Генетика мышечной дистрофии Дюшенна и Беккера у детей. Патогенез
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Дистрофинопатии представляют собой группу Х‑сцепленных нервно‑мышечных заболеваний как с мягким, так и с тяжелым течением, развитие которых обсуловлено мутациями в гене DMD, кодирующем белок дисторфин.
Тяжесть течения заболевания определяется типом мутации и функциональным доменом, в котором она локализуется.
Мягкое течение
Характеризуется бессимптомным повышением сывороточной креатинфосфокиназы и мышечными судорогами с миоглобинурией.
Тяжелое течение
Проявляется развитием классических синдромов, включающих в себя прогрессирующую мышечную дистрофию Дюшенна (ПМДД), прогрессирующую мышечную дистрофию Беккера (ПМДБ) и DMD‑ассоциированную дилатационную кардиомиопатию.
Заболевание характеризуется прогрессирующей слабостью проксимальных мышц, вызванной дегенерацией мышечных волокон. Дегенерация возникает в результате нарушения устойчивости и эластичности мышечных волокон при сокращениях. По мере развития заболевания мышечное волокно практически полностью разрушается и замещается соединительной тканью, что приводит к псевдогипертрофии мышц — увеличению их объема при утрате или значительном ослаблении функциональных возможностей. МДД входит в перечень наиболее распространенных Х-сцепленных заболеваний [1].
Прогрессирующая мышечная дистрофия, как правило, начинает проявляться с повышенной утомляемости и слабости мышц нижних конечностей.
Дифференциальный диагноз
- Мышечная дистрофия поясно-конечностная (Limb-gridle muscular dystrophy) — ген FKRP
- Дистрофия Эмери-Дрейфуса (Emery-Dreifuss muscular dystrophy) — гены EMD, FHL1, LMNA
- Спинальная мышечная атрофия (spinal muscular atrophy) — ген SMN1
- Синдром Барта (Barth syndrome) — ген TAZ
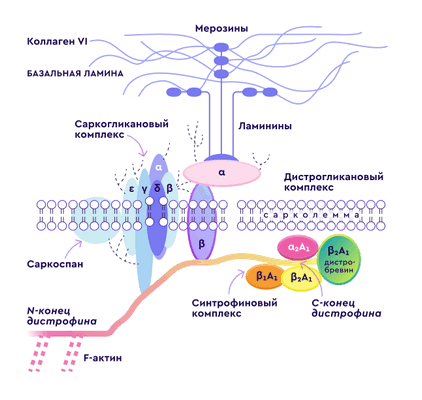
Расположение дистрофина и дистрофин-ассоциированных белков на мембране мышечного волокна.
Дистрофин является мембранным белком, а дистрофин-ассоциированный комплекс представляет собой наиболее важный элемент мышечного цитоскелета, который обеспечивает взаимодействие внутренних и внешних структур клетки, участвует в регуляции уровня кальция в мышце и передаче импульсов через мембрану мышечного волокна.
Дистрофин находится, главным образом, в мышечных клетках и некоторых нейронах. В норме в его функции входит обеспечение эластичности и устойчивости мышечного волокна при сокращении.
При отсутствии дистрофина мембрана клетки разрушается и, как следствие, мышечное волокно разрушается и замещается соединительной тканью, что приводит к значительному ослаблению функциональных возможностей.
79% от общего числа мутаций составляют крупные делеции и дупликации.
21% составляют небольшие изменения, включающие однонуклеотидные замены, небольшие вставки и делеции, а также мутации сайтов сплайсинга, причем миссенс‑варианты не характерны для ПМДД/Б [3].
Особенности клинических проявлений связывают с типом мутации в гене дистрофина:
При ПМДД
Делеции в гене в большинстве случаев приводят к сдвигу рамки считывания и преждевременному окончанию считывания информации о построении белка, в результате дистрофин не образуется.
При ПМДБ
Структурные нарушения гена не нарушают рамку считывания, в результате образуется дефектный, функционально неполноценный белок.
При DMD-ассоциированной дилатационной кардиомиопатии
Функционально активный дистрофин отсутствует в миокарде, но присутствует в скелетной мускулатуре, поскольку при данном типе дистрофинопатий патогенные варианты приводят к различным тканеспецифической транскрипции или альтернативному сплайсингу в сердечной мыщце и в скелетной [Ferlini et al 1999, Neri et al 2007].

