Галактоземия - причины, симптомы, диагностика и лечение
Добавил пользователь Алексей Ф. Обновлено: 22.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Галактоземия: причины появления, симптомы, диагностика и способы лечения.
Определение
Галактоземия - группа наследственных нарушений обмена углеводов, вызывающих в организме накопление избытка галактозы и ее метаболитов. Галактоза (от греч. galaktos - молоко) - это моносахарид, который входит в состав лактозы (молочного сахара) и содержится во всех молочных продуктах, включая грудное молоко.
В течение дня люди потребляют продукты, содержащие лактозу, из которой в кишечнике в результате гидролиза образуется галактоза.
Кроме того, галактоза может образовываться эндогенным путем, и подавляющее ее количество синтезируется в процессе ферментативных реакций.
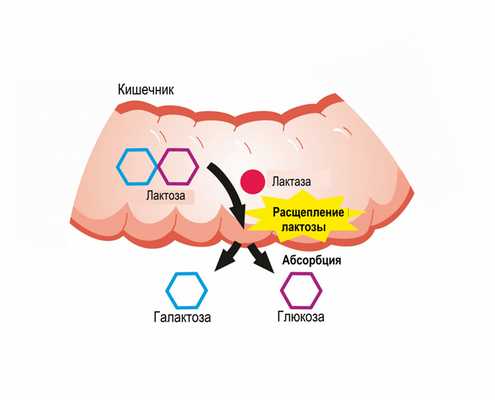
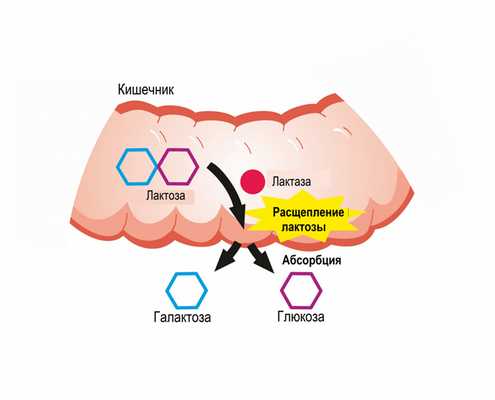
В норме молочный сахар, поступающий в организм человека, расщепляется на глюкозу и галактозу, затем галактоза всасывается и в печени превращается в глюкозу под воздействием ферментов.
В основе галактоземии лежит недостаточность одного из трех ферментов, участвующих в метаболизме галактозы: галактозо-1-фосфатуридилтраснферазы (ГАЛТ), галактокиназы (ГАЛК) или уридин-дифосфат-галактозо-4-эпимиразы (ГАЛЭ). Из-за мутации в генах, отвечающих за синтез этих ферментов, у больных галактоземией они не вырабатываются, вследствие чего в организме накапливается избыток галактозы и ее производных - метаболитов. Это оказывает токсический эффект и приводит к повреждению органов и тканей (печени, глаз, центральной нервной системы).
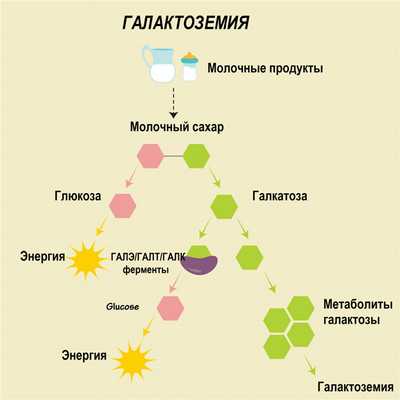
Патологические процессы обусловлены не только токсическим воздействием, но и гипогликемическим синдромом, вызванным недостаточной активностью других ферментов, участвующих в углеводном обмене.
При галактоземии в организме больного кроме галактозы может накапливаться галактозо-1-фосфат. Кроме этого, в отсутствии необходимых ферментов метаболизм галактозы протекает иначе: она превращается в галактитол. У больных всегда наблюдается накопление галактитола в крови и тканях и повышение его выделения с мочой.
Галактоземия всегда наследуется по аутосомно-рецессивному типу. Дефектный ген появляется, когда у ребенка оба родителя являются носителями заболевания. Если оба родителя здоровы, но имеют копию гена с мутацией, галактоземия у ребенка проявится с вероятностью 25%, а в 50% случаев ребенок будет просто носителем генного дефекта.
