучаемостью, судороги, двигательную рассеянность, поведенческие отклонения у ребенка. Дисгенезии головного мозга распознаются лишь в ходе проведения КТ или ядерно-магнитно-резонансной томографии. В целом, механизмы развития нарушения изучены недостаточно.
Код по МКБ-10
Эпидемиология
Дефекты развития головного мозга составляют около 20% от всех пороков развития. По наблюдениям специалистов, частота врожденных нарушений центральной нервной системы колеблется от 1 до 2 случаев на 1 тысячу родившихся младенцев. [1], [2]
Среди врожденных аномалий развития головного мозга одно из первых мест занимают корковые дисгенезии, которые становятся основными причинами появления детского эпилептического синдрома. В целом, врожденные аномалии головного мозга обнаруживаются приблизительно в 30% случаев всех дисгенезий, диагностируемых в детском возрасте.
Корковые дисгенезии наблюдаются у 25-40% пациентов с дефектами развития головного мозга и часто сопровождаются эпилептическим синдромом или другими типами симптоматической эпилепсии.
Среди врожденных аномалий головного мозга корковые дисгенезии отмечаются достаточно часто, что обусловлено широким практическим применением нейровизуализирующей диагностики - в частности, нейросонографии, магнитно-резонансной томографии, компьютерной томографии.
Причины дисгенезий головного мозга
Наиболее явными причинами появления дисгенезий головного мозга считаются:
- нарушенное внутриутробное развитие нервной системы (на стадии закладки);
- повреждение нервной системы на этапе раннего эмбриогенеза в результате генных мутаций, инфекционных заболеваний матери во время беременности, влияния радиации, травматических повреждений, воздействия на плод химических агентов и токсинов.
Среди инфекций, обладающим повреждающим действием на нервную систему плода, особенно следует выделить краснуху, токсоплазмоз, вирусный гепатит, цитомегаловирусную инфекцию.
Наиболее часто неправильное внутриутробное развитие обусловливается негативным влиянием на организм матери и малыша неблагоприятных факторов, обладающих тератогенным эффектом. Экзогенными причинами формирования дисгенезии могут стать:
- радиоактивное излучение;
- влияние химических агентов;
- повышенная температура;
- воздействие токов высокой частоты;
- неудовлетворительная экологическая обстановка, которая влечет за собой попадание в организм женщины токсических продуктов.
Помимо этого, тератогенным эффектом могут обладать и некоторые препараты лекарственного предназначения, а также гормональные средства, которые может принимать будущая мать, не зная о наступившей беременности. Имеются доказательства того, что многие медикаменты без проблем проникают через плаценту и оказываются в системе кровообращения малыша. Опасность могут представлять не только сильнодействующие вещества, но и привычные лекарственные препараты в больших дозировках, и даже поливитаминные комплексы. [3]
Спровоцировать нарушения внутриутробного развития по типу дисгенезии способны и сбои обменных процессов, вирусные и прочие инфекции, в том числе и такие, которые имеют скрытое бессимптомное течение. Особенно опасными считаются:
- гипертиреоз;
- обменные нарушения;
- сахарный диабет;
- сифилис;
- цитомегаловирусная инфекция;
- краснуха;
- листериоз;
- токсоплазмоз.
Крайне негативно влияют на протекание беременности и здоровье будущего ребенка особенности жизни беременной женщины. Тератогенное действие оказывают:
- употребление алкоголя;
- курение;
- наркомания.
Факторы риска
Предрасполагающими факторами к появлению дисгенезии головного мозга считаются такие:
- наследственная предрасположенность (родственные случаи появления патологии, по аутосомному типу наследования, либо сцепленного с Х-хромосомой);
- спонтанная мутация;
- хромосомная перестройка;
- внутриутробное инфицирование (преимущественно вирусного происхождения) или травматические повреждения;
- влияние интоксикаций, медицинских препаратов, химических веществ на этапе внутриутробного развития;
- фатальный алкогольный синдром, обусловленный материнским алкоголизмом на протяжении периода вынашивания плода;
- острая недостача питательных компонентов у плода;
- выраженные обменные расстройства у будущей матери.