Выделяют следующие клинические формы:
Прогрессирующая мышечная дистрофия Дюшенна
Является более распространенной формой данного заболевания. Первые признаки появляются в раннем возрасте (1‑5 лет) и характеризуются задержкой моторного развития. При начале ходьбы отмечаются частые падения, неловкость и утомляемость, трудности при подъеме по лестнице, беге и прыжках. Пациенты сохраняют способность к ходьбе до 10‑12 лет. Кардиомиопатия развивается практически у всех пациентов с ПМДД после 18 лет.
Заболевание быстро прогрессирует и гибель, как правило, наступает до 30‑летнего возраста в результате респираторных осложнений и прогрессии дилатационной кардиомиопатии.
Прогрессирующая мышечная дистрофия Беккера
Как правило, характеризуется более поздней манифестацией и мягкими клиническими проявлениями. Клинические признаки начинают проявляться примерно в интервале от 10 до 20 лет. Заболевание прогрессирует достаточно медленно, в большинстве случаев, пациент утрачивает способность к передвижению без инвалидной коляски не ранее 40‑летнего возраста.
Не смотря на более мягкий нервно-мышечный фенотип, сердечная недостаточность является наиболее частой причиной смерти при ПМДБ (средний возраст смерти 40‑50 лет).
DMD-ассоциированная дилатационная кардиомиопатия
По данным источников, мутации в гене DMD могут стать причиной не только ПМДД/Б, но и дилатационой кардиомиопатии 3B [2,6], которая является заболеванием сердечной мышцы и приводит к дисфункции желудочков и, как следствие, сердечной недостаточности. Женщины, гетерозиготные по патогенному варианту находятся в группе высокого риска развития данной патологии.
Прогрессирующая мышечная дистрофия Дюшенна
Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна - тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Причины
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Прогрессирующая мышечная дистрофия Беккера
Прогрессирующая мышечная дистрофия Беккера — вариант наследственной сцепленной с Х-хромосомой миодистрофии, отличающейся более замедленным и доброкачественным течением. Заболевание характеризуется постепенно усугубляющейся и распространяющейся мышечной слабостью, гипотонией и атрофией, первоначально возникающей в мышцах бедер и тазового пояса. Диагностический поиск включает неврологическое обследование, консультацию генетика и кардиолога, нейрофизиологическое тестирование нервно-мышечного аппарата, ДНК диагностику, биопсию мышц с морфологическим, иммунологическим и гистохимическим изучением полученных образцов. Лечение симптоматическое и, к сожалению, малоэффективное. Прогрессирование болезни приводит к потери больными способности самостоятельно передвигаться к возрасту 40 лет.
Прогрессирующая мышечная дистрофия Беккера впервые была описана в 1955 г. как доброкачественный вариант течения мышечной дистрофии Дюшенна. В последующем многочисленные исследования в области клинической неврологии, генетики и биохимии обнаружили существенные отличия в характере течения, биохимической и морфологической основе этих заболеваний. В результате клиническая форма Беккера была выделена как самостоятельная нозология.
Мышечная дистрофия Беккера входит в группу миопатий (миодистрофий) — заболеваний, возникающих вследствие нарушений строения и метаболизма мышечной ткани и проявляющихся мышечной слабостью. Патология наследуется рецессивно сцеплено с Х-хромосомой, поэтому болеют только лица мужского пола. Частота встречаемости составляет 1 новорожденный на 20 тыс. детей.
В основе заболевания лежит мутация в гене, ответственном за кодирование белка дистрофина. Примерно 30% от общего числа случаев мышечной дистрофии Беккера приходится на т. н. «свежие» мутации. Ген располагается в 21 локусе (в регионе Хр21.2-р21.1) короткого плеча Х-хромосомы. Примерно у 65-70% больных обнаруживаются крупные делеции указанного участка, у 5% - дупликации, у остальных — точковые мутации. Указанные структурные перестройки гена не влекут за собой полного прекращения синтеза дистрофина, как при дистрофии Дюшенна, а потенцируют синтез аномального усеченного белка, в некоторой степени способного выполнять свои функции. Это и обуславливает более доброкачественный характер дистрофии Беккера в сравнении с вариантом Дюшенна.
