Дисплазия радужной оболочки глаз, гипертелоризм с задержкой развития и глухотой
Добавил пользователь Валентин П. Обновлено: 28.01.2026
Иридодиализом принято называть локализованное разделение (разрыв), образующееся в месте прикрепления радужки к цилиарному телу.
Причины возникновения
Иридодиализ, как правило, становится следствием непроникающей или проникающей травмы глаза (бокс, струя воды под высоким давлением, фейерверк, мяч). Кроме того, он может стать осложнением при любой внутриглазной операции или быть созданным намеренно, к примеру, при интракапсулярной экстракции катаракты.
Симптомы и признаки
Незначительный иридодиализ протекает бессимптомно, не требуя какого-либо лечения. Значительные по площади иридодиализы, как правило, сопровождаются корэктопией (смещением зрачка в сторону от центра), диплопией (двоением видимых предметов), кровоизлиянием в переднюю камеру глаза (гефемой), возникновением бликов, светобоязнью. Большой иридодиализ, обычно приводит к значительному изменению нормальной анатомии радужки.
Нередко иридодиализу сопутствует уменьшение угла передней камеры, что может привести к глаукоме. Отмечались случаи развития гипотонии.
Диагностика
Выявление иридодиализа у пациентов с травмой глаза в анамнезе, в первую очередь определяется на данных световой биомикроскопии. При этом обнаруживаются одиночные или множественные разрывы сфинктера зрачка, неглубокие вдавления и разрывы по всей ширине радужки. Зрачок, обычно приобретает D-образную форму, на радужной оболочке наблюдаются небольшие геморрагии, выраженная гифема или микрогифема. Также иридодиализ может принимать вид небольшого темного цвета полулуния в периферических зонах передней камеры.
Дифференцированную диагностику иридодиализа проводят от более серьезного состояния - циклодиализа. Для этого назначается гониоскопия, которая при циклодиализе выявляет щель, образующуюся между склерой и цилиарным телом, которая уходит в супрахориоидальное пространство. Для иридодиализа характерен отрыв радужки от ее корня.
Лечение
Для иридодиализа, осложненного гифемой, требуется постельный режим и медицинское наблюдение, для предотвращения повторных кровотечений. Красные клетки крови, нередко становятся причиной уменьшения оттока водянистой влаги и повышения ВГД. С целью его снижения, назначают оральные диуретики. На глаз накладывается защитная повязка, предохраняющая от случайной травматизации глаза во сне. Временно отменяются лекарственные средства разжижающие кровь (аспирин, гепарин / варфарин). При большой гифеме может потребоваться промывание передней камеры.
Позже, при больших отрывах, которые привели к серьезной диплопии, бликовым или косметическим симптомам, может рассматриваться вопрос о хирургической операции. Хирургическое лечение обычно выполняется наложением проленового шва (10-0) на оторванный фрагмент радужной оболочки и подшиванием его к узлу цилиарного тела и шпорам склеры. Для уменьшения ощущения диплопии и улучшения косметического состояния глаза, нередко рекомендуется ношение непрозрачных мягких контактных линз.
Осложнения иридодиализа
Травматический иридодиализ (при непроникающей травме глаза, в частности) имеет высокий риск уменьшение глубины угла передней камеры, что нередко приводит к возникновению глаукомы. Травматическая глаукома вследствие иридодиализа продолжается до восстановления объема передней камеры в течение 100 дней или трех месяцев после травмы. При ее наличии может понадобиться хирургическое или медикаментозное лечение под контролем ВГД.
В медицинском центре «Московская Глазная Клиника» все желающие могут пройти обследование на самой современной диагностической аппаратуре, а по результатам - получить консультацию высококлассного специалиста. Мы открыты семь дней в неделю и работаем ежедневно с 9 ч до 21 ч. Наши специалисты помогут выявить причину снижения зрения, и проведут лечение выявленных патологий. Опытные рефракционные хирурги, детальная диагностика и обследование, а также большой профессиональный опыт наших специалистов обеспечивают благоприятный результат для пациента.