В настоящее время ученые не могут выделить какую-либо одну основную причину дисгенезии головного мозга, поэтому необходимо взвесить имеющиеся факторы риска. [4]
Развитие головного мозга человека стартует на внутриутробном этапе, активно продолжаясь после его появления на свет. Как утверждают специалисты, правое мозговое полушарие отвечает за образно-творческое мышление, за двигательную координацию, равновесие, пространственную зрительную и кинестетическую восприимчивость. Левое мозговое полушарие обусловливает математические, знаковые, логические, речевые, аналитические способности, обеспечивает восприимчивость информации на слух, целевые настройки и схематические постановки. Единый мозг представляет собой работу двух полушарий, плотно взаимосвязанных друг с другом нервно-волокнистой системой (мозолистым телом).
Мозолистое тело локализуется между мозговыми полушариями в затылочно-теменной области. Оно включает в себя 200 миллионов нервных волокон и обеспечивает скоординированную работу головного мозга и передачу информации между полушариями. При таком нарушении, как дисгенезия головного мозга, страдает познавательная функция человека. При неправильной проводимости посредством мозолистого тела доминирующее полушарие принимает на себя повышенную нагрузку, при практическом бездействии другого. Связь между двумя мозговыми «половинами» теряется. В итоге страдает пространственное ориентирование, возникает дисбаланс, больной не может правильно осознавать собственное тело, адекватно эмоционально реагировать. Нарушается функция восприятия ведущих конечностей. [5]
Дети с дисгенезией головного мозга часто не ползают, возникают сложности с ходьбой, чтением, прописью. Информация воспринимается в основном через слух и зрение. При отсутствии лечения и медицинской реабилитации у таких больных в дальнейшем появляется множество проблем, связанных с общим развитием, обучением. [6]
Симптомы дисгенезий головного мозга
Клинические симптомы у всех пациентов проявляются по-разному, поэтому и диагностика проводится в разное время. Например, тяжелые формы дисгенезии головного мозга обнаруживаются уже в раннем детском возрасте, а у взрослых нарушение может протекать скрыто и диагностироваться случайным образом.
Дети с дисгенезией головного мозга в период новорожденности могут иметь обычный и вполне здоровый вид, а показатели развития совпадают с нормальными до трехмесячного возраста. Начиная с 3-х месяцев возможно появление первых патологических признаков в виде эпилептических приступов, инфантильных спазмов и пр.
Клиническая картина может быть представлена такими признаками:
- нарушение формирования и последующего развития мозолистого тела;
- кистозное расширение полости желудочка головного мозга, аномалия мантии мозга;
- гидроцефалия;
- атрофия зрительного и слухового нервов;
- микроэнцефалия;
- опухолевые процессы (в том числе кисты) в области полушарий головного мозга;
- неполное формирование извилин;
- раннее половое развитие;
- порок развития позвоночного столба (расщепление позвоночника, незаращение позвонковой дужки);
- синдром Айкарди (ранняя миоклоническая энцефалопатия);
- липомы;
- разного рода патологии пищеварительной системы;
- заторможенное психомоторное развитие;
- интеллектуальная и физическая отсталость;
- координационные расстройства;
- дефекты со стороны других органов, в том числе и опорно-двигательной системы;
- пониженный тонус мускулатуры.
