Иммуногистохимические маркеры роста вестибулярных шванном
Добавил пользователь Владимир З. Обновлено: 28.01.2026
Метод определения Гистологическое исследование биоптата согласно гистологической классификации Всемирной организации здравоохранения (ВОЗ) с окрашиванием гематоксилином-эозином. ИГХ-исследование с применением спектра тканеспецифичных и прогностических антител (до пяти антител) (пероксидазный и авидин-биотиновый методы).
Комплексное исследование биоптатов метастатических образований различных локализаций, включающее морфологическое описание и оценку экспрессии спектра тканевых маркеров, для определения гистогенеза первичного опухолевого очага.
По статистике, в 3-15% случаев злокачественные опухоли манифестируют метастазами. Метастазы без выявленного первичного очага характеризуются случайной, нетипичной локализацией и быстрым прогрессированием процесса. Средняя продолжительность жизни пациентов с тремя и более метастатическими очагами поражения составляет три месяца. При этом локализация первичного очага у 60-70% пациентов выявляется только на аутопсии. Своевременная ИГХ-диагностика определяет тактику лечения до выявления первичного очага.
Патоморфологическая диагностика метастазов невыявленных опухолей предусматривает определение их морфологического типа, уточнение вероятного источника метастазирования, оценку злокачественного потенциала. Первая задача решается при рутинном гистологическом исследовании биопсийного материала (окрашивание гематоксилином-эозином), вторая - применением специальных методов исследования: гистохимического, иммуногистохимического (ИГХ) или молекулярно-генетического метода FISH (Fluorescence In Situ Hybridization).
При рутинной морфологической диагностике метастатические опухоли, согласно рекомендациям Европейского общества медицинской онкологии ESMO (European Society for Medical Oncology) (2004), разделяются на пять крупных категорий: аденокарцинома, плоскоклеточный рак, нейроэндокринный рак, недифференцированный рак, недифференцированная опухоль. Эти морфологические категории, наряду с данными о распространенности процесса, во многих случаях позволяют определить адекватный план обследования (и лечения). Так, например, при выявлении метастаза плоскоклеточного рака в шейных лимфатических узлах необходимо эндоскопическое обследования органов верхних дыхательных и пищеварительных путей. ИГХ-исследование, в зависимости от морфологического типа новообразования, позволяет уточнить гистогенез опухоли и/или определить вероятную локализацию первичного очага.
Для ИГХ-исследования первичных опухолей и их метастазов используется широкий спектр тканеспецифичных маркеров: -цитоспецифичные (кластеры дифференцировки лейкоцитов (CD - Clusters of Differentiation), гладкомышечный актин, миоглобин, тиреоглобулин); -маркеры пролиферации (Ki67, PCNA - Proliferating Cell Nuclear Antigen); -опухолевые маркеры - онкофетальные антигены (фетопротеин, карциноэмбриональный антиген); -гормоны (эстроген, прогестерон); -ферменты, белковые продукты клеточных онкогенов и др.
Единых алгоритмов при ИГХ-исследовании метастазов опухолей без уточненного первичного очага не разработано. Корректное определение направления дифференцировки опухолевых клеток и ряда биологических параметров опухоли само по себе является показанием для назначения определенных схем терапии. Например, обнаружение метастазов аденокарциномы в лимфоузлах подмышечной области может являться показанием к проведению терапии, аналогичной терапии рака молочной железы соответствующей стадии. Обнаружение экспрессии рецепторов эстрогенов и прогестерона в такой аденокарциноме может стать показанием к назначению антигормональной терапии вне зависимости от наличия определяемого опухолевого узла в молочной железе. Пример алгоритма, рекомендуемого для использования в дифференциальной диагностике.
Иммуногистохимические маркеры роста вестибулярных шванном
Телефон регистратуры поликлиники:
Телефон регистратуры лучевой диагностики:
Гамма-нож
 |
Вестибулярная шваннома (невринома VIII нерва, акустическая невринома) представляет собой доброкачественное новообразование, возникающее из шванновских клеток вестибулярной порции VIII нерва.
Заболеваемость невриномами слухового нерва составляет примерно 1 случай на 100000 населения в год. Каждый год в Российской Федерации появляется 1500 новых случаев неврином слухового нерва. Среди первичных интракраниальных опухолей вестибулярные шванномы вестибулярные шванномы занимают 4-е место (после глиом, менингиом и аденом гипофиза), составляя до 6-10% всех верифицированных опухолей мозга и поражая преимущественно лиц работоспособного возраста (30-60 лет), на который приходится более 80% случаев. Вестибулярные шванномы составляют около 30% опухолей задней черепной ямки и до 90% новообразований мосто-мозжечкового угла. Несколько чаще встречаются у женщин, чем у мужчин, примерное соотношение 3:2.
Клиническая картина зависит от размеров опухоли. При небольших размерах симптоматики может не быть вовсе. По мере увеличения опухоли в размерах появляется симптоматика, зависящая от того, какие структуры сдавливает опухоль:
- Поражения нервов.
- Нарушение слуховой и вестибулярной функции.
- Расстройства функции вкуса на передней 2/3 языка и слюноотделения (XIII нерв).
- Расстройства функций каудальной группы нервов.
- Расстройства функции мимической мускулатуры.
- Глазодвигательные нарушения.
- Симптомы раздражении/компрессии ствола головного мозга.
- Различные виды нистагма.
- Пирамидная симптоматика.
- Мозжечковая симптоматика.
- Гипертензионно-дислокационный синдром.
Диагностика
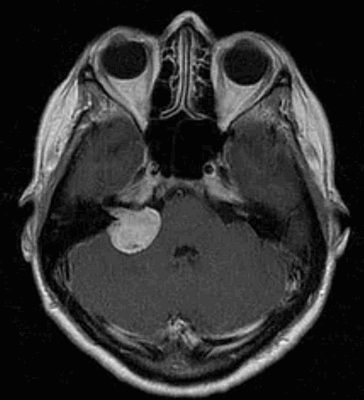
Наиболее информативным методом выявления вестибулярных шванном является МРТ головного мозга с контрастным усилением в режиме Т1 и Т2. Это исследование позволяет определить размеры опухоли, наличие перитуморозного отека, наличие признаков окклюзионной гидроцефалии, которая может быть следствием сдавления опухолью IV желудочка. Кроме этого МРТ позволяет провести дифференциальную диагностику с другими опухолями схожей локализации (чаще с менингиомой задней грани пирамиды височной кости). Еще одним стандартом диагностики является КТ в костном режиме. Независимо от снижения слуха стандартом является проведение аппаратной аудиографии.
Классификация KOOS
I стадия: опухоль находится в пределах внутреннего слухового прохода, диаметр экстраканальной части составляет 1-10 мм.
II стадия: опухоль вызывает расширение канала внутреннего слухового прохода, и выходит в мостомозжечковый угол, ее диаметр составляет, 11-20 мм.
III стадия: опухоль распространяется до ствола головного мозга без его компрессии, диаметр составляет 21 - 30 мм.
IV стадия: опухоль вызывает компрессию ствола головного мозга, ее диаметр более 30 мм.
Классификация M. Samii
Т1 - интрамеатальная опухоль.
Т2 - интра-экстрамеатальная опухоль.
Т3а - опухоль заполняет мостомозжечковую цистерну.
Т3b - опухоль распространяется до ствола головного мозга.