Аномалии развития глаза
Аномалии развития глаза - это наследственные или врожденные пороки развития органа зрения, приводящие к неправильной закладке отдельных структур и глазного яблока в целом. Характерно изменение размеров глазного яблока вплоть до его полного отсутствия, недоразвитие роговицы, радужной оболочки, стекловидного тела, хрусталика и других отделов глаза. Патология сопровождается снижением зрения, часто сочетается с другими пороками развития. Диагностируется на основании клинических признаков, офтальмоскопии и биомикроскопии глаза. Лечение аномалий развития глаза направлено на коррекцию зрения и возможное сохранение пораженных частей глаза. Также проводится терапия основного заболевания.
МКБ-10


Общие сведения
Аномалии развития глаза в совокупности встречаются достаточно часто. Многие системные пороки развития клинически проявляются различными дефектами органа зрения. Высокая актуальность данной группы патологий в практической педиатрии связана с огромной ролью зрения в повседневной жизни, особенно с рождения. В то же время, основная часть аномалий развития глаза требует длительной и сложной коррекции, которая нередко приводит лишь к частичному восстановлению зрительной функции. Кроме того, при большинстве пороков необходимо применение высокотехнологичных дорогостоящих методов хирургического лечения. В дальнейшем такие дети практически всегда имеют группу инвалидности и показания для специального обучения на дому.

Причины
Возможно наследование по аутосомно-доминантному, аутосомно-рецессивному типу, X-сцепленное наследование. Также встречаются врожденные аномалии развития глаза, возникающие под влиянием тератогенных факторов в первом триместре беременности. На 2-5 неделях эмбриогенеза происходит закладка органа зрения, воздействие радиации, алкоголя, никотина, наркотиков и запрещенных медикаментов именно в этот период приводит к нарушению правильного формирования глаза. Внутриутробные инфекции также становятся причиной врожденных пороков органа зрения. Реже аномалии формируются в составе хромосомных синдромов, например, при синдроме Дауна.
Симптомы
Патология может затрагивать глаз в целом, а может представлять собой нарушение закладки отдельных оболочек и структур органа зрения. Аномалии развития глаза в целом проявляются изменением его размеров. Чаще всего речь идет о микрофтальме различной степени вплоть до полного отсутствия глазного яблока (анофтальм). Пораженный глаз визуально меньших размеров, возможен двусторонний микрофтальм. Также встречается криптофтальм, представляющий собой недоразвитие век, которые частично или полностью срастаются с роговицей. Такие грубые аномалии развития глаза заметны сразу после рождения.
Порок может затрагивать отдельно роговицу, радужную оболочку, хрусталик иди другие отделы глазного яблока. Заметить аномалию развития может педиатр и детский офтальмолог. Врожденные патологии радужки характеризуются ее недоразвитием или отсутствием (аниридия), наличием колобом или округлых дефектов, напоминающих зрачки (поликория). Аномалии хрусталика и сетчатки могут визуально не проявляться и диагностируются только в момент осмотра.
Для всех аномалий развития глаза характерно снижение зрения различной степени выраженности. По этой причине малыш может не реагировать на свет и на окружающих. Пороки часто сочетаются с другими дефектами лицевого черепа, а также с дисплазией костной ткани, умственной отсталостью и другими заболеваниями.
Первичная диагностика возможна уже в роддоме. Грубые аномалии развития глаза, как уже было сказано, заметны при первом осмотре, диагноз не вызывает сомнения. Осмотр офтальмолога в роддоме также является обязательным для всех новорожденных. Офтальмоскопия позволяет выявить аномалии развития радужной оболочки, хрусталика. Для подтверждения диагноза часто требуется биомикроскопия глаза - осмотр при помощи щелевой лампы. Исследование дает возможность обнаружить мелкие дефекты, которые можно пропустить при обычной офтальмоскопии. Например, так можно заметить вывихи и подвывихи хрусталика, обычно связанные с заболеваниями соединительной ткани (синдром Марфана), вследствие чего мышцы хрусталика не могут держать его в правильном положении.
Лечение аномалий развития глаза
Тактика ведения пациентов зависит от вида порока, возможности восстановления утраченной функции зрения, а также от возраста ребенка и других факторов. При аномалиях глазного яблока показана стимуляция роста орбиты при помощи специальных имплантатов. В дальнейшем может быть выполнена операция по имплантации искусственного глаза. С целью улучшения зрения всем пациентам проводится плеоптическая терапия, способствующая стимуляции развития пораженного участка. В целом терапия направлена на улучшение адаптации пациентов, поскольку зрительный анализатор считается основным источником информации из внешнего мира.