Патогенез
В норме белок дистрофин поддерживает целостность сарколеммы - мембраны миоцитов (мышечных волокон), обеспечивает эластичность и устойчивость миофибрилл при мышечном сокращении. Неспособность аномального дистрофина адекватно выполнять эти функции приводит к нарушению целостности мембран мышечных волокон. В следствие этого происходят дегенеративные изменения цитоплазматических компонентов последних и повышенная транспортировка ионов калия внутрь миоцитов. Результатом таких биохимических и морфологических сдвигов является гибель миофибрилл и разрушение мышечных волокон. На месте погибших миоцитов происходит образование соединительной ткани, что обуславливает феномен псевдогипертрофии — увеличение объема и плотности мышцы при резком снижении ее сократительной способности.
Прогрессирующая мышечная дистрофия Беккера манифестирует обычно в период от 10 до 15 лет, в некоторых случаях раньше. Начальными признаками заболевания выступают чрезмерная утомляемость и мышечная слабость в тазовом поясе и нижних конечностях. У ряда пациентов первыми проявлениями являются периодические болезненные мышечные судороги (крампи), локализующиеся в ногах. Мышечная слабость обуславливает затруднение при подъеме по лестнице, при необходимости встать из положения сидя. Со временем формируется переваливающаяся «утиная походка». Для того, чтобы встать, пациент вынужден использовать вспомогательные миопатические приемы — опираться руками о расположенные рядом предметы мебели или, при отсутствии таковых, использовать в качестве опоры собственное тело (симптом Говерса).
Как и другие наследственные миопатии, заболевание Беккера характеризуется симметрично развивающимися атрофиями мышц. В первую очередь поражаются мышцы бедра и тазового пояса, затем процесс распространяется на мускулатуру плечевого пояса и проксимальных мышц рук. В начале болезни формируются псевдогипертрофии, наиболее выраженные в икроножных, дельтовидных, трех- и четырехглавых мышцах. По мере прогрессирования миодистрофии они трансформируются в мышечные гипотрофии.
Клиническая картина мышечной дистрофии Беккера во многом сходна с миодистрофией Дюшенна. Усугубление мышечной слабости с течением времени приводит к обездвиженности пациента и формированию контрактур суставов. Однако развитие дистрофического процесса в мышечной ткани при дистрофии Беккера идет гораздо медленнее, что обуславливает длительную двигательную активность больных. В среднем пациенты сохраняют способность самостоятельно передвигаться до 35-40-летнего возраста. Кроме того, дистрофия Беккера не сопровождается олигофренией, выраженным искривлением позвоночника и другими скелетными деформациями. Возможна кардиомиопатия дилятационного или гипертрофического типа, блокада ножек пучка Гисса, но сердечно-сосудистые расстройства выражены умеренно. Может наблюдаться снижение либидо, гинекомастия, атрофия яичек, импотенция.
Прогрессирующая мышечная дистрофия Беккера диагностируется неврологом на основании анамнеза, клинических данных, дополнительных обследований и генетического тестирования. В неврологическом статусе наблюдается снижение мышечной силы и умеренное снижение мышечного тонуса в проксимальных отделах конечностей, выпадение коленных рефлексов при симметричном снижении сухожильных рефлексов дистальных отделов ног и верхних конечностей, полная сохранность чувствительности.
Среди клинических анализов наибольшее значение имеет биохимический анализ крови, который выявляет многократное повышение уровня КФК. Данные электронейрографии позволяют исключить поражение нервных волокон, электромиография свидетельствует о первично-мышечном типе поражения. Биопсия мышц проводится только после отрицательных результатов генетического анализа. Морфологическое исследование полученного материала определяет диффузную разнокалиберность, дистрофические и некротические изменения мышечных волокон, разрастание соединительной ткани. Проводится специальное иммунное окрашивание образцов с последующим определением наличия в них дистрофина.
Подтвердить диагноз мышечной дистрофии Беккера позволяет консультация генетика с проведением анализа ДНК. Выявление дупликаций или делеций в гене Хр21 дает возможность установить точный диагноз. Отрицательный результат анализа ДНК не говорит об отсутствии патологии, поскольку могут иметь место точковые мутации, поиск которых представляет собой сложную и более дорогостоящую процедуру.