Т4а - опухоль вызывает компрессию ствола.
Т4b - опухоль грубо деформирует ствол мозга и IV желудочек.
Тактика лечения
В зависимости от размеров опухоли, клинической симптоматики, возраста больного, тяжести его состояния, наличия сопутствующей патологии возможны 3 варианта тактики лечения:
- динамическое наблюдение;
- хирургическое лечение;
- стереотаксическая радиохирургия.
Динамическое наблюдение
Учитывая, что вестибулярная шваннома является доброкачественной опухолью с невысокими темпами роста, при небольших ее размерах, отсутствии или минимальном неврологическом дефиците возможен выбор выжидательной тактики. Особенно такой подход оправдан у пожилых людей, или у пациентов с выраженной декомпенсированной соматической патологией. Однако, в большинстве случаев, учитывая риски, связанные с возможным ростом опухоли, более оправданным считается выбор активной тактики.
Хирургическое лечение
Наличие у пациента вестибулярной шванномы является показанием к хирургическому лечению. Большие и гигантские размеры опухоли (более 3,5 см.) являются абсолютным показанием к хирургическому лечению. При меньших размерах возможно проведение радиохирургического лечения.
Для удаления вестибулярных шванном в настоящее время используются три основных доступа: транслабиринтный, ретросигмовидный (наиболее часто используемый) и доступ через среднюю черепную ямку. Каждый из них обладает своими преимуществами и своими же недостатками. В последние годы в связи с развитием микрохирургической техники результаты хирургического лечения пациентов значительно улучшились. Летальность после удаления вестибулярных шванном колеблется в пределах (0-3%), рецидивы опухоли наблюдаются в 5-10%.
В настоящее время превалирующим фактором результативности хирургического лечения считается качество жизни пациента, поэтому интраоперационная тактика в большей степени должна быть нацелена на достижение именно этой цели. Такие послеоперационные осложнения как паралич лицевого нерва (до 25%), нарушения глотания, глазодвигательные нарушения, несут существенную угрозу инвалидизации, поэтому проведение таких операций диктуют высокие требования к профессиональной подготовке оперирующего нейрохирурга. Из послеоперационных осложнений также следует отметить ликворею у 2-20% пациентов, которая иногда может потребовать проведение повторной операции для закрытия ликворного свища; кровоизлияние в ложе операции (1-2%); отек мозжечка и ствола мозга; гемипарез; менингит (1,2%); раневую инфекцию (1,2%); парез VI черепного нерва (1-2%) и парезы других, ниже расположенных черепных нервов; подкожную гематому (3%).
Стереотаксическая радиохирургия
Целью радиохирургического метода является контроль роста опухоли. Стабилизация процесса и даже уменьшение размеров наблюдается в 85-94% случаев, что соответствует мировым данным. Другим критерием эффективности лучевой терапии является высокий (до 98%) показатель отсутствия операции после проведения облучения. Важным также является сохранение функции улиткового и других черепных нервов в случаях, когда это возможно, а также улучшение неврологического статуса пациента. Эффективность применения Гамма-ножа при лечении больных с вестибулярными шванномами создало реальную альтернативу хирургическому лечению при опухолях, размеры которых не превышают 3,5 см, при этом метод радиохирургии лишен недостатков хирургического метода лечения.
После облучения опухоли гамма-ножом возможно как уменьшение ее размеров, так и временное их увеличение. Третьим возможным вариантом развития событий может быть продолженный рост опухоли, однако, это происходит нечасто (от 0 до 7% в течение 10 лет).
Версия для слабовидящих Карта сайта
ВШ — вестибулярная шваннома
КТ — компьютерная томография
ММУ — мостомозжечковый угол
МРТ — магнитно-резонансная томография
НФ-2 — нейрофиброматоз II типа
ПНП — пальценосовая проба
ЧМН — черепно-мозговые нервы
Вестибулярная шваннома (ВШ) — это доброкачественная опухоль, развивающаяся из верхней вестибулярной порции вестибулокохлеарного нерва в месте перехода центрального миелина в периферический (зона Oberstain-Redlich) в результате гиперпродукции шванновских клеток. Это самая распространенная опухоль в задней черепной ямке у взрослых, составляющая 80% опухолей мостомозжечкового угла (ММУ) [1].
ВШ в 95% случаев поражает только одно ухо. В 5% случаев невриномы имеют двусторонний характер роста и являются проявлением нейрофиброматоза II типа (НФ-2). Эти новообразования развиваются вследствие генетических нарушений в 22-й хромосоме, при этом изменяется продукция белка шванномина, который контролирует рост шванновских клеток [2, 3]. Заболевание диагностируется чаще в трудоспособном возрасте (30—60 лет). ВШ несколько чаще встречается у женщин, что объясняется наличием у опухоли рецепторов к прогестерону. С этим же связан более интенсивный рост опухоли у женщин [4].
Темп роста опухоли варьирует от 1 до 30 мм/год. По течению заболевания можно выделить три группы ВШ: не растущие или с очень медленным ростом (менее 0,2 см в год), с медленным ростом (0,2—1,0 см в год), с быстрым ростом (более 1,0 см в год) [5]. Известно, что большинство опухолей имеет медленный рост, но есть также быстрорастущие образования, удваивающие свой размер всего за 6 мес. Несмотря на то что большинство новообразований можно отнести к одному из перечисленных видов, у небольшого числа пациентов периоды отсутствия или медленного роста опухоли чередуются со значительным увеличением размера в течение короткого срока. Особенно это относится к кистозным шванномам, которые способны относительно быстро увеличиваться в размере именно за счет кровоизлияния в строму опухоли и формирования кистозного компонента [6].
Для диагностики ВШ используют магнитно-резонансную томографию (МРТ) головного мозга с введением контрастных веществ. Отоневрологическое обследование, наряду с методами нейровизуализации, также является ведущим в диагностике, наблюдении и оценке динамики симптоматики после лечения. Это объясняется в первую очередь тем, что прогрессирующее снижение слуха и субъективный ушной шум являются, как правило, первыми и основными проявлениями болезни [7].
В зависимости от величины опухоли и ее соотношения с окружающими мозговыми и костными структурами наиболее используемыми и удобными являются классификации по Koos и Ганноверская классификация, созданная под руководством проф. M. Samii.
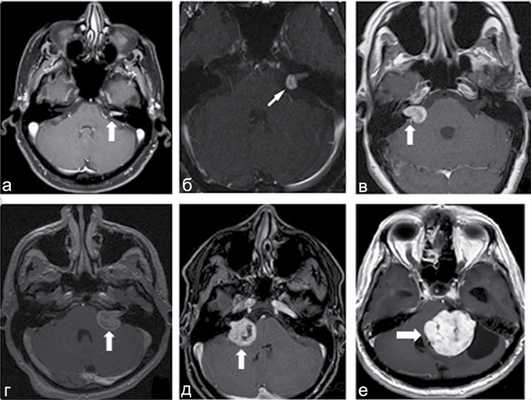
Классификация по Koos: I стадия: опухоль находится в пределах внутреннего слухового прохода, диаметр внутриканальной части составляет 1—10 мм; II стадия: опухоль вызывает расширение канала внутреннего слухового прохода и выходит в ММУ, ее диаметр составляет 11—20 мм; III стадия: опухоль распространяется до ствола головного мозга без его компрессии, диаметр составляет 21—30 мм; IV стадия: опухоль вызывает компрессию ствола головного мозга, ее диаметр более 30 мм. Ганноверская классификация по M. Samii: Т1 — интрамеатальная опухоль; Т2 — интра-экстрамеатальная опухоль; Т3а — опухоль заполняет мостомозжечковую цистерну; Т3b — опухоль распространяется до ствола головного мозга; Т4а — опухоль вызывает компрессию ствола; Т4b — опухоль грубо деформирует ствол мозга и IV желудочек (рис. 1) Рис. 1. МРТ головного мозга с контрастом. Примеры градации ВШ по Ганноверской классификации. a — Т1, интрамеатальная опухоль; б — Т2, интраэкстрамеатальная опухоль; в — Т3а, опухоль заполняет мостомозжечковую цистерну; г — Т3b, опухоль распространяется до ствола головного мозга; д — Т4а, опухоль вызывает компрессию ствола; е — Т4b, опухоль грубо деформирует ствол и IV желудочек. [7, 8].