Хирургическое лечение включает операции, например, по замене хрусталиков в случае врожденной катаракты, вмешательства на роговице и другие микрохирургические манипуляции. Коррекция зрения также проводится с помощью очков и контактных линз. При недоразвитии радужной оболочки или полном ее отсутствии показаны специальные линзы, выполняющие функции радужки.
Поскольку аномалии развития глаза часто сочетаются с другими пороками, необходима их хирургическая коррекция, а также лечение основного заболевания. Многие врожденные обменные нарушения, например, мукополисахаридозы, являются причиной глазных аномалий, лечение ферментами в подобных случаях также улучшает прогноз для органа зрения.
Прогноз и профилактика
Грубые пороки плохо поддаются коррекции. При одностороннем анофтальме зрение на втором глазу обычно снижено. Методы плеоптической терапии имеют лишь незначительный эффект, качество жизни улучшается только после имплантации глаза. Однако даже в этом случае инвалидность неизбежна. Кроме того, аномалии развития глаза редко встречаются изолированно, что также оказывает негативное влияние на прогноз. Профилактика заключается в исключении воздействия тератогенных факторов на ранних сроках гестации. Необходимо планирование беременности и генетическая консультация в случае наследственных заболеваний у родителей и других родственников.
1. Большие и малые аномалии развития органа зрения при хромосомной патологии/ Шерстнев Г. Е.// Вестник Совета молодых учёных и специалистов Челябинской области. - 2017.
2. Морфологические особенности клинических проявлений некоторых видов врожденных аномалий хрусталика и стекловидного тела/ Харлап С. И., Салихова А. Р., Аветисов К. С., Аветисов С. Э. // Вестник офтальмологии. - 2017. - 133(2).
Аномалии развития сетчатки глаза (колобома, дисплазия, гемангиома)
Аномалии оболочек глаза обнаруживаются сразу после рождения ребенка. Они могут возникать из-за мутации генов, хромосомных сбоев, токсического воздействия во внутриутробном периоде экзогенных или эндогенных факторов. Кроме того, огромное значение имеют инфекционные заболевания, которыми мать болеет в период беременности. Также влияние оказывают воздействующие на эмбрион негативные факторы окружающей среды, токсины, лекарственные препараты, радиация и пр. Особенно грубые изменения глазных сред выявляются при воздействии на плод вредных факторов на начальных сроках беременности. Как правило, негативно влияющими, становятся следующие инфекции: краснуха, сифилис, токсоплазмоз, цитомегаловирус, герпес, ВИЧ. Среди лекарственных средств и веществ, к наиболее опасным относятся: кокаин, талидомид, этанол.

Что такое колобома глаза
Аномалии сетчатки
К врожденным аномалиям сетчатки принято относить: колобому аплазию, гипоплазию, дисплазию, альбинизм. Кроме того, врожденными дефектами являются: гиперплазия пигментного эпителия, сосудистые аномалии, миелиновые нервные волокна, факоматозы.
Колобома сетчатки — это отсутствие ретинальной ткани на ограниченном участке. Как правило, она сопровождается колобомами радужки и хориоидеи, может локализоваться в центре глаза или на периферии нижней его половины. Причина возникновения колобомы — неполное закрытие эмбриональной щели. При офтальмоскопии, данная аномалия выглядит белой ограниченной областью овальной либо круглой формы недалеко от диска зрительного нерва,иногда прилегающей к нему. При отсутствии сетчатки и хориоидеи, склера обнажается. Нередко колобома сопровождается микрофтальмом, дефектами развития скелета и др.
Дисплазия сетчатки — аномалия, возникающая в процессе развития плода, как нарушение соотношения элементов клеток. Дисплазия может проявляться неприлеганием сетчатки. Это редкая патология, возникающая из-за недостаточной инвагинации оптического везикула, является одним из признаков трисомии 13, а также синдрома Вокера — Варбурга. Выявляется в сочетании с иными дефектами глаза, мышечной ткани, мозжечка.