Классификация заболевания
Выделяются следующие варианты заболевания:
- I тип - классическая галактоземия - обусловлена дефицитом фермента ГАЛТ. Вариант классической галактоземии - форма Дуарте. Это более легкая форма, активность фермента ГАЛТ при таком варианте может достигать 25%, а иногда и выше;
- II тип - недостаточность галактокиназы - характеризуется мутацией в гене, кодирующем фермент ГАЛК;
- III тип - дефицит эпимеразы - характеризуется мутацией в гене, кодирующем фермент ГАЛЭ. Выделяют доброкачественный и тяжелый фенотипы. Легкая форма считается доброкачественной и связана с дефицитом фермента только в циркулирующих клетках крови. При тяжелой форме недостаточность фермента наблюдается во всех тканях организма.
В зависимости от степени тяжести выделяются такие виды заболевания:
- легкий тип, который обычно диагностируется случайно и проявляется непереносимостью молока;
- средний тип - симптомы появляются сразу после кормления грудным молоком либо молочными смесями;
- тяжелый тип - характеризуется серьезными поражениями органов и тканей.
При галактоземии симптомы отличаются в зависимости от степени тяжести заболевания. Если болезнь протекает легко, то может наблюдаться плохая переносимость молока и достаточно ранний отказ ребенка от грудного вскармливания.
При классической галактоземии в течение нескольких дней или недель потребления грудного молока или продуктов, содержащих лактозу, у ребенка могут появиться:
- симптомы отравления: тошнота, рвота, диарея, вялость;
- быстрое снижение веса;
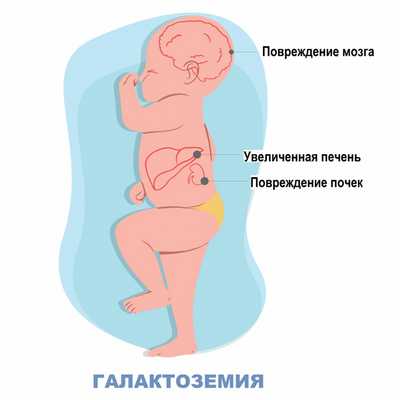
- желтуха и гепатомегалия, свидетельствующие о развитии поражения печени;
- скопление жидкости в брюшной полости;
- мышечные судороги;
- непроизвольные движения глазных яблок;
- снижение тонуса мышц;
- отставание в физическом и психическом развитии;
- катаракта;
- кровоточивость, связанная с гипокоагуляцией (нарушением свертываемости крови);
- в редких случаях - гемолитическая анемия;
- геморрагическая сыпь на коже в результате повреждения стенок сосудов;
- отеки в результате нарушения функции почек.
Пациенты с вариантом Дуарте имеют более легкие проявления, включающие желтуху, увеличение печени и задержку физического развития.
У пациентов с дефицитом галактокиназы (галактоземия типа II) основным симптомом является катаракта из-за образования галактитола, который повреждает волокна хрусталика. Идиопатическая внутричерепная гипертензия (псевдоопухоль мозга) встречается редко. Симптомы выражены слабее, чем при классической галактоземии.
При дефиците эпимеразы (галактоземия типа III) доброкачественная форма заболевания ограничивается изменениями в эритроцитах и лейкоцитах и не вызывает каких-либо клинических проявлений. Симптомы при тяжелой форме те же, что и при классической галактоземии, но иногда присоединяется потеря слуха.
Диагностика галактоземии
Диагноз классической галактоземии обычно ставится в течение первой недели жизни ребенка по анализу крови в рамках стандартного скрининга новорожденных. Уровень тотальной галактозы (сумма концентраций галактозы и галактозы-1-фосфата) в сыворотке крови не должен превышать 7,2 мг/дл. Если показатели выше, проводят подтверждающую диагностику:
- определение активности фермента ГАЛТ;
- ДНК исследование состоит из двух этапов:
Синонимы: Анализ кала на углеводы; Содержание сахаров в кале; Анализ на содержание углеводов в кале. Fecal sugar; Reducing Substance, Feces; Stool sugar; Reducing substances faeces; Stool (or faecal) reducing subs.