Принципиально важна ранняя диагностика шванном, так как на ранних стадиях развития (при размере ВШ менее 2 мм) возможно тотальное удаление опухоли с сохранением функции лицевого нерва и слуха у слышащих пациентов. Запущенные опухоли приводят к инвалидизации, удалить их тотально без возникновения грубого неврологического дефицита зачастую невозможно. Парез лицевого нерва разной степени выраженности при тотальном удалении опухоли может развиваться у 90% пациентов [9].
Пациенты с опухолями на стадии T1—T3 обращаются, как правило, к оториноларингологу, поскольку снижение слуха и/или субъективный ушной шум могут быть единственными жалобами на этот момент. Однако, несмотря на то что данная патология хорошо изучена и легко диагностируется при наличии современных методов нейровизуализации (КТ, МРТ), на сегодняшний день все равно большинству пациентов диагноз ставят только на стадии Т4а, Т4b. Ежегодно в ФГАУ НМИЦН им. Н.Н. Бурденко оперируют около 150 пациентов. Среди них большинство с большими и гигантскими опухолями. В 2010 г. было прооперировано 75 больных, из них 58 (77%) с ВШ на стадии Т4а, Т4b по Samii. В 2012 г. прооперированы 133 пациента, из которых 103 (77%) пациента с большими и гигантскими образованиями. В 2015 г. оперированы 192 пациента, из них 137 (71%) имели опухоль на стадии Т4а, Т4b по Samii. При получении этих данных мы заинтересовались данной проблемой и провели исследование, посвященное причинам поздней диагностики.
Цель исследования — выявить причины поздней диагностики ВШ и разработать рекомендации для решения этой проблемы на основании анализа данных историй заболеваний пациентов, проходивших лечение в ФГАУ НМИЦН им. Н.Н. Бурденко с этим заболеванием.
Материал и методы
Проведен ретроспективный обзор 192 наблюдений больных с ВШ, проходивших лечение в ФГАУ НМИЦН им. Н.Н. Бурденко за 2015 г.
Оценивались следующие параметры: пол, возраст пациентов, анамнестические и клинические данные (отоневрологическая симптоматика, время между началом клинических проявлений и постановкой диагноза ВШ, размер опухоли по Koos/Samii).
Результаты
Среди больных, находившихся на лечении, было 55 (29%) пациентов с небольшими опухолями (Koos I, II Samii T1—T3), 137 (71%) пациентов — с большими и гигантскими опухолями, 63 (33%) пациента с ВШ стадии Т4а по классификации Samii, 74 (38%) пациента с ВШ стадии Т4b; 55 (29%) пациентов с ВШ стадии III по классификации Koos, 82 (43%) — с ВШ стадии IV по Koos. Среди общего числа больных с большими и гигантскими ВШ жители Москвы и Московской области составляли 45 человек. Длительность заболевания с момента появления первых клинических проявлений до установления диагноза составляла в среднем 41 мес (табл. 1, рис. Таблица 1. Длительность заболевания с момента появления первых симптомов до установления диагноза в зависимости от стадии роста опухоли 2). Рис. 2. Длительность заболевания с момента появления первых симптомов до установления диагноза в зависимости от стадии роста.
Из табл. 1 видно, что у 28 пациентов период с появления первых симптомов до постановки диагноза составил от 6 мес до 1 года. 23 пациентам не могли установить правильный диагноз на протяжении 2 лет с момента появления жалоб. 77 пациентов имели анамнез более 2 лет. Самая большая длительность заболевания в представленной серии составляет 26 лет.
Из анамнеза известно, что 117 больных обращались за помощью к различным специалистам, и диагноз ВШ также был поставлен не сразу (табл. 2, рис. Таблица 2. Причины поздней диагностики ВШ в зависимости от стадии роста опухоли 3). Рис. 3. Причины поздней диагностики вестибулярной шванномы в зависимости от стадии роста.
Таким образом, из табл. 2 следует, что 37 больных не обращали внимания на одностороннее снижение слуха, 46 пациентов обращались за помощью к терапевтам и неврологам. Самым частым ошибочным диагнозом в этой группе являлись остеохондроз шейного отдела позвоночника (30 пациентов — 65%) и старческая тугоухость (10 пациентов — 22%)
Самая большая группа больных (56 пациентов) длительное время находилась на амбулаторном наблюдении ЛОР-врачей и сурдологов, самым частым диагнозом являлась сенсоневральная тугоухость (34 пациента — 61%), однако в некоторых случаях ЛОР-врачи ставили диагноз «отосклероз» (2 пациента — 4%), «евстахиит» (12 пациентов — 21%), «хронический экссудативный отит» (8 пациентов — 14%). Пациенты получали длительную консервативную терапию препаратами с недоказанной эффективностью, включающую «сосудистые» и ноотропные средства, физиотерапевтические методы лечения.
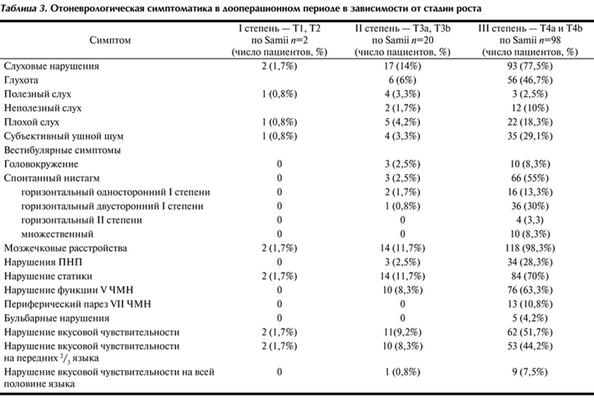
120 пациентам было проведено полное отоневрологическое обследование. Оценивали размер опухоли и сопутствующую симптоматику: нарушение слуха, вестибулярные симптомы в виде головокружения и спонтанного нистагма, мозжечковые расстройства (пальценосовая проба, нарушение статики), нарушение функции V, VII черепных нервов, бульбарные нарушения, нарушения вкусовой чувствительности (табл. 3). Таблица 3. Отоневрологическая симптоматика в дооперационном периоде в зависимости от стадии роста
Из приведенных в табл. 3 данных видно, что у пациентов с I степенью роста опухоли отоневрологическая симптоматика была скудной, что подтверждает тот факт, что диагноз на этой стадии поставить трудно. У пациентов со II степенью выявляли выраженное снижение слуха, шум в ухе, вестибулярные симптомы, нарушения вкуса, гипестезию на лице, бульбарные нарушения. Несмотря на это, данным пациентам диагноз ВШ был поставлен не сразу. Самая большая группа — пациенты с III степенью (98 пациентов) — в 47% случаев имели глухоту на одно ухо, в 98% — мозжечковые расстройства, в 63% — гипестезию на участках лица, в 30% — нистагм. Появление неврологической симптоматики, по-видимому, стало причиной выполнения МРТ и выявления ВШ.