Альбинизм — генетически обусловленное нарушение развития системы зрения, возникающее из-за нарушения синтеза меланина. Люди с альбинизмом также могут страдать нистагмом, аномалиями рефракции с астигматизмом, иметь слабую пигментацию глазного дна, дисплазию центральной зоны сетчатки, изменение перекреста зрительного нерва. Указанной аномалии также присущи дефекты цветового зрения, изменение яркости и контрастности, межполушарная асимметрия ЗВП сверхнормальная ЭРГ.
Альбинизм бывает тирозиназонегативный и тирозиназопозитивный. В первом случае, возникновение заболевания связано с отсутствием выработки фермента тирозиназы, а также пигмента меланина. Люди с таким заболеванием имеют белую кожу и волосы, не загорают. Их светлая радужка легко просвечивается, рефлекс с глазного дна окрашен в ярко-розовый тон, виден на расстоянии. При тирозиназопозитивной форме альбинизма, наоборот, способность к выработке меланина сохраняется, но его нормальное накопление невозможно. Кожа таких пациентов способна загорать, но слабо пигментирована. Волосы имеют очень светлый или желтоватый оттенок, нарушения зрения — меньшую степень выраженности.
До настоящего времени альбинизм не лечится. Медицинская помощь заключается в очковой коррекции нарушений рефракции оптическими стеклами со светофильтрами от повреждающего сетчатку воздействия яркого света.
Гиперплазия пигментного эпителия врожденного характера, характеризуется очаговой гиперпигментацией сетчатки. Группировка пигментных пятен имеет форму медвежьего следа с единичными и множественными очагами. Изменения на сетчатки вокруг них отсутствуют. Увеличение очагов пигментации происходит редко, они практически не подвержены малигнизации.
Также к аномалиям развития относят и миелиновые нервные волокна. Их могут относить, как к дефектам развития сетчатки, так и к дефектам развития зрительного нерва.
Миелиновое покрытие волокон зрительного нерва в норме должно заканчиваться у дальнего края решетчатой пластинки. В некоторых случаях оно продолжается вплоть до нервных волокон ретинальных нейронов второго порядка, минуя диск зрительного нерва. При проведении офтальмоскопии волокна выглядят белыми радиально расположенными блестящими полосами, которые идут к периферии от диска зрительного нерва. Иногда с диском зрительного нерва они не связаны. Как правило, подобная аномалия не сопровождается какими-либо симптомами, хотя в некоторых случаях может происходить возникновение скотом.
Аномалии сосудов сетчатки врожденного характера проявляются как гроздьевидная ангиома, капиллярная гемангиома Гиппеля—Линдау, болезнь Коатса, а также, в виде кавернозной гемангиомы, ретинопатии недоношенных, просовидных (милиарных) аневризм Лебера, капиллярной гемангиомы, парафовеальных телеангиоэктазий и пр.

Болезнь Коатса диагностика и лечение
Гроздьевидная ангиома — это, как правило, односторонний дефект, который при офтальмоскопии имеет свои характерные признаки: расширенные и извитые в значительной степени артерии и вены, артериовенозные шунты. При наличии церебральной сосудистой патологии со снижением центрального зрения, говорят о «синдроме Вабурна—Мазона». Лечение в большинстве случаев не проводится, так как болезнь не прогрессирует.
Болезнь Коатса — врожденные сосудистые аномалии с характерными , телеангиэктазиями сетчатки, различного размера аневризмами с отделением экссудата, что со временем приводит к отслойке сетчатки. Еще одно название заболевания - «наружный геморрагический ретинит». Как правило, оно имеет одностороннее течение, выявляется у детей (обычно мальчиков) в раннем возрасте.
При офтальмоскопии обнаруживается отложение твердого ярко-желтого экссудата в субретинальном пространстве заднего глазного полюса. На поздних стадиях болезни возникает катаракта, глаукома, происходит субатрофия глаза. При средней тяжести патологии отмечаются только телеагиэктазии.
При диагностике, такую аномалию необходимо дифференцировать с опухолевыми процессами, скрытыми экссудатом или отслоенной сетчаткой, а также с ретинопатией недоношенных.
Лечение - устранение экссудации посредством облитерации аномальных сосудов лазеркоагуляцией или криопексией. Если наблюдается масштабная экссудативная отслойка сетчатки, назначается оперативное лечение.
Факоматозы - пороки развития врожденного характера с офтальмологическими и системными проявлениями. Наиболее распространенными среди них являются: гемангиомоподобные образования, гумартомы, узлы.