С целью выявления сердечной патологии назначается электрокардиография, Эхо-КГ, консультация кардиолога. Кардиологическое обследование может обнаружить нарушение внутрижелудочковой проводимости, АВ-блокаду, дилатацию желудочков, гипертрофические изменения миокарда, кардиомиопатию, сердечную недостаточность.
Пренатальная диагностика рекомендована, когда мать является носителем патогенного гена. Если ребенок мужского пола, то вероятность развития заболевания у него составляет 50%. Биопсия хориона может проводиться в сроке 11-14 нед. беременности, амниоцентез — после 15-й недели, забор пуповинной крови (кордоцентез) — на сроке больше 18 нед.
Дифференциальная диагностика
Дифференциальная диагностика проводится с прогрессирующей мышечной дистрофией Дрейфуса, миодистрофией Дюшена, мышечной дистрофией Эрба-Рота, метаболической миопатией, полимиозитом и дерматомиозитом, воспалительной миопатией, спинальной амиотрофией, наследственной полиневропатией.
Лечение миодистрофии Беккера
На современном этапе несколькими группами ученых ведутся настойчивые исследования в области поиска эффективных методов лечения прогрессирующих миодистрофий. В настоящее время пациенты получают в основном метаболическую и симптоматическую терапию. Разработаны различные схемы лечения, позволяющие улучшить двигательные возможности больного и несколько замедлить прогрессирование болезни.
Терапия глюкокортикоидами используется для снижения скорости прогрессирование атрофии мышечной ткани. Пациентам назначают метаболические средства (витамины группы В, левокарнитин), витамин D и препараты кальция для профилактики остеопороза, β-адреноблокаторы и ингибиторы АПФ для предупреждения кардиомиопатии, диуретики при сердечной недостаточности.
Наблюдения показали, что постельный режим усугубляет мышечную слабость. Поэтому пациентам рекомендуется умеренная физическая активность, занятия плаванием. Поддержание мышечной эластичности и силы, а также профилактика контрактур проводится средствами массажа, физиотерапии и лечебной гимнастики. Применение различных ортопедических средств (ходунков, инвалидных колясок, фиксаторов для ног, экзоскелетов) позволяет расширить двигательные возможности пациентов и их способность к самообслуживанию. По показаниям проводится хирургическое лечение контрактур.
Прогрессирующая мышечная дистрофия Беккера имеет неблагоприятный прогноз. Хотя обездвиженность у пациентов наступает гораздо позже, чем при дистрофии Дюшенна, в конечном итоге поражение сердечной мышцы и дыхательной мускулатуры приводят к гибели пациентов от сердечной или дыхательной недостаточности. Продуманный уход, адекватная терапия, вентиляционная поддержка дыхания, применение ортопедических средств могут лишь увеличить продолжительность и улучшить качество жизни пациента. Профилактика заключается в предупреждении рождения ребенка с патологией путем генетического консультирования будущих родителей и проведение пренатальной диагностики.
Миодистрофия Дюшенна-Беккера

Миодистрофия Дюшенна-Беккера или дистрофинопатия относится к группе генетических Х-сцепленных рецессивных заболеваний детского возраста. Данная миопатия чаще встречается у представителей мужского пола и проявляется мышечной слабостью, вызванной синтезом аномального белка дистрофина, в норме поддерживающего целостность и метаболизм клеток скелетной мускулатуры. Существует более злокачественная форма заболевания — миодистрофия Дюшенна, вызванная полной остановкой синтеза белка-дистрофина.
Частота встречаемости - 1 на 30 тыс новорожденных мальчиков. Продолжительность жизни при таком заболевании - в среднем 20-30 лет, хотя известны случаи пациентов с миодистрофией, проживших дольше.
Патология была названа в честь французского невролога Гийома Бенжамена Армана Дюшенна, который подробно описал болезнь в 1861 г. Более редкий, но мягкий вариант болезни в 1955 г изучил Беккер, имя которого и носит доброкачественный тип данной псевдогипертрофической миодистрофии, при котором синтезируется небольшое количество дистрофина.