Галактоземия
Галактоземия - наследственная ферментопатия, характеризующаяся нарушением нормального процесса углеводного обмена, а именно - метаболизма галактозы. Признаками галактоземии являются непереносимость грудного молока и молочных смесей, рвота, анорексия, гипотрофия, желтуха, цирроз печени, спленомегалия, отеки, катаракта, задержка психомоторного развития. Скрининг на галактоземию проводится всем новорожденным; дополнительное обследование включает определение уровня галактозы в крови и моче, проведение нагрузочных проб с галактозой и глюкозой, генетическое тестирование, УЗИ брюшной полости, ЭЭГ и др. Основу терапии галактоземии составляет безлактозная диета, назначаемая с первых дней жизни ребенка.
Общие сведения
Галактоземия - наследственная патология обмена веществ, обусловленная недостаточностью активности ферментов, принимающих участие в метаболизме галактозы. Неспособность организма утилизировать галактозу приводит к тяжелым поражениям пищеварительной, зрительной и нервной системы детей в самом раннем возрасте. В педиатрии и генетике галактоземия относится к редким генетическим заболеваниям, встречающимся с частотой один случай на 10 000 - 50 000 новорожденных.
Впервые клиника галактоземии была описана в 1908 году у ребенка, страдавшего сильным истощением, гепато- и спленомегалией, галактозурией; при этом заболевание исчезло сразу после отмены молочного питания. Позднее, в 1956 г. ученый Герман Келкер определил, что в основе заболевания лежит нарушение метаболизма галактозы.
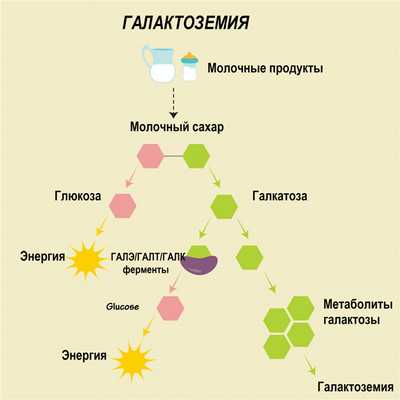
Причины галактоземии
Галактоземия является врожденной патологией, наследуемой по аутосомно-рецессивному типу, т. е. заболевание проявляется только в том случае, если ребенок наследует две копии дефектного гена от каждого из родителей. Лица, гетерозиготные по мутантному гену, являются носителями заболевания, однако у них тоже могут развиваться отдельные признаки галактоземии в легкой степени.
Превращение галактозы в глюкозу (метаболический путь Лелуара) происходит при участии 3-х ферментов: галактоза-1-фосфатуридилтрансферазы (GALT), галактокиназы (GALK) и уридиндифосфат-галактозо-4-эпимеразы (GALE). В соответствии с дефицитом этих ферментов различают 1 (классический вариант), 2 и 3 тип галактоземии.
Выделение трех типов галактоземии не совпадает с порядком действия ферментов в процессе метаболического пути Лелуара. Галактоза поступает в организм с пищей, а также образуется в кишечнике в процессе гидролиза дисахарида лактозы. Путь метаболизма галактозы начинается с ее превращения под действием фермента GALK в галактозо-1-фосфат. Затем при участии фермента GALT галактозо-1-фосфат преобразуется в УДФ-галактозу (уридилдифосфогалактозу). После этого с помощью GALE метаболит превращается в УДФ - глюкозу (уридилдифосфоглюкозу).
При недостаточности одного из названных ферментов (GALK, GALT или GALE) концентрация галактозы в крови значительно повышается, в организме накапливаются промежуточные метаболиты галактозы, которые вызывают токсическое поражение различных органов: ЦНС, печени, почек, селезенки, кишечника, глаз и др. Нарушение метаболизма галактозы и составляет суть галактоземии. Наиболее часто в клинической практике встречается классический (1 тип) галактоземии, обусловленный дефектом фермента GALT и нарушением его активности. Ген, кодирующий синтез галактоза-1-фосфатуридилтрансферазы, находится в околоцентромерном участке 2-ой хромосомы.
Симптомы галактоземии
По тяжести клинического течения выделяют тяжелую, среднюю и легкую степени галактоземии.
Первые клинические признаки галактоземии тяжелой степени развиваются очень рано, в первые дни жизни ребенка. Вскоре после кормления новорожденного грудным молоком или молочной смесью возникает рвота и расстройство стула (водянистый понос), нарастает интоксикация. Ребенок становится вялым, отказывается от груди или бутылочки; у него быстро прогрессируют гипотрофия и кахексия. Ребенка могут беспокоить метеоризм, кишечные колики, обильное отхождение газов.