К факоматозам относят нейрофиброматоз Реклингаузена, болезнь Гиппеля - Линдау, туберозный склероз, возникающие по ауто-сомно-доминантному принципу. Кроме того, к ним относят синдром Стерджа - Вебера - Краббе.
Причина болезни - мутация гена, идентифицированного во всех случаях доминантных заболеваний.
Для нейрофиброматоза Реклингаузена характерна опухоль шванновских клеток, с кожным проявлением — возникновением множественных фибром. Ответственный за развитие заболевания ген находится в 17-й хромосоме. Болезнь проявляется деформирующей нейроматозной слоновостью, которую провоцирует нейрофиброматозная инфильтрация.
При диагностике, обращают внимание на пятна на коже цвета кофе с молоком. Их должно быть не менее 6, размером свыше 1,5 см.
Среди офтальмологических проявлений нейрофиброматозов отмечают: плексиформную нейрофиброму глазницы и век, врожденную глаукому, S-образную форму глазной щели, гамартомы радужки и гамартомную инфильтрацию в сосудистую оболочку, конъюнктивальную нейрофиброму, проминирование и утолщение нервов роговицы, глиому зрительного нерва, буфтальм, пульсирующий экзофтальм.
Гамартома - опухоль ткани эмбриона с задержкой дифференцировки, относительно дифференцировки органа, на котором она развивается. У клеток такой опухоли нет аномалий в структуре, но произошли изменения в плотности клеточных популяций.
Меланоцитарные гамартомы, носящие название узелков Пиша, выявляются на радужке взрослых больных еще до кожных проявлений заболевания и являются диагностическим признаком.
Плексиформная нейрофиброма - клубок гипертрофированных, переплетенных между собой нервов. Из-за пролиферации шванновских клеток, они выглядят бугристыми.
Особенно частое осложнение нейрофиброматоза первого типа - сосудистые нарушения. К ним относят: окклюзии сосудов или сужение их просвета, которые в дальнейшем приводят к периваскулярной фиброглиальной пролиферации. Одними из признаков кислородного голодания сетчатки при этом принято считать аваскулярные зоны по ее периферии, преретинальные фиброглиальные мембраны, артериовенозные шунты, атрофию зрительного нерва.
Если опухоль стала причиной деформации прилежащих тканей или нарушения функции органа, ее необходимо удалить.
Нейрофиброматоз второго типа - заболевание редкое. Его типичным симптомом является швантома (обычно, двусторонняя) восьмой пары черепных (слуховых) нервов. Офтальмологическое осложнение - это комбинированные гамартомы пигментного эпителия сетчатки, менингиома зрительного нерва, глиома.
Что такое гамартома глаза
Болезнь Гиппеля-Линдау представляет собой генетически обусловленное заболевание с дефектом в 3р25 хромосоме. Как правило, обнаружение ее происходит случайно, когда ребенок проходит диагностическое обследование на предмет косоглазия.
Для ангиом сетчатки характерна форма черешен, которые снабжены развитыми сосудами, питающими и дренирующими ее. Подобные образования - гемангиобластомы сетчатки, поскольку гистологически они подобны последним, формирующимся в мозжечке. Для гемангиобластом сетчатки характерен экзофитный и эндофитный рост с вовлечением в процесс диска и самого зрительного нерва. Нередко выявляется их сочетание с макулопатиями. Ангиоматоз сетчатки также может сопровождать кистоз почек, карциному почки, феохромоцитому и пр.
Из-за повышенной проницаемости сосудов, внутри может скапливаться ретинальный экссудат с большим количеством липидов, что приводит к развитию экссудативной отслойки сетчатки на последних стадиях заболевания.
При диагностике артериовенозной ФАГ, выявляется концентрация в ангиоме контрастного вещества. На поздних стадиях заболевания, флюоресцеин выходит за пределы сосудов в окружающие ткани, что обусловлено дефективностью опухолевых сосудов.
В качестве лечения, применяют лазерную коагуляцию, криолечение, хирургическое удаление опухоли.
Туберозный склероз, получивший название болезни Бурне-Вилля, - редкое аутосомно-доминантное заболевание, возникающее из-за сбоя двух генов в 9-й и 16-й хромосомах.