В относительно легких случаях дисгенезии, при нормальном умственном и двигательном развитии, могут наблюдаться признаки нарушенного информационного обмена между мозговыми полушариями. [7]
Первые признаки
Дисгенезия головного мозга у младенцев обнаруживается чаще всего после трех месяцев жизни, хотя возможна диагностика патологии ещё на этапе внутриутробного развития. Первые признаки неполадки у малышей обычно такие:
- появление судорог, инфантильных спазмов;
- судорожные припадки;
- ослабление крика;
- проблемы со зрением, обонянием и/или осязанием;
- нарушения коммуникации; [8]
- признаки мышечной гипотонии (понижение рефлекторной активности, обильное выделение слюны, торможение физического развития, слабая двигательная активность, нарушенная хватательная функция).
В старшем возрасте при дисгенезии обращают на себя внимание ухудшение слуховой и зрительной памяти, нарушение двигательной координации и терморегуляции (гипотермия).
Инфантильные спазмы представляют собой судорожные внезапные сгибания-разгибания конечностей. Гипотония мускулатуры характеризуется пониженным мышечным тонусом (может сочетаться с потерей мышечной силы).
Более распространенными считаются несиндромные типы дисгенезии, которые продолжительное время остаются бессимптомными и выявляются практически случайно - например, в ходе диагностики задержки умственного развития, судорог или большого размера головы. Макроцефалия бывает частично обусловлена присутствием гигантских кистозных образований, локализованных кзади от третьего желудочка. Реже могут отмечаться эндокринологические нарушения.
Синдромные формы дисгенезий головного мозга бывают следующими:
- Синдром Айкарди - встречается преимущественно у девочек и характеризуется инфантильными спазмами, специфическими хориоидальными лакунами, позвонково-реберными дефектами. Исход патологии чаще неблагоприятный: у пациентов сохраняются судороги, отмечается глубокая умственная отсталость.
- Семейный синдром с генитальной патологией, который может проявляться микроцефалией и прочими пороками центральной нервной системы.
- Синдром Андерманна характеризуется поражением периферической нервной системы в сочетании с дисгенезией головного мозга (или гипотрофией).
- Синдром периодического гипергидроза и гипотермии (обратный синдром Шапиро).
Дисгенезия мозолистого тела
Дисгенезия является врожденной патологией головного мозга, так как начинает свое развитие ещё на внутриутробном этапе под влиянием разных факторов. Вторичная дисгенезия мозолистого тела рассматривается в виде вторичной деструкции прозрачной перегородки: такой дефект можно визуализировать при коронарной проекции в ходе проведения нейросонографии. Многие патологии взаимосвязаны с дисгенезией прозрачной перегородки, в том числе стеноз водопровода на фоне вторичной гидроцефалии, агенезия мозолистого тела, аномалия Киари II, порок миграции и септооптическая дисплазия. При септооптической дисплазии отмечается дисгенезия прозрачной перегородки и гипоплазия зрительных каналов, нервных волокон и хиазмы. Большая часть пациентов с подобной патологией дополнительно страдают нарушениями со стороны системы гипофиза-гипоталамуса. [9]
Дисгенезия мозолистого тела у ребенка поражает нервные сплетения, соединяющие два мозговых полушария, а именно - мозолистое тело, имеющее уплощенную форму и локализованное под корой мозга. Заболевание может иметь разную степень тяжести:
- Легкая степень дисгенезии отмечается при сохранении интеллектуальных способностей и моторной активности. Присутствуют лишь нарушения передачи импульсных сигналов между полушариями.
- Сложная степень сопровождается не только общими признаками дисгенезии, но и другими аномалиями мозгового развития. Отмечаются выраженные сбои в нейронных связях, судорожные приступы, явное торможение умственного развития.
Осложнения и последствия
Исходы врожденных мозговых аномалий можно разделить на три варианта:
- Практически полномерное выздоровление, с отсутствием видимых нарушений и возвращением пациента к нормальной жизненной активности.
- Остаточные явления, не угрожающие жизни пациента, но в определенной степени ограничивающие его бытовую и социальную деятельность.
- Грубые расстройства, значительные нарушения интеллектуального развития.