В процессе обследования ребенка с галактоземией неонатологом выявляется угасание рефлексов периода новорожденности. При галактоземии рано появляется стойкая желтуха различной степени выраженности и гепатомегалия, прогрессирует печеночная недостаточность. К 2-3 месяцу жизни возникают спленомегалия, цирроз печени, асцит.
Нарушения процессов свертывания крови приводит к появлению кровоизлияний на коже и слизистых оболочках. Дети рано начинают отставать в психомоторном развитии, однако степень интеллектуальных нарушений при галактоземии не достигает такой тяжести, как при фенилкетонурии. К 1-2 месяцам у детей с галактоземией выявляется двусторонняя катаракта. Поражение почек при галактоземии сопровождается глюкозурией, протеинурией, гипераминоацидурией. В терминальной фазе галактоземии ребенок погибает от глубокого истощения, тяжелой печеночной недостаточности и наслоения вторичных инфекций.
При галактоземии средней тяжести также отмечается рвота, желтуха, анемия, отставание в психомоторном развитии, гепатомегалия, катаракта, гипотрофия. Галактоземия легкой степени характеризуется отказом от груди, рвотой после приема молока, задержкой речевого развития, отставанием ребенка в массе и росте. Однако даже при легком течении галактоземии продукты обмена галактозы токсическим образом воздействуют на печень, приводя к ее хроническим заболеваниям.
Галактоземия может протекать в моносимптомной форме, при которой обнаруживается только поражение ЦНС, катаракта или диспепсические расстройства. Описан вариант бессимптомной (асимптоматической) галактоземии Дюарте, при которой недостаточность ферментов выявляется только при биохимическом исследовании крови.
Осложнения галактоземии включают цирроз печени, сепсис, кровоизлияния в стекловидное тело, первичную аменорею, синдром истощения яичников. При галактоземии у 50% детей дошкольного возраста выявляется моторная алалия, характеризующаяся трудностью организации и координации речевых движений, бедностью словарного запаса, обилием парафазий и персевераций при сохранном понимании обращенной речи.
Диагностика
Для снижения риска развития осложнений при галактоземии необходимо как можно более раннее выявление патологии. Возможна пренатальная диагностика галактоземии, включающая проведение биопсии хориона, амниоцентеза с последующим исследованием ворсин и амниотической жидкости. В России, согласно современным стандартам, осуществляется скрининг новорожденных на следующие наследственные заболевания: фенилкетонурию, врожденный гипотиреоз, галактоземию, адрено-генитальный синдром и муковисцидоз. Неонатальный скрининг проводится на 3-5 сутки у доношенных детей и 7-10 сутки - у недоношенных. С этой целью производится забор капиллярной крови, которая переносится на фильтровальную бумагу и виде высушенных пятен отправляется в генетическую лабораторию.
Если при неонатальном скрининге у ребенка выявляется подозрение на галактоземию, проводится повторное решающее тестирование. В случае повторного обнаружения высокого уровня галактозы в крови или низкого уровня исследуемого фермента, ребенку устанавливается диагноз галактоземии. Сведения о таком ребенке сообщаются участковому педиатру, а семья новорожденного приглашается на консультацию генетика в медико-генетическую консультацию. Врач-генетик проводит подробный анализ родословной, выполняет генетическое тестирование для выявления мутантного гена, объясняет специфику питания ребенка с галактоземией.
Иногда для диагностики галактоземии прибегают к определению уровня галактозы в моче, проведению нагрузочных проб с галактозой и глюкозой. Мониторинг биохимических показателей крови и общего анализа крови и мочи при галактоземии позволяет определить степень повреждения внутренних органов (почек, печени и др.).
Дети с галактоземией нуждаются в консультации детского невролога, детского офтальмолога, проведении электроэнцефалографии, УЗИ органов брюшной полости, биомикроскопии глаза. В некоторых случаях показана пункционная биопсия печени. Галактоземию следует дифференцировать от других гликогенозов, сахарного диабета I типа, врожденной атрезии желчных протоков, гепатита, гемолитической болезни новорожденных.