Большинство заболеваний органа слуха и равновесия периферического генеза не угрожают жизни пациента, однако ухудшают ее качество. Особое значение уделяется ВШ, поскольку данное заболевание приводит к инвалидизации, а при отсутствии лечения — и к гибели пациентов [10].
Поэтому у всех больных с односторонней нейросенсорной тугоухостью, особенно в сочетании с вестибулярными нарушениями, необходимо исключить опухоль ММУ, в частности невриному.
В настоящее время для диагностики ВШ «золотым стандартом» считают МРТ-исследование с контрастным усилением сигнала, которое проводят пациентам при одностороннем снижении слуха [11].
Отоневрологическое исследование, как показали работы Н.С. Благовещенской, иногда имеет решающее значение в постановке диагноза, оценке результатов лечения и прогнозировании течения заболевания [12].
Проявления ВШ следует дифференцировать в первую очередь с острой и хронической сенсоневральной тугоухостью улиткового происхождения, чаще всего сосудистого или (реже) токсического генеза. Кроме того, ВШ могут иметь признаки, схожие с болезнью Меньера и арахноидитом ММУ. Во II и III стадии развития ВШ схожи по симптомам с менингиомами и холестеатомами ММУ, опухолями пирамиды височной кости. Кроме того, возможно наличие нетипичной клинической картины в виде оталгии, головной боли или чувствительных нарушений на половине лица [13, 14]. У всех больных с односторонней нейросенсорной тугоухостью, особенно в сочетании с вестибулярными нарушениями, необходимо исключить опухоль ММУ, в частности В.Ш. Таким образом, все пациенты с подозрением на данное заболевание должны быть обследованы отоневрологом и аудиологом (с проведением тональной пороговой и надпороговой аудиометрии, импедансометрии, дополнительных ретрокохлеарных тестов, вестибулометрии, позволяющей диагностировать гипо- или арефлексию лабиринта).
Ежегодно в ФГАУ НМИЦН им. акад. Н.Н. Бурденко оперируют большое число пациентов с ВШ, среди которых многие с большими и гигантскими образованиями. Анализируя данные истории болезней пациентов, можно выделить несколько причин поздней диагностики:
1) низкая онконастороженность по поводу заболевания среди врачей и пациентов (19% пациентов не обращались к специалистам);
2) незнание патологии врачами общей практики и оториноларингологами, так как ВШ достаточно редко встречается в повседневной практике. По данным нашего исследования, самый большой процент запущенных неврином обнаруживается у пациентов, которые длительное время лечились у ЛОР-врачей, — 21,4%. Чуть меньше процент в группе пациентов, наблюдавшихся у терапевтов и неврологов, — 20,3%;
3) низкий уровень использования врачами МРТ и аудиометрии при наличии жалоб на снижение слуха. 7,6% пациентов выполнили МРТ самостоятельно. Некоторым пациентам было рекомендовано проведение обследования, но из-за недоступности МРТ (дороговизны и/или отсутствия необходимого оборудования) рекомендация не была соблюдена.
В Нидерландах M. Kleijwegt и соавт. [16] в 2016 г. проводили исследование по эпидемиологии В.Ш. По данным их серии наблюдений с 2001 по 2012 г., у 34,9% пациентов диагностирована ВШ на стадии Koos I, у 59,9% больных — ВШ стадии II/III по Koos и только 14% был поставлен диагноз ВШ на стадии IV по Koos. Авторы сообщают об увеличении случайных находок при выполнении МРТ.
Однако в России, несмотря на эволюцию методов диагностики опухолей головного мозга, в настоящее время имеется ряд проблем на этапе скрининга пациентов с подозрением на ВШ.
В 2018 г. на конгрессе нейрохирургов A. Sweeney и соавт. [17] представили клинические рекомендации по скринингу ВШ: на основе аудиограммы пациентам с односторонней разностью ≥10 децибел (дБ) на 2 или более непрерывных частотах или ≥15 дБ на 1 частоте рекомендуется проведение МРТ-скрининга, чтобы минимизировать частоту недиагностированных В.Ш. По мнению авторов, скрининг пациентов с односторонней разностью ≥15 дБ при 3000 Гц может свести к минимуму частоту не диагностированных по данным МРТ ВШ.
1. На сегодняшний день существует серьезная проблема, связанная с поздней диагностикой ВШ, причины которой — малая онконастороженность населения и врачей, незнание патологии врачами общей практики и оториноларингологами и недоступность диагностических тестов (аудиометрии, МРТ).
2. Необходимо внедрение в повседневную практику отоневрологического, аудиологического исследований, а также методов нейровизуализации, в первую очередь МРТ с контрастным усилением сигнала, у всех пациентов с односторонним снижением слуха.
3. Необходимо проведение тематических лекций, семинаров, конференций, посвященных проблеме диагностики ВШ, предназначенных для практикующих врачей широкого профиля: оториноларингологов, неврологов и терапевтов в поликлиническом звене.
ФГБНУ «НИИ нейрохирургии им. акад. Н.Н. Бурденко», Москва, Россия
ФГБУ "НИИ нейрохирургии им. акад. Н.Н. Бурденко" РАМН, Москва
Злокачественная опухоль оболочек периферических нервов, развившаяся из слухового нерва: случай из практики и обзор литературы
Журнал: Журнал «Вопросы нейрохирургии» имени Н.Н. Бурденко. 2017;81(4): 95‑100
НИИ нейрохирургии им. акад. Н.Н. Бурденко РАМН, Москва
Авторами представлен редкий случай наблюдения пациентки со злокачественной опухолью периферического нерва, развившейся из слухо-вестибулярного нерва, а также обзор литературы, включающий 30 известных случаев данной патологии.
ФГАУ «НИИ нейрохирургии им. акад. Н.Н. Бурденко» Минздрава России, Москва, Россия
Список сокращений:
MPNST — злокачественная опухоль оболочек периферических нервов
Злокачественная опухоль оболочек периферических нервов — malignant peripheral nerve sheath tumor (MPNST) относится к редкой и гетерогенной группе мезенхимальных злокачественных новообразований, рассматриваемых в большинстве публикаций как саркоматозный процесс. Эта группа опухолей составляет 5—10% всех сарком мягких тканей, с примерной частотой встречаемости 1 человек на 1 млн населения в год [1—4].