Выраженность последствий дисгенезии зависит от объема патологических изменений головного мозга, а также от причины, спровоцировавшей эту патологию. Большое значение имеет своевременность постановки диагноза и адекватность лечебных манипуляций. [10]
В целом, у многих больных дисгенезией головного мозга отмечается умеренное или выраженное нарушение интеллектуального развития, имеется отставание и в физическом плане.
Диагностика дисгенезий головного мозга
В тяжелых случаях дисгенезии головного мозга диагностику возможно проводить уже при визуальном осмотре новорожденного малыша. Дополнительные обследования могут назначаться при мышечной гипотонии в периоде новорожденности, при появлении судорог, при задержке психического развития.
Распространенными способами диагностики считаются такие:
- скриннинговое и акушерское ультразвуковое исследование в ходе беременности;
- нейросонография через область родничка в первые 12-18 месяцев жизни малыша;
- электроэнцефалография с возможным видеомониторингом;
- магнитно-резонансная томография. [11]
Для распознавания нарушений, сопутствующих дисгенезии, соматических патологий выполняют ультразвуковое исследование почек, сердца, органов брюшной полости. Дополнительно может потребоваться генетическая консультация. Лабораторные анализы проводятся в рамках общей оценки состояния организма: выполняют общие анализы крови и мочи, реже - исследование цереброспинальной жидкости.
Инструментальная диагностика в детском возрасте, в период открытых родничков, чаще всего представлена нейросонографией - доступной, мобильной, безопасной и информативной процедурой. Нейросонография может применяться как при врожденных, так и при инфекционных, нейропластических или травматических поражениях головного мозга. [12]
Дифференциальная диагностика
В ходе постановки диагноза дисгенезии головного мозга в периоде новорожденности следует проводить дифференциацию с такими патологическими состояниями:
Аплазия мозолистого тела
Порок развития в виде полного или практически полного отсутствия спайки нервных волокон, соединяющих полушария головного мозга, определяется как аплазия мозолистого тела, которая синонимична его агенезии, то есть несформированности в процессе развития. [1]
На врожденные пороки развития головного мозга приходится не менее 25% всех аномалий эмбрионального периода онтогенеза.
По некоторым данным, аномалии мозолистого тела, включая его агенезию (аплазию), выявляются у 0,3-0,7% пациентов, которым пои показаниям проводится МРТ головного мозга.
Обособленная аплазия мозолистого тела у детей - редкий врожденный дефект, но в составе генетически обусловленных синдромов считается достаточно частой врожденной аномалией, распространенность которой оценивается в 230 случаев на 10 тыс. детей с проблемами развития.
В трети случаев аплазии или частичной агенезии мозолистого тела отмечаются нарушения со стороны психики.
Причины аплазии мозолистого тела
Аплазия мозолистого тела (corpus callosum), обеспечивающего связь между полушариями большого мозга и их скоординированное функционирование, является врожденным дефектом, и в большинстве случаев ее точные причины медики определить не могут. Но чаще всего это хромосомные нарушения, влияющие на внутриутробное формирование церебральных структур плода, или наследственно обусловленная аномалия, которая является частью генетических синдромов с пороками развития головного мозга. [2]
Так, мозолистое тело не формируется у плода при синдромах лишней хромосомы (трисомиях), к которым относятся синдромы Варкани, Патау, Эдвардса.
Отсутствие мозолистого тела выявлено при генетически обусловленном синдроме Мовата-Вильсона, синдроме Айкарди, синдроме Мардена-Уокера; синдромах Донна-Барроу, Андермана, Прауда, Аперта, синдроме Х-сцепленной гидроцефалии. А частичная аплазия мозолистого тела характерна для синдромов Питта-Хопкинса, Денди-Уокера, Сенсенбреннера.