Также отдельно выделена лицелопаточно-плечевая миопатия Ландузи-Дежерина, которая является еще более редким заболеванием и встречается чаще (примерно в 3 раза) — у представительниц женского пола. Тип наследования — аутосомно-доминантный. Для болезни Ландузи-Дежерина характерен более поздний дебют - в 20 и более лет. Она также вызывает слабость и атрофию мышц, помимо этого у больных снижается слух.
Патогенез обусловлен делециями либо дупликациями в одном или нескольких экзонах, а также точечными мутациями в генетическом коде дистрофина спектрин актининового суперсемейства, содержащегося в клетках поперечно-полосатой мускулатуры, придающего им гибкости и участвующего в соединении белков цитоскелета мышечных волокон и окружающего внеклеточного матрикса. Его полифункциональность заключается в поддержании целостности мембран мышечных волокон во время их сокращения и расслабления, а также в формировании нейромышечного синапса.
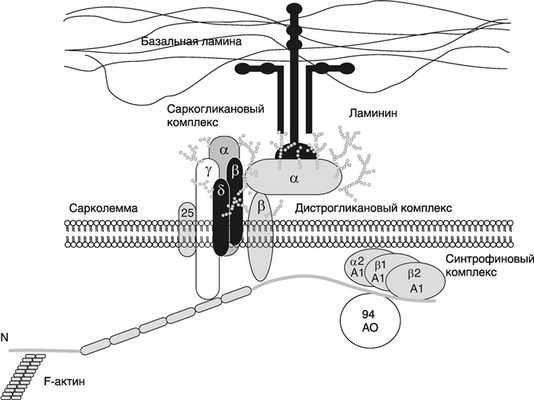
Структура дистрофина и дистрофин-ассоциированного комплекса белков
Если кодируется и соответственно синтезируется патологический аномальный дистрофин, то происходят необратимые клеточные изменения в мембране и обмене веществ, усиливается вход К+ в клетки, что ведет к гибели мышечных клеток и замещению скелетных мышц соединительной или жировой тканью.
Процесс атрофии прогрессирующий и может иметь восходящий характер или два направления - в сторону мускулатуры бедра и тазового пояса или плечевого пояса и рук. Нарушения начинают проявляться в проксимальных отделах, затем достигают и дальних - дистальных отделов скелета. Замещение мышечных тканей соединительной приводит к уменьшению размеров мышц и влечет за собой костные деформации.
Классификация
В зависимости от выраженности мышечной дистрофии, причин и прогноза заболевание бывает доброкачественное и злокачественное, лопаточно-перонеальное с ранними контрактурами, дистальное, плечелопаточно-лицевое, лопаточно-малоберцовое, конечностно-поясное, поражающее глазные мышцы, глазоглоточное (окулофарингеальное), а также напоминающая дистрофию аутосомно-рецессивного детского типа.
Мутации (делеции либо дупликации) гена миодистрофии, расположенного в коротком плече Х-хромосомы 21.2, которые передаются от родителей (вероятность - 50%) или примерно в 30% могут быть спонтанными - возникать de novo в яйцеклетках матери, даже не являющейся носителем заболевания. Также причиной может быть гонадный мозаицизм матери.
Симптомы миодистрофии Дюшенна
Как миопатия миодистрофия Дюшенна вызывает мышечную слабость и дистрофию, проявляющуюся:
- уменьшением массы мышц и затруднением движений, например, подъема рук, при беге, с детства и с прогрессированием с возрастом;
- сгибательными мышечными контрактурами - ограничение подвижности бедренных, коленных суставов верхних конечностей, которые могут приводить к обездвиженности;
- снижением глубоких сухожильных рефлексов или развитием полной арефлексии;
- специфической походкой и осанкой (в виде поясничного гиперлордоза, кифоза, сколиоза, «осиной талии», крыловидных лопаток, симптома «свободных надплечий», развитие седловидной или килевидной грудной клетки);
- задержкой моторного развития и поздним началом ходьбы (не ранее чем в 1 год и 2 мес.) и трудности при попытке встать с пола;
- миалгиейв ногах;
- нарушениями речи;
- повышением риска и частоты переломов;
- псевдогипертрофией икр, а в некоторых случаях и ягодичных, дельтовидных мышц, а также живота.