Последняя редакция морфологической классификации ВОЗ определяет два подтипа злокачественных опухолей оболочки периферического нерва: эпителиоидная MPNST и MPNST с периневральной дифференциацией, которые признаются достаточно различными по клиническому течению. Другие подтипы MPNST, такие как опухоль Тритона, железистая MPNST и др., которые ранее рассматривались отдельно, в настоящее время расцениваются как гистологические варианты [5]. MPNST могут развиваться как первично из клеток оболочки периферического нерва, так и при малигнизации нейрофибром и шванном [3]. Большинство MPNST развивается в толще мягких тканей из проксимальных частей периферических нервов верхних и нижних конечностей. Наиболее часто поражаются седалищные нервы, плечевое сплетение, крестцовое сплетение. Черепные нервы являются источником роста опухоли в менее чем 5% случаев [3, 6, 7]. Из интракраниальных MPNST чаще поражаются лицевой и слуховой нервы [2, 4]. На 1041 вестибулярную шванному приходится 1 случай MPNST слухового нерва [1]. До 50% MPNST развиваются у пациентов с нейрофиброматозом [8, 9].
Стоит отметить, что литературы по интракраниальным MPNST, в частности по опухолям, происходящим из акустико-фациальной группы нервов, крайне мало. В основном публикации представляют «случай из практики», и лишь в единичных работах анализируются серии наблюдений [1, 4].
Основными факторами риска возникновения MPNST являются нейрофиброматоз 1-го типа и предшествующее радиологическое лечение. В 8—13% случаев у пациентов с НФ 1-го типа в течение жизни развивается MPNST [10]. Большинство источников литературы [8, 9] представляют случаи малигнизации доброкачественных неврином или нейрофибром. Риск трансформации вестибулярной шванномы в MPNST после проведения радиологического лечения составляет от 1/500 до 1/2000 случаев [11—13]. В единичных исследованиях [4, 13—15] сообщается о развитии MPNST без предшествующего лучевого лечения.
Дифференциальный диагноз MPNST включает невриному, параганглиому, гемангиому, эпендимому, ангиолипому, солитарную фиброзную опухоль, лимфому, гемангиоперицитому, каверному, атипичную или анапластическую менингиому, бифросаркому, синовиальную саркому, меланому.
Метастазирование MPNST осуществляется как гематогенным путем, так и с током ликвора [6].
Лечение пациентов с данной патологией включает в первую очередь хирургическое удаление опухоли. Цель хирургического вмешательства — тотальное удаление, но и оно не предотвращает рецидива. В большинстве случаев радикальное удаление выполнить не удается ввиду инфильтративного роста опухоли. Радикальность в этом случае приводит к грубой инвалидизации или летальному исходу. Оперативное лечение необходимо дополнять лучевой терапией. Применение химиотерапии и ее эффективность оспариваются частью специалистов [1, 6].
Наиболее современное исследование, посвященное MPNST, представляет анализ 24 случаев MPNST слухового нерва, возникших без предшествующего хирургического и лучевого лечения [1]. Распределение пациентов по полу в серии наблюдений было равнозначное. Средний возраст пациентов на момент установления диагноза составил 44 года, что было примерно на 10 лет меньше, чем при спорадической вестибулярной шванноме [1, 16]. В клинической картине заболевания, помимо кохлеовестибулярного синдрома, выявлялся паралич лицевого нерва. По данным нейровизуализации определялись такие признаки, как нечеткость границ опухоли, отек ствола головного мозга, полости распада в опухоли [1].
Прогноз у пациентов с MPNST крайне неблагоприятный. Медиана продолжительности жизни с момента установления диагноза составляет 3 мес. Наиболее значимыми факторами, влияющими на эффективность лечения, признаются тотальное удаление, радиотерапевтическое лечение, женский пол [17].
Клиническое наблюдение
Пациентка К., 19 лет, поступила 02.03.16 в ННПЦН им. акад. Н.Н. Бурденко с жалобами на головокружения, боль в лице и области уха, слабость лицевой мускулатуры, сильную головную боль, тошноту и рвоту, двоение предметов перед глазами, онемение на лице.
Появление первых симптомов — сильной стреляющей боли в правом ухе и головокружений — отмечено 17.11.15 на фоне острой респираторной инфекции. Через 3 дня присоединилась сильная головная боль с тошнотой, рвотой, стал быстро снижаться слух на правое ухо. Впервые выполненная МРТ головного мозга 27.11.15 показала изменения в области правого внутреннего слухового прохода.
В результате выполненной 02.12.15 МРТ головного мозга с контрастным усилением, показавшей наличие интраканальной опухоли, распространяющейся в сторону мостомозжечковой цистерны, заподозрена невринома слухового нерва. Было принято решение проводить динамическое наблюдение. Однако 14.12.15 остро развилась слабость лицевой мускулатуры, стала нарастать интенсивность головокружений. Внепланово была выполнена контрольная МРТ головного мозга 13.01.16, где отмечалось увеличение экстраканальной части опухоли почти в 2 раза. В течение подготовки к хирургическому лечению отмечалось значительное ухудшение состояния: нарастание статодинамических нарушений, увеличение выраженности симптоматики со стороны правого мостомозжечкового угла, появление бульбарных нарушений, выраженных общемозговых симптомов. По МРТ головного мозга от 22.02.16 отмечалась прогрессия экстраканальной части опухоли до 3 см. На фоне терапии глюкокортикоидами, обезболивающими и противорвотными препаратами состояние пациентки стабилизировалось.
Пациентка поступила в ННПЦН им. акад. Н.Н. Бурденко в тяжелом состоянии. В клинической картине заболевания выявлялась развернутая симптоматика поражения правого мостомозжечкового угла в виде недостаточности тройничного, лицевого (паралич лицевой мускулатуры справа), слухового (глухота) нервов, бульбарных нарушений, правосторонней гемиатаксии. Также выявлялась выраженная общемозговая симптоматика, представленная головной болью, тошнотой и рвотой, локальными болями в области уха.
Учитывая темп развития симптомов, был заподозрен злокачественный характер опухоли. В день поступления (02.03.16) пациентке выполнена МРТ головного мозга и спинного мозга с контрастным усилением. Была выявлена диссеминация процесса с появлением опухоли в левом внутреннем слуховом проходе, множественных очагов патологического накопления контрастного вещества оболочек спинного мозга. Динамика распространения первичного очага и метастазирование представлены на рис. 1 и 2.
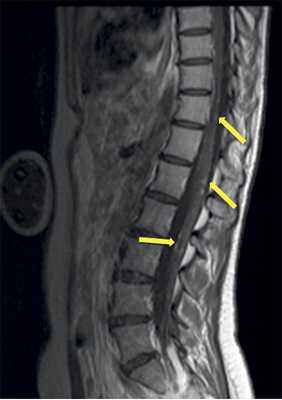
Рис 2. МРТ от 02.03.2016 - метастазирование опухоли по оболочкам спинного мозга (указаны стрелками).
Учитывая распространенность патологического процесса, низкий уровень статуса по шкале Карновского (40 баллов), было принято решение о паллиативной операции — расширенной декомпрессии задней черепной ямки от правого сигмовидного синуса до середины левой гемисферы мозжечка и краниовертебрального перехода, частичном удалении опухоли с экспансивной пластикой твердой мозговой оболочки.
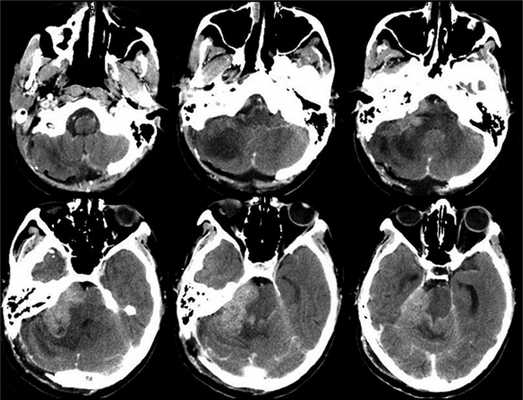
Рис. 3. Рис 3. КТ головного мозга, выполненная на 6-е сутки после операции. Признаки быстрого продолженного роста опухоли.
Таким образом, от появления симптомов до выявления опухоли на МРТ головного мозга прошло 10 дней, а длительность заболевания до летального исхода составила 136 дней.
Морфологическое исследование выявило злокачественную опухоль с кровоизлияниями и некрозами. Опухоль состояла из биполярных клеток с удлиненными отростками и овальными полиморфными ядрами, клетки местами формировали альвеолярные структуры. Фигуры митозов были видимы даже при относительно небольшом увеличении (рис. 4). Иммуногистохимическое исследование выявило: положительную экспрессию эпителиальных маркеров — цитокератина АЕ1/3 (+++) и эпителиального мембранного антигена EMA (++), гладкомышечного актина SMA (++), синаптофизина syn (+) в опухолевых клетках; экспрессию CD34 (+) в эндотелии сосудов. Экспрессии S100, глиофибриллярного кислого белка GFAP, общего лейкоцитарного антигена CD45 в опухолевых клетках не выявлено. Также в опухолевых клетках отсутствовала экспрессия INI1 при сохранной экспрессии антитела в эндотелии сосудов (рис. 5). Индекс мечения пролиферативного маркера Ki-67 — более 90% (рис. 6). Проводился дифференциальный диагноз между атипической тератоидно-рабдоидной опухолью, эпителиоидной саркомой, эпителиоидной злокачественной опухолью оболочек периферического нерва. Заключение: учитывая возраст, характер роста и распространение опухоли, иммуногистохимические данные более соответствуют диагнозу «эпителиоидная злокачественная опухоль периферического нерва (WHO grade IV)». Аутопсия не производилась.
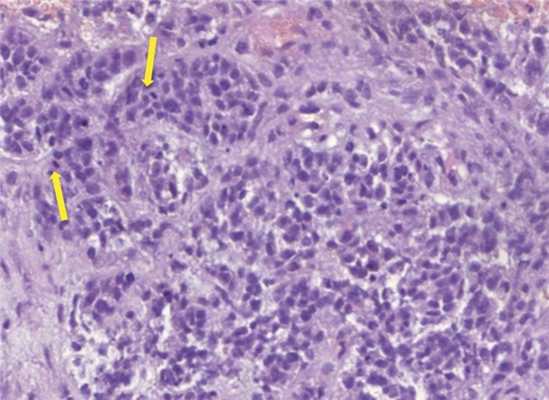
Рис. 4. Гистологический препарат опухоли. Окраска гематоксилином и эозином, увеличение Х300. Альвеолярные структуры и митозы (указаны стрелками).
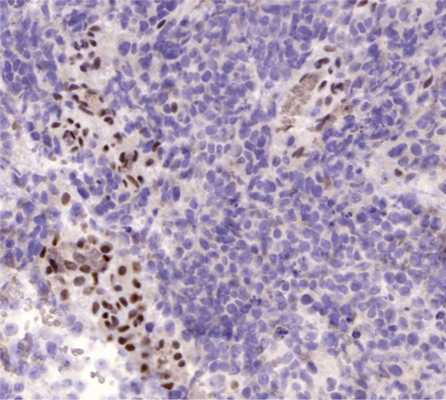
Рис. 5. Гистологический препарат опухоли. Иммуногистохимическое исследование, увеличение Х300. Отсутствие экспрессии INI1 в ядрах опухолевых клеток при сохранной экспрессии в эндотелии сосудов (внутренний контроль корректной работы антитела).
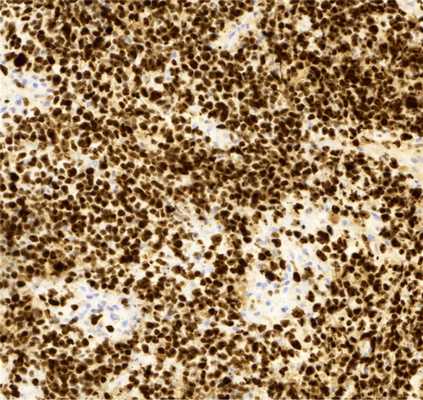
Рис. 6. Гистологический препарат опухоли. Иммуногистохимическое исследование, увеличение Х200. Высокий индекс мечения пролиферативного маркера Ki-67.
MPNST являются малоизученными опухолями, что связано с низкой частотой встречаемости (очень редко встречается MPNST слухо-вестибулярного нерва) и крайне неблагоприятным исходом. Выраженность симптоматики уже на ранних стадиях заболевания зачастую помогает диагностировать опухоль, когда она небольших размеров. Дифференциальный диагноз, в частности с невриномой слухового нерва, создает трудности в планировании оперативного лечения. К моменту операции пациенты имеют опухоли большого размера и признаки метастазирования по центральной нервной системе. Оперативное лечение на ранних этапах при условии радикального удаления и проведения последующей лучевой терапии увеличивает продолжительность жизни от 1 года до 3 лет. При отсутствии оперативного лечения в среднем через 3 мес после начала заболевания наступает летальный исход. Применение химиотерапии в настоящее время оспаривается.
Заключение
MPNST, развивающаяся из VIII пары черепных нервов, является редкой опухолью, представляющей большие трудности в хирургическом удалении и последующем адъювантном лечении. Отсутствие хирургического лечения приводит к летальному исходу в короткие сроки. Даже тотальное удаление опухоли не предотвращает развития рецидива. Прогноз заболевания неблагоприятный.
Авторы заявляют об отсутствии конфликта интересов.
Комментарий
Джесси Ли Кресак, Мегген Уолш, Кафедра патологии, иммунологии и лабораторной медицины, Университет Флориды, Гейнсвилл, Флорида, Соединенные Штаты Америки
Нейрофиброматоз - гетерогенная группа наследственных онкологических синдромов, приводящих к опухолям центральной и периферической нервной систем.
На сегодняшний день наиболее распространенной формой является нейрофиброматоз 1-го типа (NF1, 96%), за которым следует нейрофиброматоз 2-го типа (NF2, 3%) и менее известная форма - шванноматоз. Нейрофиброматоз не имеет половой или расовой предрасположенности.
Нейрофиброматоз 1 типа
Введение. NF1, также известный как болезнь фон Реклингхаузена или периферический нейрофиброматоз, является синдромом аутосомно-доминантной предрасположенности к опухоли, наиболее часто характеризующимся развитием множественных нейрофибром периферических нервов.
Заболеваемость NF1 составляет примерно 1 на 2500 - 3000 новорождённых (1).
Ожидаемая продолжительность жизни сокращается в среднем до 54 лет, часто из-за злокачественных новообразований (2).
Злокачественные новообразования, связанные с NF1, включают злокачественные опухоли оболочек периферических нервов, глиомы, лейкемию, феохромоцитомы, стромальные опухоли желудочно-кишечного тракта ( GI ) и другие. NF1 вызван мутацией в гене-супрессоре опухоли нейрофибромина, расположенном на хромосоме 17.
Он имеет высокую пенетрантность, а частота мутаций гена NF высока, причем 80% имеют наследование по линии отца (3). Самое раннее известное изображение предполагаемого нейрофиброматоза датируется 13 веком с рисунками цистерцианского монаха (4).
В 1862 году Вирхов сослался на наследственный компонент, когда описал человека, у которого “тело было полностью покрыто шишками размером от булавочной головки до голубиного яйца”, и он отметил, что “проявления наследовались уже в течение трех поколений».
Однако именно студент Вирхова Фридрих фон Реклингхаузен в 1882 году дал наиболее полное клиническое и гистологическое описание этого заболевания и ввел термин “нейрофиброма”.
Диагностические критерии. Симптомы NF1 могут значительно различаться у пациентов, и поэтому своевременный и конкретный диагноз трудно установить.
Хотя клинический диагноз может быть заподозрен очень рано в детстве или младенчестве, основные признаки могут полностью отсутствовать до более старшего возраста.
Приблизительно 30% пациентов с NF1 будут соответствовать одному из приведенных ниже критериев к возрасту 1 год, 97% пациентов будут соответствовать двум критериям к возрасту 8 лет; в ретроспективном обзоре пациентов с NF1, все пациенты соответствовали критериям к возрасту 20 лет (1).
В 1987 году Национальные институты здравоохранения (NIH) США разработали консенсус в отношении диагноза из-за клинических различий.
Клинический диагноз может быть установлен, если критерии, перечисленные в таблице 1, удовлетворяются без возможного альтернативного диагноза.
Патогенез и генетические особенности. Ген, ассоциированный с NF1, был идентифицирован в 1990 году, и было обнаружено, что он является одним из крупнейших генов в геноме человека, охватывающим 280 kbp ДНК (6). Ген NF1 расположен на хромосоме 17q11.2, которая кодирует белок, известный как нейрофибромин (7).
Нейрофибромин относится к активирующему GTPase семейству белков-супрессоров опухоли, которые регулируют функцию передачи сигналов RAS/MAPK и механистическую мишень рапамицина (mTOR). Примерно 50% мутаций происходят de novo у пациентов без семейного анамнеза.
Для семей с унаследованной мутацией наблюдается полная пенетрантность; однако клинические проявления у членов семьи могут сильно различаться. Вариабельность фенотипической экспрессии, вероятно, является результатом эпигенетической модификации (8).
Исследования, посвященные корреляции генотипа и фенотипа, показали, что делеция всего гена, известная как микроделеция 17q11.2, связана с более тяжелой формой заболевания, в то время как мозаицизм может привести к легкому или даже сегментарному изменению (6, 7).
Было идентифицировано почти 1500 различных мутаций гена NF1, и эти мутации были обнаружены по всей длине большого локуса гена (6).
Нонсенс, сдвиг рамки считывания и точечные мутации были идентифицированы с большинством мутаций, приводящих к укороченной форме белка.
Генетическое тестирование часто проводится у членов семьи пациента, у которого была выявлена патологическая мутация, и, таким образом, конкретная мутация может быть проверена у их родственников.
В противном случае, учитывая удивительно высокую степень подверженности мутациям и отсутствие “горячих точек” на гене, NF1 не поддается мутационному анализу в качестве практического диагностического инструмента (6).
Диагноз NF1 чаще всего ставится на основании клинических данных, рассмотренных выше.
Клинические проявления
Нейрофибромы. Нейрофибромы являются наиболее распространенным типом опухолей NF1, встречающейся примерно у 60% пациентов. Гистологически нейрофибромы NF1 неотличимы от спорадических опухолей, хотя первые часто бывают крупнее.
Нейрофибромы могут быть кожными или внутренними, затрагивающими глубокие мягкие ткани. Кожные формы могут быть пятнистыми, узловатыми или бляшкообразными, развивающимися в позднем детстве и увеличивающимися в количестве во взрослом возрасте (2).
Внутренние или глубокие нейрофибромы могут возникать по всему телу, включая периорбитальные, забрюшинные области, желудочно-кишечный тракт и средостение (9).
Патогномоничные для NF-1 плексиформные нейрофибромы - это внутренние нейрофибромы, которые вместо того, чтобы расти интраневрально в пределах одного нерва, разрастаются, вовлекая несколько пучков или ветвей нерва или сплетения.
Эта модель роста соответствует характерному описанию “мешка с червями”, данному при пальпации или хирургическом исследовании этих опухолей. Плексиформные нейрофибромы часто развиваются в детстве и быстро растут, оказывая массовое воздействие на соседние структуры.
В отличие от кожной формы, плексиформные нейрофибромы имеют повышенный риск трансформации в злокачественные опухоли оболочек периферических нервов (MPNST). MPNST являются редкими агрессивными веретеноклеточными саркомами, на долю которых приходится лишь около 5% всех сарком мягких тканей.
Около 50% MPNST развивается при NF1, поскольку у этих пациентов риск развития одного из них в течение жизни составляет от 8 до 13% (3).
Внезапное изменение или рост плексиформной нейрофибромы при наблюдении за визуализацией, а также повышенное поглощение фтордезоксиглюкозо-позитронно-эмиссионной томографии (ПЭТ-сканирование) должны вызывать подозрение на злокачественную трансформацию.
Лечение нейрофибром включает хирургическую резекцию и лазерную терапию, в то время как цель хирургии плексиформной нейрофибромы часто заключается в ее удалении. Недавние клинические испытания с участием ингибитора тирозинкиназы иматиниба показали уменьшение объема опухоли более чем на 20% у подгруппы пациентов (10).
В настоящее время проводятся дальнейшие исследования, посвященные этой терапии, а также ингибиторам mTOR (10).
MPNST являются значительной причиной смертности у пациентов с NF1, и, несмотря на радикальное иссечение с широким хирургическим доступом с последующим химиолучевым лечением, 5-летняя выживаемость остается низкой из-за частых метастазов в легких и костях, а также местных рецидивов (2).
Пигментные аномалии. Пятна кофе с молоком - это доброкачественные коричнево-бежевые пигментированные макулы, которые могут встречаться в любом месте тела и часто являются признаком NF1. К возрасту 1 года у 99% пациентов с диагнозом NF1 будет шесть или более макул цвета кофе с молоком размером более 5 мм (препубертатные критерии в соответствии с NIH) (1, 9).
Хотя пятна с кофе с молоком являются общей особенностью NF1, они неспецифичны, поскольку их можно увидеть примерно у 10% населения в целом, а также при других генетических синдромах - карликовости Сильвера Рассела, MEN IIb, синдроме Легиуса и синдроме Маккьюна-Олбрайта.
Количественное ограничение, используемое в диагностических критериях, основано на исследовании Кроу и др. в 1956 году, в котором 78% из 203 проанализированных пациентов с NF1 имели по крайней мере шесть пятен от кофе с молоком размером более 15 мм (11).
Это количество больше, чем указано для населения в целом. В ретроспективном обзоре младенцев с родимыми пятнами Mihm и соавт. показали, что у 1,8% афроамериканских младенцев будет три или более пятен цвета кофе с молоком, в то время как у кавказских младенцев редко бывает два или более (12).
Гистологически эти высыпания представляют гиперпигментацию базального эпидермиса с присутствием макромеланосом (9).
Хотя возможности для злокачественной трансформации нет, иногда требуется косметическое лечение.
Отчеты о случаях показывают, что у некоторых пациентов может быть хороший ответ на дерматологическую лазерную терапию для депигментации; однако для получения ответа может потребоваться несколько процедур, и у части пациентов рецидив пигментации произойдет в течение 6 месяцев (13).
Пигментные пятна в подмышечной впадине и паху являются еще одним определяющим симптомом NF1. Только примерно 40% пациентов будут иметь пигментные пятна в младенчестве, а у 90% пациентов с NF1 они появятся к 7 годам (1).
Меланоцитарные узелки также могут встречаться в радужной оболочке у пациентов с NF1. Эти небольшие, часто множественные гамартоматозные поражения, известные как узелки Лиша, встречаются у 93% взрослых с NF1. Они протекают бессимптомно (14).
Глиомы. Пациенты с NF1 подвергаются повышенному риску развития глиом низкой и высокой степени злокачественности. Наиболее часто встречающейся глиомой в условиях NF1 является глиома зрительного нерва низкой степени злокачественности.
Эти глиомы зрительного нерва встречаются примерно у 15% пациентов с NF1 и обычно появляются в возрасте 7 лет (2).
Как правило, эти опухоли являются пилоцитарными астроцитомами I степени по классификации Всемирной организации здравоохранения и гистологически эквивалентны пилоцитарным астроцитомам общей популяции с двухфазным ростом, волосоподобными отростками и волокнами Розенталя.
Учитывая вялотекущий характер роста этих опухолей, лечение обычно состоит из наблюдения. Когда острота зрения снижается, может быть применена химиотерапия (2).
Расположение этих опухолей не позволяет проводить хирургическое вмешательство, и у пациентов с NF1 лучевой терапии избегают из-за повышенного риска развития злокачественных новообразований, вызванных радиацией (15).
Глиомы ствола головного мозга являются второй по частоте глиомой NF1, и опять же, эти опухоли обычно являются пилоцитарными астроцитомами.
По тем же причинам, что и при глиомах зрительного нерва, химиотерапия является единственным доступным методом лечения с целью уменьшения симптомов и повышения длительности выживания.
Наконец, пациенты с NF1 имеют пятикратный риск развития злокачественных глиом, особенно глиобластом, по сравнению с общей популяцией со средней выживаемостью около 1 года.
Скелетно-мышечные нарушения. У детей с NF1 значительно повышен риск развития рабдомиосаркомы, что примерно в 20 раз выше, чем у населения в целом (2).
Эти рабдомиосаркомы могут возникать в любом месте; однако систематический обзор показал преобладание локализации в тканях мочевого пузыря и предстательной железы (16). Протоколы лечения, применяемые при несиндромных рабдомиосаркомах, также могут быть применены к пациентам с NF1.
У пациентов с NF1 часто отмечаются различные аномалии скелета, включая остеопению, сколиоз или врожденную дисплазию большеберцовой кости. Кроме того, многие пациенты с NF1 невысокого роста, хотя пропорции их тела остаются нормальными (17).
Механистическая связь NF1 и деформаций скелета в значительной степени неизвестна; однако сообщалось, что у пациентов с NF1 низкая минеральная плотность костей и низкая концентрация витамина D (2).
Исследования показали, что риск переломов у детей, страдающих NF1, увеличивается в три раза, а у взрослых-в пять раз (17, 18). Повторные переломы у этих пациентов могут привести к псевдоартрозу.
Желудочно-кишечные проявления. Желудочно-кишечный тракт может быть поражен нейрофибромами и злокачественными опухолями оболочек периферических нервов, аналогично другим участкам тела; однако также существуют различные другие злокачественные новообразования желудочно-кишечного тракта, связанные с нейрофиброматозом.
Что касается нейрофибром, желудочно-кишечный тракт может быть поражён очаговыми нейрофибромами или диффузной нейрофиброматозной пролиферацией, локализованной в собственной пластинке. В обоих случаях ганглиозные клетки могут пролиферировать без клинического значения.
Стромальные опухоли желудочно-кишечного тракта (GIST) не являются редким явлением у пациентов с NF1 и встречаются до 25% случаев (19). В отличие от GIST в общей популяции, GIST, ассоциированные с NF1, редко сочетаются с мутациями в KIT или PDGFRA.
В большинстве случаев GIST протекает бессимптомно и доброкачественно. Гистологически GIST имеет сходства с несиндромными опухолями, состоящие из веретенообразных клеток и скейноидных волокон.
Эндокринные опухоли желудочно-кишечного тракта также наблюдаются у пациентов с NF1, и они имеют склонность к локализации в периампулярной области.
Наиболее распространенной эндокринной опухолью, о которой сообщается, является соматостатинома; однако в этой ситуации также были описаны гастринома, инсулинома, карциноиды и параганглиомы (19).
Другие злокачественные новообразования. Пациенты с NF1 также имеют предрасположенность к злокачественным новообразованиям вне тканей нервной системы.
У детей с NF1 в семь раз повышен риск злокачественных новообразований кроветворной системы, особенно миелоидного лейкоза, по сравнению с их сверстниками. Лечение и прогноз аналогичны для общей популяции.
Пациенты с NF1 также подвержены повышенному риску развития рака молочной железы, особенно у женщин в возрасте до 50 лет.
Проявление феохромоцитомы часто включает покраснение, учащенное сердцебиение и гипертонию, и хирургическое вмешательство часто является основным лечебным мероприятием.
Нейрофиброматоз 2 типа
Введение. NF2, также известный как двусторонний акустический нейрофиброматоз или центральный нейрофиброматоз, представляет собой наследственный опухолевый синдром, характеризующийся преимущественно развитием шванном, наряду с менингиомами, эпендимомами и аномалиями зрения.
Несмотря на название, нейрофибромы встречаются относительно редко.
NF2 наследуется по аутосомно-доминантному типу с предполагаемой частотой 1 на 25 000, распространенностью 1 на 60 000 и пенетрантностью приблизительно 0,95 (20).
Пациенты обычно находятся в возрасте около 20 лет, и прогностические аспекты включают возраст на момент постановки диагноза, фазу развития менингиомы и доступ к специализированным медицинским центрам (21).
Заболевание вызвано мутацией в гене NF2 на 22-й хромосоме, который кодирует белок мерлин. Более половины случаев вызваны мутациями гена de novo у пациентов без семейного анамнеза заболевания.
Диагностические критерии. Двусторонние шванномы верхней вестибулярной ветви восьмого черепного нерва (вестибулярная шваннома или акустическая неврома) являются патогномоничными для NF2.
Однако, поскольку у 41% пациентов, у которых в конечном итоге было доказано наличие NF2, на начальном этапе не было двусторонних вестибулярных шванном, было создано несколько диагностических стандартов для NF2.
К ним относятся широко признанные критерии Манчестера, а также дополнительные критерии NIH, приведенные в таблице 2. Бейзер и соавт. недавно предложили систему оценки для замены критериев Манчестера с якобы повышенной чувствительностью при сохранении 100% специфичности (21, 22).
• Не менее шести пятен с кофе с молоком (диаметр> 5 мм у препубертатных лиц и > 15 мм у постпубертатных пациентов)
Читайте также:
- Смешанно-клеточный вариант лимфомы Ходжкина
- Температура тела новорожденного. Потребность новорожденного в витаминах и микроэлементах
- Примеры семейной эксудативной витриоретинопатии симулирующей ретинобластому
- Роль вредных привычек в деформации зубов. Искусственное вскармливание при деформации зубов.
- Подготовка к зондированию желудка у ребенка. Фракционное зондирование желудка у детей