Нарушается формирование мозолистого тела при аномалиях извилин мозга, например, шизенцефалии, или при врожденных энцефалоцеле и кистах мозговых структур (как в случае синдрома Чадли-Маккалоу), а также мальформации или синдроме Арнольда-Киари. [3]
Среди вероятных факторов риска аплазии мозолистого тела и других врожденных церебральных пороков отмечаются тератогенные воздействия на эмбрион повышенной радиации и различных токсинов; употребляемых в период беременности алкоголя и наркотических средств; применение некоторых лекарственных препаратов и вирусные инфекции матери.
Также повышен риск данного дефекта у ребенка при наличии нарушений развития и дисгенезии головного мозга в семейном анамнезе.
Мозолистое тело начинается формироваться на шестой-восьмой неделе беременности, но нарушения этого процесса может происходить в промежутке между третьей и пятнадцатой неделями гестации. В эмбриологии патогенез отсутствия мозолистого тела связывают с двумя биологическими механизмами.
Во-первых, это может объясняться дефектами генов, которые регулируют и координируют дорсолатеральную миграцию - перемещение эмбриональных клеток нервного гребня (экзодермального клеточного тяжа на краях нервной трубки) или мезендодермы головы к местам формирования структур головного мозга. Большинство эмбриональных пороков и врожденных дефектов - результат нарушения данного процесса.
Другой механизм агенезии мозолистого тела может заключаться в том, что аксоны нейронов неокортекса не пересекают среднюю линию между полушариями головного мозга эмбриона, и вместо формирования волокнистого тракта между правым и левым полушариями происходит образование аномальных пучков нервных волокон, которые располагаются продольно - не соединяя полушария мозга. [4]
Симптомы аплазии мозолистого тела
Какие симптомы вызывает нарушение взаимодействия между полушариями мозга при отсутствии связи между ними, которую должно обеспечивать мозолистое тело?
У младенцев первые признаки могут проявляться проблемами с кормлением и частыми судорогами. Но при наличии врожденных синдромов клиника включает их симптоматику, в том числе, аномалии черепа (микроцефалию), лицевого скелета (микрогнатию) и черт лица; олигодактилию или отсутствие пальцев; спастичность и контрактуры суставов и т.д. [5]
При аплазии мозолистого тела у ребенка могут отмечаться нарушения зрения и слуха, пониженный мышечный тонус и плохая координация движений - со значительной задержкой начала ходьбы и развития моторики. Присутствуют и когнитивные нарушения (с неспособностью воспринимать информацию, дефицитом внимания и проблемами освоения речи), и отклонения в поведении (нередко схожие с аутизмом). [6]
Аплазия мозолистого тела имеет последствия и осложнения, которые варьируются в зависимости от связанных аномалий головного мозга. У детей с наиболее тяжелыми пороками развития головного мозга могут быть судороги, спастичность, гидроцефалия, нарушения физического и умственного развития.
Диагностика аплазии мозолистого тела
Для установления генетического диагноза проводятся молекулярно-генетические исследования, хромосомные и субтеломерные анализы. В пренатальной педиатрии практикуется генетическое тестирование во время беременности - с помощью амниоцентеза (анализа околоплодной жидкости). [7]
Выявить патологию способна только инструментальная диагностика:
Визуализация позволяет установить наличие таких конкретных структурных особенностей частичной аплазии, как рудиментарный рострум (передней отдел corpus callosum) или аплазия задних отделов мозолистого тела - radiatio corporis callosi и splenium. [8]
Чтобы идентифицировать и отличить другие патологии мозолистого тела - дисгенезию (дефектное развитие), гипоплазию (частичное недоразвитие), атрофию или гипоплазию мозолистой оболочки, а также подтвердить наличие генетического синдрома - проводится дифференциальная диагностика. [9]
Шизэнцефалия
Шизэнцефалия — врожденный порок ЦНС в виде расщелины головного мозга, возникающий в результате поздней нейрональной миграции. Основными факторами риска болезни служат генетические дефекты, тератогенные влияния в антенатальном периоде, внутриутробная гипоксия и нейроинфекции. Состояние проявляется полиморфными судорожными приступами, задержкой психомоторного развития, очаговой неврологической симптоматикой. Пренатальная диагностика выполняется на плановом УЗ-скрининге беременности, постнатальная — с помощью церебрального МР-сканирования, нейросонографии, ЭЭГ. Лечение поддерживающее: антиконвульсанты, ноотропы, проведение комплексной реабилитации. При наличии осложнений выполняются нейрохирургические вмешательства.
Первые описания патологии были сделаны Вилмартом еще в 1887 году, после чего в 1946 г. ученые Яковлев и Вадсфорф выделили два морфологических подтипа заболевания. Первое описание ультразвуковой картины порока принадлежит У. Клингенсмиту, Д. Койффи-Рагану. Шизэнцефалия встречается с частотой 1,5:100000 живорожденных новорожденных. Намного чаще патология регистрируется среди пациентов с эпилептическими приступами — 1 случай на 1650 больных. Расовых и половых различий среди заболевших не выявлено.
Этиологические факторы аномалии изучены недостаточно. В современной неврологии существуют разные мнения по поводу порока: одни специалисты связывают его развитие с церебральной ишемией в эмбриональном периоде, другие — с дизрупцией (деструктивным процессом в первоначально правильно сформированном органе), возникшей под влиянием неизвестных причин. Выделяют следующие факторы риска формирования расщелины мозга:
- Генные мутации. У ряда новорожденных присутствует мутантный ген EMX2 (ген гомеобокса), который участвует в росте и дифференцировке нейробластов. Изредка шизэнцефалия выявляется при мутации COL4A1. Поскольку генный дефект есть не у всех пациентов, в этиопатогенезе играют роль и другие факторы.
- Внутриутробная гипоксия. Такое состояние встречается при наличии у матери экстрагенитальных заболеваний (анемия, сердечная недостаточность, болезни легких), патологий беременности (резус-конфликт), различных генетических синдромах у плода.
- Токсические воздействия. Нарушения эмбрионального развития часто провоцируются негативным влиянием табачного дыма (в том числе при пассивном курении), алкоголя, бытовых и промышленных химических токсинов. Некоторые лекарства (антикоагулянты, антибиотики, цитостатики) также могут нарушать формирование ЦНС у плода.
- Внутриутробные инфекции. Возникновение шизэнцефалии возможно при первичном заражении или активации латентной инфекции в первом триместре беременности. Наибольшей тропностью к нервной ткани обладают вирусы простого герпеса, цитомегаловирус, токсоплазма.
Ряд исследователей основным звеном патогенеза называют внутриутробный инсульт в бассейне средней мозговой артерии, что приводит к образованию участка ишемизированной ткани, в котором нарушаются процессы структурной организации клеток. Церебральной гипоксии при формировании шизэнцефалии обычно сопутствует хронический воспалительный процесс, вызванным нейроинфекцией.
Существует два типа болезни с учетом патоморфологических особенностей. Для 1 типа характерна сомкнутая расщелина, края которой разделены узкой бороздкой, покрытой сверху эпендимой и паутинной оболочкой. При 2 типе патологии формируется разомкнутая расщелина с далеко отстоящими друг от друга краями. При этом желудочки сообщаются с субарахноидальным пространством, происходит циркуляция ликвора.
Макроскопически патология у новорожденных представляет собой щель головного мозга с преимущественной локализацией в области латеральной борозды. Микроскопически при таком пороке ЦНС наблюдается дисплазия серого вещества, покрывающего расщелину, нарушение структуры слоев мозговой коры в зоне поражения. Зачастую, кроме шизэнцефалии, у новорожденных, детей другого возраста выявляются другие пороки: врожденные аномалии прозрачной перегородки, мозолистого тела, зрительного перекреста.