Лечение галактоземии
Основная роль в лечении галактоземии принадлежит диетотерапии. Особенность питания при галактоземии заключается в пожизненном исключении из рациона продуктов, содержащих лактозу и галактозу: любого молока (женского, коровьего, козьего, детских молочных смесей, низколактозных смесей и пр.), всех молочных продуктов, хлеба, выпечки, колбас, конфет, маргаринов и др. При галактоземии запрещается употребление растительных и животных продуктов, содержащих потенциальные источники галактозы - галактозиды (бобовые, соя) и нуклеопротеины (почки, печень, яйца и др.).
Дети, страдающие галактоземией, обеспечиваются специальными смесями на основе изолята соевого белка, гидролизата казеина, синтетических аминокислот, а также безлактозными казеинпредоминантными молочными смесями. С 4-х месячного возраста вводятся фруктовые и ягодные соки; с 4,5 месяцев - фруктовое пюре; с 5 месяцев - овощное пюре; с 5,5 месяцев - безмолочные каши из кукурузной, гречневой или рисовой муки в разведении специализированной смесью; с 6 месяцев - мясной прикорм на основе мяса кролика, цыпленка, индейки, говядины; с 8 месяцев - рыба. Альтернативным источником углеводов для пациентов с галактоземией служат продукты на основе фруктозы.
Для улучшения метаболических процессов назначаются поливитамины, кокарбоксилазу, АТФ, оротат калия. Лицам с галактоземией противопоказан прием спиртовых настоек и гомеопатических препаратов, поскольку последние содержат лактозу. Дети с речевыми нарушениями нуждаются в консультации логопеда и целенаправленной работе по коррекции ОНР.
Прогноз и профилактика галактоземии
Лечение галактоземии, начатое с первых дней жизни позволяет избежать развития цирроза, катаракты, олигофрении. Если лечение начато в более поздние сроки, когда уже произошло поражение печени и ЦНС, с помощью рациональной диетотерапии прогрессирование заболевания можно замедлить. При тяжелых формах галактоземии может быть летальный исход. Диспансерное наблюдение ребенка с галактоземией осуществляется педиатром, генетиком, диетологом, детским окулистом и детским неврологом. Детям с галактоземией присваивается инвалидность.
Учитывая наследственную обусловленность галактоземии, медико-генетическое консультирование рекомендуется пройти будущим родителям, в чьих семьях есть родственники или дети с данным заболеванием. Беременным с высоким риском рождения ребенка с галактоземией, следует ограничить употребление молочных продуктов.
Нарушения обмена галактозы
Галактоземия (ГАЛ) - группа наследственных нарушений обмена углеводов, при котором в организме накапливается избыток галактозы и ее метаболитов, что обусловливает клиническую картину заболевания и формирование отсроченных осложнений. Тип наследования всех форм галактоземии - аутосомно-рецессивный.
Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
E74.2 - Нарушения обмена галактозы (Недостаточность галактокиназы. Галактоземия)

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
В зависимости от дефекта одного из трех основных ферментов, участвующих в обмене галактозы выделяют три типа ГАЛ:
I. Классический - галактоземия I типа, обусловленная дефицитом фермента галактозо-1-фосфат-уридилтрансферазы (ГАЛТ) и наличием гомозиготных или компаунд-гетерозиготных мутаций в гене GALT (G/G). Отдельно выделяют вариант Дуарте (D/D) и галактоземию- Дуарте (G/D), при которых полиморфный вариант находится в гомозиготном состоянии или компаунд-гетерозиготном состоянии с патогенной мутацией.
Этиология и патогенез
Галактоза (от греческого слова galaktos - молоко) - моносахарид из группы гексоз, изомер глюкозы. Несмотря на большое сходство молекул глюкозы и галактозы, превращение последней в глюкозу требует нескольких ферментативных реакций, которые протекают в цитоплазме клетки (рис. 1). Галактоза имеет важнейшее значение для роста и развития организма и является компонентом грудного молока.
Галактоза не только является значимым источником энергии для клетки, она играет важную пребиотическую роль, служит необходимым пластическим материалом для образования гликопротеидов, гликолипидов и других сложных соединений, используемых организмом для формирования клеточных мембран, нервной ткани, процессов миелинизации нейронов и др. Нарушение метаболизма галактозы, наблюдаемое при галактоземии, неизбежно приводит к расстройству функционирования многих органов и систем организма.