Особенности походки у ребенка
Миодистрофия Дюшенна-Беккера характеризуется отличительной «утиной походкой детей», при этом у них широко расставлены стопы, разведены носки, есть тенденция ходьбы на носочках, при этом плечи отведены назад и приподнят подбородок. При первых признаках «нестандартной походки», а также частых падениях, неловкости в движениях, трудностях при вставании, беге, ходьбе, быстрой утомляемости — родителям следует незамедлительно обратиться к врачу.
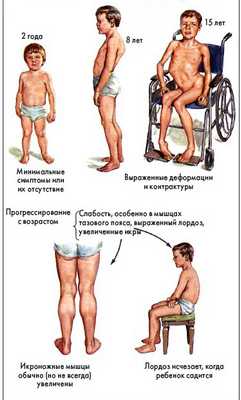
Внешние проявления мышечной дистрофии Дюшенна
В результате прогрессирования патологии возникают симптомы и нарушения со стороны других органов и систем - страдает сердце, развивается кардиомиопатия, проявляющаяся гипертрофией левого желудочка и аритмией, а также примерно в 25% случаев происходит снижение интеллекта вплоть до олигофрении, до сих пор невыясненного происхождения. В редких случаях развиваются нейро-эндокринные расстройства, например, гипогенитализм или атрофия яичек.
Отличия доброкачественной миодистрофии Беккера
Более доброкачественной вариант миодистрофии обычно начинает проявляется с 10-20 лет и провоцирует более слабо выраженные симптомы мышечной слабости, а также:
- болезненные мышечные спазмы;
- развитие кардиомиопатии лишь у 50-60% пациентов;
- прогрессирующий поясничный лордоз;
- «утиная» походка;
- более поздняя инвалидизация - по достижению 40-летнего возраста;
- сохранение интеллекта и возможности к воспроизведению потомства.
Анализы и диагностика
Помимо выявления признаков мышечной слабости для подтверждения диагноза миодистрофии Дюшенна-Беккера необходимо проведение генетических исследований (ПЦР) и иммуногистохимического анализа дистрофина биоптата мышечной ткани, а также лабораторной диагностики, в том числе:
- определение концентрации сывороточных ферментов, которое позволяет выявить повышение уровня альдолазы, креатинфосфокиназы обычно в 10-100 раз, причем чем ниже этот показатель тем больше вероятность доброкачественности патологии и сохранения трудоспособности в течение жизни;
- ультразвуковое, морфологическое исследование, КТ скелетных мышц, изучение биоптатов мышц, где можно выявить некротизированные мышечные волокна с регенерацией, фагоцитоз и жировое перерождение мышечной ткани;
- на электромиограмме (ЭМГ) удается обнаружить признаки миопатии;
- на электрокардиограмме (ЭКГ) — обнаруживаются нарушения проводимости и патологические изменения миокарда.
Важно! Основным методом предупреждения миодистрофии является пренатальная диагностика патологии.
Лечение
До сегодняшнего дня не было разработано радикальных методов лечения миодистрофии Дюшенна-Беккера. Основная терапия - симптоматическая, включающая:
X Международная студенческая научная конференция Студенческий научный форум - 2018

Наследственность всегда представляла собой одно из наиболее труднообъяснимых явлений в истории человечества. Современное состояние науки о наследственности не дает никаких оснований для безучастного наблюдения над проявлением тяжелых наследственных пороков у человека, как это имело место еще недавно. И такое заболевание, как прогрессирующая мышечная дистрофия Дюшенна, не осталось без внимания.
Целью является изучение заболевания прогрессирующая дистрофия Дюшенна: узнать этиологию и патогенез, клинические признаки, частоту встречаемости, способы диагностики и лечения.
•выяснить этиологию и патогенез заболевания;
•узнать клинические проявления заболевания;
•выяснить частоту встречаемости;
•охарактеризовать методы диагностики и лечения.
Этиология и патогенез
Мышечная дистрофия Дюшенна представляет собой наследственную прогрессирующую мышечную дистрофию, характеризующаяся началом в раннем возрасте, симметричной атрофией мышц в сочетании с сердечно-сосудистыми, костно-суставными и психическими нарушениями, злокачественным течением; наследуется по рецессивному Х-сцепленному типу. Встречаемость составляет приблизительно 1 на 3500 новорожденных мальчиков.