Классическая триада патологии - эпилепсия, ангиофибромы лица, умственная отсталость. Сетчатка поражена беловатыми опухолевыми образованиями, локализующимися возле диска зрительного нерва, отдаленно напоминающие плоды шелковицы. На диске зрительного нерва могут выявляться астроцитомы, которые получили название гигантских друз. Иногда их ошибочно ассоциируют с ретинобластмой.
В качестве лечения выбирают помещение пациентов в неврологическую клинику. Прогноз заболевания серьезный: при нарастании неврологических проявлений вскоре наступает летальный исход.
Синдром фронтоназальной дисплазии. Возможности пренатальной диагностики и особенности медико-генетического консультирования
1 Медико-генетическое отделение Московского областного НИИ акушерства и гинекологии,
2 Кафедра медицинской генетики ФГБОУ ДПО РМАНПО Минздрава России, Москва.
Фронтоназальная дисплазия (ФНД) - порок развития средней части лица, заключающийся в нарушении перемещения глаз по направлению к носу в процессе эмбриогенеза. Впервые был описан в 1967 г. W. De Myer [1]. Автор изучал больных с единообразным пороком развития и назвал это заболевание синдромом срединной расщелины лица. В 1970 г. S. Sedano и соавт. предложили переименовать этот синдром в синдром фронтоназальной дисплазии, так как расщелина лица не была облигатным признаком, строго определяющим этиопатогенез синдрома [2]. Этими же авторами был описан эмбриопатогенез ФНД: порок формируется с 19-го по 21-й день эмбрионального развития из-за нарушения миграции мезодермы, обусловленного мутацией в ALX3 гене [3].
Синдром ФНД характеризуется аутосомно-доминантным типом наследования с различной пенетрантностью (проявляемостью) и экспрессивностью (степенью выраженности). При этом чаще всего встречаются спорадические случаи, как проявление мутации de novo [4].
Манифестные (часто встречаемые) признаки синдрома ФНД или синдромальное "ядро" заболевания (рис. 1):
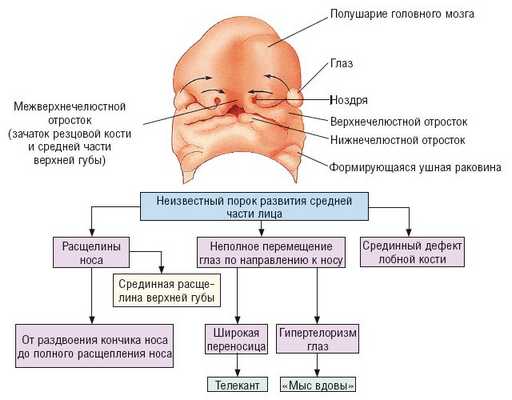
Рис. 1. Патогенез синдрома ФНД [6].
Глаза: узкие глазные щели, гипертелоризм, эпи-, телекант, катаракта, дегенерация сетчатки, колобома нижнего века.
Лоб: клиновидный рост волос на лбу ("мыс вдовы"), срединный дефект лобной кости (скрытая расщелина черепа, лобное менингоэнцефалоцеле).
Нос: расщелины разной степени тяжести (от раздвоенного кончика носа до полной расщелины, возможно, в сочетании с широкой срединной расщелиной верхней губы), расщелины крыльев носа, широкая переносица, отсутствие кончика носа, назальные кожные привески.
Патология центральной нервной системы: агенезия мозолистого тела.
К редким симптомам, описанным при ФНД, относят: носовые и ушные привески, микрофтальм, низко расположенные уши, кондуктивную тугоухость, липомы на лбу и в мозолистом теле. В исследовании, посвященном синдрому ФНД [6], было установлено, что агенезия мозолистого тела встречалась в 57% случаев, липома мозолистого тела - в 19% случаев. Также описаны дефект межжелудочковой перегородки, тетрада Фалло, поли-, син-, брахидактилия, расщелина позвоночника, омфалоцеле, крипторхизм [7].
Прогноз для жизни и здоровья при синдроме ФНД зависит от наличия и тяжести сопутствующих аномалий. Пороки развития лица устраняются обычно серией пластических операций. По данным литературы [6, 7], интеллект у больных ФНД обычно сохранен. Однако W. De Myer отмечает умственную отсталость у 8% и легкое снижение интеллекта у 12% больных [1].