Большое количество потребляемых в течение дня пищевых продуктов (в первую очередь молочные продукты) содержат лактозу, из которой в кишечнике в результате гидролиза образуется галактоза; некоторые продукты питания содержат галактозу в чистом виде. У человека галактоза может образовываться эндогенным путем, подавляющее ее количество синтезируется в процессе ферментативных реакций между уридиндифосфатглюкозой (УДФ-глюкозой) и УДФ-галактозой, а также в процессе обмена гликопротеинов и гликолипидов.
Галактоземия относится к наследственным болезням углеводного обмена и объединяет несколько генетических форм заболеваний. Галактоземия тип I (ГАЛ I) обусловлена мутациями в гене GALT, картированном на 9p13.3, что приводит к дефициту галактозо-1-фосфатуридилтрансферазы (ГАЛТ). Галактоземия тип II (ГАЛ II) обусловлена мутациями в гене GALK1, картированном на 17q25.1, что приводит к дефициту галактокиназы (ГАЛК). Галактоземия тип III (ГАЛ III) обусловлена мутациями в гене GALE, картированном на 1р36.11, что приводит к дефициту уридин-дифосфат (УДФ)-галактозо-4-эпимеразы (ГАЛЭ) (рис 1).
В результате недостаточности любого из трех ферментов - ГАЛТ, ГАЛК или ГАЛЭ - в крови повышается концентрация галактозы. При снижении активности ферментов ГАЛТ и ГАЛЭ, помимо избытка галактозы, в организме пациента накапливается также избыточное количество галактозо-1-фосфата, что на сегодняшний день считается основным патогенетическим фактором, обусловливающим большинство клинических проявлений ГАЛ и формирование отсроченных осложнений. Избыток галактозы в организме может метаболизироваться в других биохимических путях: она может превращаться в галактитол. Накопление галактитола в крови и тканях и повышение его экскреции с мочой наблюдается при всех формах ГАЛ; в хрусталике глаза избыток галактитола способствует формированию катаракты. Имеются сведения о том, что высокое содержание галактитола в тканях мозга способствует набуханию нервных клеток и формированию псевдоопухоли мозга у отдельных пациентов.
Патологические процессы при ГАЛ обусловлены не только токсическим действием указанных продуктов, но и их тормозящим влиянием на активность других ферментов, участвующих в углеводном обмене (фосфоглюкомутазы, глюкозо-6-фосфатдегидрогеназы), следствием чего является гипогликемический синдром. Предполагается также, что предрасположенность к сепсису у новорожденных с ГАЛ тип I обусловлена ингибированием бактерицидной активности лейкоцитов.
Наиболее частой формой ГАЛ является ГАЛ I. Ген GALT состоит из 11 экзонов, охватывающих 4,3 т.п.н. хромосомы 9p13. В базе данных по мутациям человека Human Genetic Mutaton Database (HGMD Professional, Version 2020.3) описано 346 патогенных вариантов в гене GALT [47]. К наиболее распространенным патогенным вариантам относятся две мутации: c.563A>G (p.Q188R) и c.855G>T (p.K285N), обуславливающие развитие ГАЛ I. Среди пациентов европейского происхождения наиболее частой является мутация c.563A> G (p.Q188R), составляющая 64% всех наблюдаемых патогенных вариантов. Вариант c.404C> T (p.S135L) связан с «мягким» фенотипом ГАЛ I и встречается почти исключительно у пациентов африканского происхождения. Другие распространенные патогенные варианты включают: c.855G> T (p.K285N), c.626A> G (p.Y209C), c.413C> T (p.T138M), c.584T> C (p.L195P) и IVS2-2А> G. У евреев-ашкенази часто встречается делеция размером около 5,5 kb. Все эти варианты связаны с почти или полной потерей активности фермента.
Галактоземии II типа (ГАЛ II) обусловлена мутациями в гене GALK1, картированном на 17q25.1. Описано около 50 мутаций, частых мутаций не описано. Известна одна мутация, p.P28T, которая часто встречается у цыган [17].
Галактоземия тип III (ГАЛ III) обусловлена мутациями в гене GALE, картированном на 1р36.11. Одна из самых редких форм ГАЛ. Известно около 30 мутаций, частых мутаций не описано.
Читайте также:
- Диагностика односторонней вестибулярной гипофункции при головокружении. Методика
- Аускультативная картина сердечного порока. Морфология сердечных шумов
- Радиационные поражения крупных сосудов. Стенки сосудов при лучевой болезни
- Значение нейтрофилов. Механизмы фагоцитоза
- Диагностика, лечение и профилактика сывороточной болезни