Значительная часть генов, связанных с наследственными болезнями мышщ, кодирует белки, ассоциированные с мембранами мышечных волокон. Одной из основных функций подобных белков является стабилизация мембраны за счет связывания цитоскелета мышечной клетки с внеклеточным матриксом. Это, в первую очередь, относится к стержневидному белку дистрофину.
Мышечная дистрофия Дюшенна обусловлена мутацией в гене дистрофин, локус которого Xp21. Дистрофин отвечает за соединение цитоскелета каждого мышечного волокна с основной базальной пластинкой (внеклеточного матрикса) через белковый комплекс, который состоит из многих субъединиц. Отсутствие дистрофина приводит к проникновению избыточного кальция в сарколему (клеточную мембрану). Как следствие изменения этих сигнальных путей, вода наполняет митохондрии, которые после этого разрываются. При дистрофии скелетных мышц, митохондриальная дисфункция приводит к усилению стресса вызванного цитозольным-кальциевым сигналом и усилению производства стресс-индуцированных активных форм кислорода (АФК). В этом сложном каскадном комплексе, который включает в себя несколько реакций еще до сих пор не понятно до конца, почему из-за повреждения сарколеммы увеличиваются проявления окислительного стресса, который в итоге приводит к смерти клетки. Мышечные волокна подвергаются некрозу и, наконец, происходит замена мышечной ткани жировой, а также соединительной.
Причиной нарушения является синтез дефектного белка дистрофина, вследствие протяженных делеций в гене миодистрофине Дюшенна (DMD), локализованном на X-хромосоме в сегменте Xp21 (метод позиционного клонирования), кодирующем этот самый белок белок. Делеции возникают чаще всего при мейотической рекомбинации либо в процессе пролиферации зародышевых клеток, следствием чего является гонадный мозаицизм — появление нескольких генераций половых клеток (ооцитов) с мутантными и нормальными аллелями.
В соответствии с современными представлениями огромный ген DMD находится под контролем сложной системы регуляции транскрипции и сплайсинга. По крайней мере, восемь независимых промоторов осуществляют альтернативную специфическую транскрипцию первых экзонов в разных тканях и на разных стадиях эмбрионального развития. Один промотор мышечного типа и два мозгового, активные в кортикальном отделе мозга и в клетках Пуркинье соответственно, экспрессируют полноразмерную молекулу дистрофина, в то время как пять других промоторов обеспечивают экспрессию последних доменов взаимоисключающим способом, главным образом в немышечных и в немозговых тканях.
Высококонсервативные последовательности шести экзонов, кодирующих С-конец дистрофина, альтернативно сплайсируются, образуя несколько структурно различающихся изоформ дистрофина, осуществляющих различные функции.
Делеции захватывают от одного до нескольких соседних экзонов и сосредоточены обычно в двух «горячих» районах — в области 5'-конца гена (экзоны 6—19) и в З'-конце (экзоны 40-53). Результатом этого является появление дефектного белка дистрофина.
Считается, что в 30% семей с единичными случаями миодистрофии Дюшенна/Беккера болезнь развивается вследствие спонтанного возникновения в гаметах мутаций в DMD-гене и матери таких больных не являются гетерозиготными носителями этих мутаций. В подобных семьях риск повторного рождения больного ребенка минимален и не превышает общепопуляционной частоты. В остальных 70% семей матери больных мальчиков гетерозиготны по мутациям DMD-гена, и при каждой беременности таких женщин риск повторного рождения больного сына составляет 50%.
1) Мужчины с мышечной дистрофией Дюшенна. Миодистрофия Дюшенна — прогрессирующая миопатия, приводящая к дегенерации и слабости мышц. Начинаясь с мышц тазобедренного пояса и сгибателей шеи, мышечная слабость прогрессивно захватывает плечевой пояс и дистальные мышцы конечностей и туловища. Хотя изредка и выявляют случайно больных в период новорожденности за счет гипотонии или задержки развития, обычно больных мальчиков диагностируют в возрасте от 3 до 5 лет при появлении аномалий походки.