Симптомы шизэнцефалии
Клиническая картина заболевания варьирует в зависимости от локализации и размеров расщелины. Средний возраст манифестации симптоматики составляет 4 года, хотя в тяжелых случаях отдельные признаки выявляются уже у новорожденных. Для шизэнцефалии, как и для других кистозных полостей головного мозга, типично запаздывание клинических проявлений, что затрудняет диагностику, если она не была выполнена в ходе пренатального скрининга.
Основным признаком болезни являются разнообразные эпилептические пароксизмы: сложные фокальные приступы (нарушения сознания в сочетании с подергиванием одной из конечностей), простые фокальные припадки, фокальные пароксизмы с вторичной генерализацией. Интенсивность пароксизмальных явлений колеблется от единичных приступов до 10-30 пароксизмов в сутки. У новорожденных и детей первого года жизни отмечаются единичные тонические или миоклонические приступы.
К непароксизмальным симптомам болезни относят нарушения иннервации лицевой мускулатуры, что проявляется асимметрией лица, обеднением мимики. Также развиваются парезы мышц, иннервируемых бульбарной группой нервов, что манифестирует расстройствами глотания, произношения звуков. У новорожденных, а также младенцев возможна персистенция безусловных рефлексов, у детей постарше — гемипаретическая форма ДЦП.
Более редкими признаками расщелины мозга служат нарушения сна, ощущения шума или пульсации в голове, расстройства зрения (яркие вспышки света перед глазами, ухудшение остроты зрения, сужение зрительных полей). Такие жалобы в основном предъявляют дети школьного возраста. Возможны нарушения координации движений, неустойчивость походки, мелкоразмашистый тремор.
Расщелина мозга чревата формированием гидроцефалии, которая проявляется увеличением и деформацией головы ребенка, нарушением оттока ликвора. Без своевременной медицинской помощи существует риск гипертензионно-гидроцефального синдрома. У новорожденного, младенца возникают генерализованные судороги, выбухание родничка, церебральная рвота. При поражении ствола мозга на фоне гидроцефалии возможен летальный исход.
Тяжелый неврологический дефицит наблюдается при сочетании шизэнцефалии и септо-оптической дисплазии, гетеротопии серого вещества. Дети с расщелиной головного мозга страдают от умственной отсталости, задержки речевой функции, различных психических отклонений. Опасность представляют фармакорезистентные судорожные приступы, которые чреваты развитием эпилептического статуса.
На современном этапе развития ультразвуковой диагностики в большинстве случаев шизэнцефалия диагностируется антенатально во время планового скрининга беременности. Намного лучше визуализируется расщелина сомкнутого типа, тогда как открытая шизэнцефалия может остаться незамеченной. Для постнатального подтверждения диагноза детскому неврологу требуются следующие методы исследования:
- МРТ головного мозга. Магнитно-резонансная томография — безопасный метод даже для новорожденных. При 1 типе аномалии обнаруживается выпячивание стенки желудочков с узким каналом. 2 тип характеризуется открытой расщелиной, которая заполнена цереброспинальной жидкостью. При необходимости выполняется МРТ мозговых сосудов.
- Нейросонография. УЗИ головного мозга используется как альтернатива МРТ у новорожденных, младенцев до закрытия большого родничка. Сонография достаточно информативна при шизэнцефалии I типа. Метод также применяется для выявления сопутствующих аномалий строения церебральных структур. Для уточнения диагноза нейросонография дополняется УЗДГ церебральных сосудов.
- ЭЭГ. По результатам электроэнцефалографии у больных разных возрастных групп, включая новорожденных, определяется замедление основной фоновой активности, региональные эпилептиформные паттерны в лобно-центрально-височной зоне. Реже наблюдается вторичная билатеральная синхронизация, мультифокальная эпилептиформная активность.
- Офтальмоскопия. При исследовании глазного дна обнаруживается важный офтальмологический симптом гидроцефалии — отечность дисков зрительных нервов. Метод применим для больных разного возраста, в том числе, новорожденных. Для оценки работы зрительного анализатора рекомендованы визометрия, периметрия, биомикроскопия глаза.
- Генетическое тестирование. Поскольку аномалия сочетается с мутациями EMX2, COL4A1, показано проведение автоматического секвенирования экзона. Однако информативность такого метода остается невысокой ввиду полиэтиологичности заболевания.
Лечение шизэнцефалии
Консервативная терапия
Медицинская помощь детям с шизэнцефалией ограничивается поддерживающим симптоматическим лечением. Больным показано динамическое наблюдение у детского невролога, регулярный КТ- или МРТ-контроль состояния расщелины, определение наличия или отсутствия вторичных структурных изменений мозговых тканей. Основные группы препаратов, которые включаются специалистом в индивидуальную схему лечения шизэнцефалии:
- Антиконвульсанты. Назначаются различные комбинации противосудорожных препаратов для купирования эпилептических приступов, предупреждения рецидивов.
- Нейрометаболиты. Препараты, улучшающие трофику головного мозга, используются для стимуляции психоречевого и моторного развития, уменьшения неврологического дефицита.
- Дегидратанты. Для нормализации ликвородинамики могут рекомендованы осмотические диуретики, салуретики, онкодегидратанты. Лечение проводится под контролем диуреза и объема вводимых инфузионных растворов.
Оперативное лечение
Хирургическое лечение применяется при осложненном течении заболевания. Обычно операции проводятся новорожденным, младенцам для ликвидации нарастающей гидроцефалии. Чаще всего выполняются эндоскопическая вентрикулостомия III желудочка и ликворошунтирующие вмешательства. При учащении эпилептических пароксизмов, появлении и нарастании у новорожденного либо пациента другого возраста грубой очаговой неврологической симптоматики производится аспирация содержимого крупных кист.
Реабилитация
Важную роль играет комплексная реабилитация пациентов, включая новорождённых и детей младенческого возраста. Для повышения мышечной силы, объема произвольных движений используется массаж, рефлексотерапия, механотерапия и ЛФК. Для развития речи ребенку требуется помощь логопеда-дефектолога. С целью повышения обучаемости и социализации больных прибегают к услугам нейропсихолога, коррекционного педагога.
Благоприятное течение характерно для новорожденных с сомкнутой односторонней шизэнцефалией, которая не сопровождается грубым неврологическим дефицитом. Внушает опасения разомкнутая билатеральная форма порока, вызывающая раннюю инвалидизацию и смерть больных. Профилактические меры включают рациональное ведение беременности, исключение тератогенных влияний на организм будущей матери, усовершенствование методики пренатальных УЗ-скринингов.
1. Шизэнцефалия (обзор литературы и клинические наблюдения/ О.А. Милованова, И.М. Мосин, Л.В. Калинина// Неврологический журнал. — 2011. — №3.
2. Пренатальная диагностика шизэнцефалии/ А.Е. Волков, Е.Н. Андреева// SonoAce Ultrasound. — 2010. — №21.
3. Лучевые методы исследования, МРТ головного мозга у больных с шизэнцефалией/ М.И. Пыков, К.В. Ватолин, О.А. Милованова, Н.В. Чернышева// Эффективная фармакотерапия. Педиатрия. — 2010. — №5.
4. Шизэнцефалия, тип II: случай пренатальной диагностики/ Е.Н. Андреева// Пренатальная диагностика. — 2008. — №4.
Читайте также:
- Разномодальные агрессивно-оборонительные реакции. Механизмы аффективной и холодной атаки
- Врожденные миопатии у ребенка
- Принципы и схема лечения больных острой лучевой болезнью
- Рентгенограмма, КТ, МРТ, УЗИ при дисгенезии гонад
- Антигены аденовирусов. Патогенез поражений аденовирусов. Клиника, диагностика, лечение и профилактика аденовирусных инфекций.