Пренатальная диагностика синдрома фронтоназальной дисплазии
Несмотря на то что изменения фенотипа при синдроме ФНД, казалось бы, очевидны: расщелина лица, агенезия мозолистого тела, патология мягких тканей носа, гипертелоризм и др. и не должны вызывать затруднений у врача ультразвуковой пренатальной диагностики, в литературе встречается ограниченное количество публикаций, посвященных пренатальной диагностике синдрома ФНД. Редкие работы о пренатальной диагностике единичных случаев синдрома посвящены в основном применению новых технологий 3D/4D с методиками поверхностной реконструкции 9. Этот факт объясняется, скорее всего, тем, что врачи выставляют диагноз отдельных симптомов данного заболевания (чаще всего это расщелина губы/неба, лобная черепномозговая грыжа, агенезия мозолистого тела, патология развития мягких тканей носа) с перечислением всех найденных при ультразвуковом исследовании пороков без попытки провести клинико-синдромальный поиск. Такой "однобокий" подход к диагностике найденных аномалий не позволяет выставить правильный клинический диагноз синдрома ФНД, что в дальнейшем приведет к неполному и неадекватному медико-генетическому консультированию (МГК) семьи, которое заключается как в определении прогноза на данную беременность, так и в формировании тактики репродуктивного поведения семьи в дальнейшем и выработке специфических мер профилактики патологии.
Медико-генетическое консультирование при синдроме ФНД
При диагностике патологии с аутосомнодоминантным типом наследования изучение фенотипа/генотипа родителей позволяет установить, явилось ли данное заболевание следствием новой мутации (de novo) либо патологический ген унаследован от кого-то из родителей.
Если у одного из родителей находят даже малейшие признаки синдрома ФНД (учитывая различную пенетрантность и экспрессивность генов, которые определяют клиническую выраженность симптомов), риск повтора данной патологии составит 50%, в случае возникновения мутации de novo этот риск не превышает уровень общепопуляционного (1%), так как члены семьи здоровы.
Клиническое наблюдение 1
При проведении пренатальной эхографии в 34 нед беременности (настоящая беременность вторая, в семье один здоровый ребенок) в медико-генетическом отделении МОНИИАГ были выявлены лицевые дизморфии у плода женского пола - гипертелоризм, раздвоенный кончик широкого носа, образование в области переносицы (лобное менингоцеле малых размеров). Выставлен пренатальный диагноз синдрома ФНД, имеющей аутосомно-доминантный тип наследования, полностью подтвержденный после родов при осмотре новорожденного генетиком-синдромологом (рис. 2).
Колобома зрительного нерва
Основной причиной врожденной колобомы зрительного нерва, а также дефекта оболочек глазного яблока является неполное (или аномальное) сопоставление краев при закрытии эмбриональной щели. В норме процесс этот завершается к 4-5 неделе гестации. Колобома может быть локализована на любом участке края глазной щели, то есть может быть как со стороны радужки и хориоидеи, так и со стороны сетчатки и зрительного нерва.
Клиническая картина
Колобома зрительного нерва может быть расположена как с одной стороны, так и с обеих. При офтальмоскопии доктор определяет шарообразное углубление сребристо-белого цвета с четкими границами. Экскавации подвержен незначительно увеличенный в размере диск зрительного нерва. Обычно она смещена книзу, поэтому нейроретинальный край нередко отсутствует, верхний край при этом выглядит стандартно. Децентрализации экскавации обусловлена положением щели относительно примитивного диска во время зародышевого развития. Колобома может распространяться к нижней части и захватывать примыкающие сетчатку и хориоидею на значительном протяжении.
Острота зрения может быть сохранена либо соответствовать правильной светопроекции. Во время периметрии врач определяет увеличение размера слепого пятна, а также наличие больших центральных или центроцекальных скотом. Иногда колобома зрительного нерва возникает совместно с колобоматозными дефектами в области хориоидеи и сетчатой оболочки. В этом случае нарушение поля зрения зависит от конкретной локализации этих дефектов.
При колобоме зрительного нерва часто имеются и другие аномалии в строении органов оптической системы. К ним относят задний лентиконус, остатки гиалоидной артерии, ямка диска зрительного нерва, задний эмбриотоксон.
Довольно часто колобома зрительного нерва осложняется нерегматогенной отслойкой сетчатой оболочки. Патогенез этого осложнения до конца не выяснен, но чаще оно развивается после 20 лет.
Еще одним осложнением, которое ухудшает течение колобомы зрительного нерва, является перипариллярная неоваскулярная субретинальная мембрана. Ее патогенез также неизвестен, но осложнение это чаще формируется в возрасте 40-50 лет. Однако, в литературе описан случай формирования такой мембраны у пациента четырех лет, который страдал от изолированной колобомы зрительного нерва.
При гистологическом исследовании тканей глаза при колобоме зрительного нерва в области дистального ее конца были обнаружены ориентированные концентрически гладкомышечные клетки. Эта особенность строения объясняет феномен сокращения при офтальмоскопии колобомы зрительного нерва.
Практически у всех пациентов с подобной аномалией имеется косоглазие, миопия высокой степени и миопический астигматизм.
При проведении КТ и В-сканирования выявляется глубокий дефект в задней зоне глаза, а также незначительное увеличение диаметра зрительного нерва в области контакта его со склеральной оболочкой. Также у пациентов с колобомой можно выявить МРТ-признаки ипсилатеральной гипоплазии внутричерепного отрезка зрительного нерва.
Амплитулда ЭРГ чаще сохранена. Иногда при значительных размерах колобомы зрительного нерва, когда в процесс вовлечены большие области сетчатки, при ЭРГ выявляют аномалии. Отмечается перманентное снижение амплитуды компонента Р100, удлинение латентности и изменение конфигурации ответа. ЗВП могут оставаться нормальными при реакции на вспышку, но иногда отмечается удлинение латентности Р100 и уменьшение амплитуды этого компонента.
У детей колобома зрительного нерва часто встречается совместно с другими аномалиями развития организма (очаговая гипоплазия кожи Гольтца, синдром эпидермального невуса, синдром Гольденхара (окулоаурикуловертебральная дисплазия), синдром Дауна, Уокера-Варбурга, Эдвардса). Иногда у пациентов с базальным энцефалоцеле также выявляют колобому зрительного нерва.
У 11% пациентов с колобомой зрительного нерва имеются проявления синдрома CHARGE. Это заболевание включает следующие симптомы: порок сердца, гипотрофия, задержка роста, глухота из-за нарушения работы слухового анализатора, гипоплазия гениталий.
Синдром COACH включает колобому диска зрительного нерва, атаксию и другие проявления патологии мозжечка.
Необходимо отличать изолированную колобому от синдрома северного сияния.
При наличии хориоретинальной колобомы нужно исключить атрофические фокусы, связанный с токсоплазмой или другими патологиями нижнего отдела глазного дна. В последнем случае очаги атрофии обычно пигментированы в центральной зоне.
При колобоме зрительного нерва острота зрения снижена, а при хориоретинальном распространении выявляют дефекты поля зрения, которые соответствуют расположению очага.
Среди осложнений наиболее серьезными является отслойка сетчатки из-за разрыва мембраны, которая выстилает зону хориоретинальной колобомы.
Способы превентивного лечение колобомы зрительного нерва в настоящее время не разработаны. При развитии осложнений (отслойка сетчатки) проводят оперативное вмешательство в виде витрэкстомии, тампонирования газом и ограничительной фотокоагуляции в краевой области разрыва. Если имеется хориоидальная неоваскуляризация, то проводят фотокоагуляцию непосредственно неоваскулярной мембраны.
В медицинском центре «Московская Глазная Клиника» все желающие могут пройти обследование на самой современной диагностической аппаратуре, а по результатам - получить консультацию высококлассного специалиста. Клиника консультирует детей от 4 лет. Мы открыты семь дней в неделю и работаем ежедневно с 9 ч до 21 ч. Наши специалисты помогут выявить причину снижения зрения, и проведут грамотное лечение выявленных патологий.
Читайте также:
- Как выбрать визажиста на свадьбу? Визажист для свадебного макияжа
- Причины и механизмы развития гистиоцитозов из клеток Лангерганса. Этиология, патогенез
- Диагностика спонтанного внутричерепного кровоизлияния по КТ, МРТ
- Случай достижения стойкой ремиссии плантарного фасциита при комплексном консервативном лечении плоскостопия
- Детский ботулизм