К 5 годам большинство пораженных детей используют приемы Говерса и имеют псевдогипертрофию мышц голеней, т.е. увеличение голеней вследствие замены мышц жировой и соединительной тканью. К возрасту 12 лет основная часть больных обездвижены в инвалидном кресле и имеют контрактуры и сколиоз. Большинство пациентов умирают от нарушения легочной функции и пневмонии; средний возраст смерти — 18 лет.
Почти 95% больных миодистрофией Дюшенна имеют те или иные кардиологические отклонения (дилатационная кардиомиопатия или электрокардиографические аномалии), а 84% имеют видимые поражения мышцы сердца при вскрытии. Хронические нарушения сердца бывают почти у 50% пациентов, изредка сердечная недостаточность вызывает у них жалобы. Хотя дистрофии также присутствует в гладких мышцах, гладкомышечные осложнения встречаются редко и включают расширение желудка, заворот кишок и гипотонию мочевого пузыря.
Больные миодистрофией Дюшенна имеют IQ примерно на 1 среднеквадратичное отклонение ниже обычного, и почти треть имеет ту или иную степень умственной отсталости. Причины этого не установлены.
2) Женщины с мышечной дистрофией Дюшенна Возраст начала и тяжесть миодистрофии Дюшенна у женщин зависят от степени смещения инактивации Х-хромосомы. Если Х-хромосома, несущая мутантный аллель DMD, активна в большинстве клеток, у женщины развиваются признаки миодистрофии Дюшенна; если преимущественно активна Х-хромосома, несущая нормальный аллель DMD, женщины имеют только несколько или не имеют вообще симптомов данного заболевания.
Независимо от того, есть ли у них клинические симптомы скелетной мышечной слабости, женщины-носительницы имеют отклонения в функции сердечной мышцы, например дилатационную кардиомиопатию, дилатацию левого желудочка и электрокардиографические изменения.
Особенности фенотипических проявлений дистрофии Дюшенна:
• Возраст начала: детство;
• Небольшая интеллектуальная недостаточность;
• Высокий уровень креатинкиназы сыворотки.
Клиническая картина очень яркая. Часто заболевание ставится после выяснения генетического анамнеза (наличие случаев в семье), неврологического осмотра. В неврологическом статусе отмечается пропадание коленных рефлексов, чуть позже исчезают рефлексы с бицепса, трицепса. Ахилловы рефлексы долгое время сохранны.
Внешне может выявиться деформация суставов стопы, имеются признаки кардиомиопатии: нарушение пульса, глухость сердечных тонов, расширение полостей сердца по ЭхоКГ, изменения на электрокардиограмме.
Важным фактором является повышение биохимических показателей креатинфосфокиназы (фермент-показатель распада мышц). Активность данного фермента увеличивается в десятки раз. Имеется прямая корреляция между степенью увеличения активности фермента и выраженностью проявлений дистрофии Дюшенна. В сложных диагностических ситуациях проводят цитологическое исследование.
Терапия, применяемая в клинической практике, пока включает лишь необходимые симптоматические мероприятия. Для улучшения метаболизма мышечной ткани возможно назначение анаболических стероидов (метандиенона, нандролона деканоата), АТФ, актопротекторов (этилтиобензимидазола); для облегчения нервно-мышечной передачи — неостигмина. Внедряются в практику различные ортопедические приспособления для облегчения передвижения.
Итак, мы изучили такое заболевание, как мышечная дистрофия Дюшенна. Выяснили, что данное заболевание возникает в результате нарушения синтеза белка дистрофина. Клиническое проявление заключается в дегенерации и слабости мышц. Диагностика производится с помощью неврологических тестов, так же собирается генетический анамнез. Важным фактором является повышение биохимических показателей креатинфосфокиназы. Лечение мышечной дистрофии Дюшенна симптоматическое.
Таким образом, мы видим, что заболевание является тяжелым, поэтому его необходимо предупреждать. Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Гаусманова - Петрусевич И. Мышечные заболевания. Польск. Госуд. Мед. Изд. - Варшава, 2001. - 365с.
Страхова О.С., Белозерова Ю.М., Темин П.А. Кардиомиопатия при прогрессирующей мышечной дистрофии Дюшенна.- Ст-Петербург - 1999.- 423с.
Читайте также: