Истинная полицитемия (ИП, эритремия, болезнь Вакеза) - эпидемиология, распространенность
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Ph-негативные миелопролиферативные заболевания (истинная полицитемия
эссенциальная тромбоцитемия
первичный миелофиброз) у взрослых
- Национальное гематологическое общество European Hematology Association The American Society of Hematology European Society of Pathology European Association for Haematopathology Российское общество патологоанатомов Российское онкогематологическое общество
Оглавление
Ключевые слова
Список сокращений
Алло-ТГСК - аллогенная трансплантация гемопоэтических стволовых клеток
Ауто-ТГСК - аутологичная трансплантация гемопоэтических стволовых клеток
БФ - бластная фаза
ВОЗ - Всемирная организация здравоохранения
ИП - истинная полицитемия
КТ - компьютерная томография
МДС - миелодиспластический синдром
МПЗ - миелопролиферативное заболевание
МПЗн - миелопролиферативное заболевание неклассифицированное
МРТ - магнитно-резонансная томография
НМГ - низкомолекулярный гепарин
ОВ - общая выживаемость
ОМЛ - острый миелоидный лейкоз
ПМФ - первичный миелофиброз
Пост-ИП МФ - постполицитемический миелофиброз
Пост-ЭТ МФ - посттромбоцитемический миелофиброз
ПЦР - полимеразная цепная реакция
УЗИ - ультразвуковое исследование
ХМЛ - хронический миелоидный лейкоз
ХФ - хроническая фаза
ЭТ - эссенциальная тромбоцитемия
P32 - радиоактивный фосфор
DIPSS (Dynamic International Prognostic Scoring System) - Динамическая международная шкала оценки прогноза
ELN (European Leukemia Net) - Европейская организация по изучению и лечению лейкозов
EORTC - Европейская организация по исследованиям в области лечения рака
G-CSF (granulocyte colony stimulating factor) - гранулоцитарный колониестимулирующий фактор
IPSET-thrombosis (The International Prognostic Score for ET) - Международный Прогностический Индекс рисков тромбоза при эссенциальной тромбоцитемии
IPSS (International Prognostic Scoring System) - Международная шкала оценки прогноза
IWG-MRT (The international working group for myeloproliferative neoplasms research and treatment) - Международная рабочая группа по изучению и лечению миелопролиферативных заболеваний
NSSN (National Comprehensive Cancer Network®) - Национальная Онкологическая Сеть США
PVSG (Polycythemia Vera Study Group) - группа по изучению истинной полицитемии
PUVA (psoralen + UVA treatment) - ПУВА - терапия; ультрафиолетовая фототерапия в комбинации с псораленом
?? - препараты, включенные в перечень жизненно необходимых и важнейших лекарственных препаратов для медицинского применения.
Термины и определения
Трепанобиопсия - диагностическая процедура забора образцов костного мозга путем пункции гребня подвздошной кости.
Миелофиброз - морфологический термин, который характеризует фиброз стромы костного мозга с вытеснением элементов гемопоэза, встречается при метастатическом и лимфомном поражениях костного мозга, острых лейкозах, миелодиспластических синдромах, МДС/МПЗ, хроническом миелолейкозе BCR-ABL1+, волосатоклеточном лейкозе. Среди неопухолевой патологии миелофиброз в костном мозге может встречаться при инфекционных и аутоиммунных заболеваниях, на поздних этапах некроза костного мозга различной этиологии вследствие фиброгистиоцитарной пролиферации с замещением фиброзной тканью некротизированного костного мозга, или выявляется при повторной трепанобиопсии из этого же участка подвздошной кости. При дифференциальной диагностике следует принимать во внимание возможность миелофиброза реактивной природы при терапии G-CSF, с расширением и омоложением гранулоцитарного ростка, усилением ретикулинового каркаса стромы. Как и во всех случаях диагностики миелопролиферативных заболеваний, необходим тщательный сбор клинико-анамнестических, лабораторных данных. Миелофиброз может возникнуть в результате трансформации ИП и ЭТ. Таким образом, следует различать ПМФ, миелофиброз, возникший в результате прогрессии/трансформации ИП/ЭТ и морфологический термин «миелофиброз».
1. Краткая информация
1.1 Определение
Миелопролиферативные заболевания (МПЗ) представляют собой клональные заболевания, возникающие на уровне стволовой кроветворной клетки. МПЗ характеризуются пролиферацией одной или более клеточной линии миелопоэза в костном мозге с признаками сохранной терминальной дифференцировки, и сопровождаются изменением показателей периферической крови.
Истинная полицитемия (ИП) (синонимы: эритремия, болезнь Вакеза, истинная красная полицитемия) - клональное МПЗ, которое характеризуется пролиферацией эритроидного, гранулоцитарного, мегакариоцитарного ростков миелопоэза, с преимущественной пролиферацией эритроидного ростка кроветворения (панмиелоз), увеличением числа эритроцитов и повышением уровня гемоглобина, тромбоцитозом, лейкоцитозом в периферической крови (панцитоз), независимостью эритропоэза от нормальных механизмов регуляции. Почти все больные являются носителями мутации JAK2V617F или другой функционально сходной мутации.
Эссенциальная тромбоцитемия (синонимы: первичный тромбоцитоз, идиопатический тромбоцитоз, геморрагическая тромбоцитемия) - клональное МПЗ с неконтролируемой пролиферацией мегакариоцитов, характеризующееся повышенным числом крупных и гигантских мегакариоцитов в костном мозге, тромбоцитозом в периферической крови (>450 х 109/л), высоким риском тромбозов и/или кровотечений.
Миелопролиферативное заболевание, неклассифицируемое (МПЗн) Согласно рекомендациям ВОЗ 2008 г., данный диагноз следует использовать при наличии клинических, лабораторных и гистологических (в трепанобиоптате костного мозга) признаков МПЗ, не соответствующих какой-либо определенной нозологической форме классических Ph-негативных МПЗ. Чаще всего эта категория используется: При ранних стадиях заболевания (манифестация) - при расхождении между клиническими, лабораторными и морфологическими данными, позволяющими верифицировать ту или иную нозологическую форму МПЗ. При бластной фазе заболевания, без предшествующего анамнеза и установленного ранее диагноза миелопролиферативного заболевания. При сочетании МПЗ с воспалительными, метаболическими или опухолевыми заболеваниями, маскирующими основные признаки той или иной нозологичесой формы. МПЗ неклассифицируемое не диагностируется: при объеме трепанобиоптата костного мозга недостаточном для адекватного анализа; при отсутствии предоставленных клиницистами клинических и лабораторных данных, при наличии предшествующей терапии цитостатиками или колониестимулирующими факторами; при наличии реаранжировки генов PDGFRA, PDGFRB, FGFR1, выявлении химерного гена BCR-ABL1.
Первичный миелофиброз (ПМФ) (синонимы: хронический идиопатический миелофиброз, агногенная миелоидная метаплазия, миелосклероз с миелоидной метаплазией, сублейкемический миелоз, хронический гранулоцитарно-мегакариоцитарный миелоз) возникает de novo, характеризуется клональной пролиферацией стволовых клеток, аномальной экспрессией цитокинов, фиброзом костного мозга, гепатоспленомегалией как следствие экстрамедуллярного гемопоэза, симптомами опухолевой интоксикации, кахексией, лейкоэритробластозом в периферической крови, лейкемической прогрессией, невысокой выживаемостью.
1.2 Этиология и патогенез Ph - негативных МПЗ
Этиология МПЗ до сих пор не установлена. Ведущей гипотезой является многоэтапность возникновения заболевания, где предрасположенность к болезни реализуется под воздействием внешних факторов, повреждающих геном нормальной клетки и приводящих к ее злокачественной трансформации. Несмотря на то, что в последние годы достигнуты значительные успехи в расшифровке молекулярно-генетических механизмов Ph-негативных МПЗ, первоначальная мутация, приводящая к малигнизации гемопоэтической клетки неизвестна [1].
Открытие мутации V617F в гене JAK2 в 2005г явилось значительным шагом в понимании биологических особенностей Ph-негативных МПЗ. Практически у всех пациентов с ИП выявляется мутация гена JAK2: в 96% случаев мутация JAK2V617F (14 экзон), в 2% наблюдений мутация в экзоне 12 гена JAK2 [2, 3]. Мутация JAK2V617F выявляется при ЭТ в 55% наблюдений и присутствует примерно в 45 - 68% случаев при ПМФ. Тогда как мутация в 12 экзоне гена JAK2 практически не встречается при ЭТ и ПМФ [4,5].
Помимо мутации гена JAK2 у больных МПЗ выявляют мутации и в других генах. Мутации гена MPL встречаются в 4% наблюдений при ЭТ, в 8% наблюдений при ПМФ, и редко при ИП. Причем наиболее частые мутации MPLW515L/K в экзоне 10 [6, 7, 8]. Мутация MPLS505N выявляется как при ЭТ, так и при наследственной тромбоцитемии [9, 10]. Данные мутации не являются строго специфичными для МПЗ и имеют вторичный генез в цепи генетических событий.
Не так давно появились данные о диагностической значимости соматических мутаций в 9 экзоне гена CALR, кодирующего белок кальретикулин. Выявлены 36 разных видов мутаций в этом гене, которые приводят к образованию дефектного белка. В исследованиях in vitro клетки, экспрессирующие мутированный ген, обладали способностью цитокин-независимого роста в культуре, что вероятно связано с активацией белков STAT5. У пациентов без мутаций в генах JAK2 и MPL мутации в данном гене были выявлены в 67% случаев при ЭТ и 88% - при ПМФ. Другие авторы также выявили крайне высокую частоту мутаций гена CALR у пациентов с МПЗ (в 70-84% случаев при отсутствии мутации гена JAK2). При этом мутации CALR были обнаружены в 8% случаев при миелодиспластическом синдроме и в единичных наблюдениях при других миелоидных неоплазиях. Важно, что ни в одном случае заболеваний не миелоидной природы, мутации в данном гене не были выявлены [11,12].
Мутации в генах JAK2, MPL, CALR имеют важное диагностическое значение. Их выявление свидетельствует о клональном характере заболевания и помогает в дифференциальной диагностике ИП, ЭТ, ПМФ от ряда других миелоидных неоплазий, а также вторичных эритроцитозов и тромбоцитозов. Наряду с этим активно изучается значимость данных мутаций в прогнозе МПЗ. Несмотря на ряд проведенных исследований, не представляется возможным сделать однозначное заключение в отношении прогностической значимости аллельной нагрузки JAK2V617F при ИП, ЭТ, ПМФ. Вопрос влияния аллельной нагрузки на выживаемость или прогрессирование ИП и ЭТ с исходом в миелофиброз требует дальнейшего изучения [13, 14].
При ИП и ЭТ выявляются и другие мутации: TET2, IDH, ASXL1, DNMT3A и др. [1]. Ни одна из них не специфична для классических Ph-негативных МПЗ, а их патогенетическая значимость исследуется.
Молекулярно-генетические нарушения при Ph-негативных МПЗ приводят к активации JAK-STAT сигнального пути. Результатом этого является повышение пролиферации и увеличение количества эритроцитов, лейкоцитов и тромбоцитов периферической крови при ИП или изолированный тромбоцитоз при ЭТ. Патогенез МПЗ, в частности ПМФ, сложен и состоит из цепи событий, первичным из которых является появление патологического клона. Известно, что лейкемические моноциты и мегакариоциты активно продуцируют множество цитокинов (TGF-?, FGF, VEGF, ANG1, OPG, BMP4), избыток которых стимулирует фиброз, неоангиогенез и приводит к остеосклерозу. Наряду с этим нарушается связь стволовых клеток с микроокружением, что способствует появлению экстрамедуллярных очагов гемопоэза, прежде всего в селезенке и печени. Массивный выброс цитокинов - одна из причин возникновения симптомов опухолевой интоксикации, что приводит к значительному ухудшению качества жизни пациентов с ПМФ [15].
Клональная миелопролиферация при Ph-негативных МПЗ также может сопровождаться вторичным воспалением с изменениями стромы костного мозга и патологической выработкой цитокинов. В развитии миелофиброза, как первичного, так и вторичного, остеосклероза и ангиогенеза вовлечены трансформирующий фактор роста бета (TGF-?) миелоидных предшественников, ростовой фактор вырабатываемый тромбоцитами (PDGFR) и эндотелиальный сосудистый фактор роста (VEGF) [16]. Патологическая выработка цитокинов, хемокинов и металлопротеиназ может участвовать в патологическом межклеточном взаимодействии нейтрофилов, моноцитов и мегакариоцитов, приводя к выходу CD34+ миелоидных предшественников и эндотелиальных клеток в периферическую кровь [17, 18].
1.3 Эпидемиология
ЭТ - редкое / орфанное заболевание. Популяционные эпидемиологические данные о заболеваемости и распространенности в России отсутствуют. Литературные данные о заболеваемости по данным зарубежных регистров составляют приблизительно 1,5 - 2,53 : 100 000 населения [19]. При анализе десятилетней динамики заболеваемости в Санкт-Петербурге ежегодная первичная заболеваемость колебалась от 0,60 до 2,10 и составила в среднем 1,30 на 100 000 населения в год [20].
1.4 Кодирование по МКБ 10
D47.4 - первичный миелофиброз
D45 - истинная полицитемия
D47.3 - эссенциальная тромбоцитемия
1.5 Классификация
В соответствии с классификацией ВОЗ 2008г группа хронических МПЗ объединяет восемь нозологических форм:
Полицитемия
Полицитемия у новорожденных - синдром увеличенной концентрации клеточных элементов крови (в большей степени эритроцитов) [1].
Код(ы) МКБ-10:
| МКБ-10 | |
| Код | Название |
| Р61.1 | Полицитемия новорожденного |
Дата разработки/пересмотра протокола: 2017 год.
Сокращения, используемые в протоколе:
| ВПС | - | Врожденный порок сердца |
| ЭхоКГ | - | Эхокардиография |
| ОПН | - | Острая почечная недостаточность |
| ОЦК | - | Объем циркулирующей крови |
| РКИ | - | Рандомизированное контролируемое испытание |
| МКБ | - | Международной классификации болезней |
Пользователи протокола: неонатологи, педиатры, акушеры-гинекологи.
Категория пациентов: новорожденные дети.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или высококачественное (++) когортное или исследование случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное, или исследование случай-контроль, или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+), результаты которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GPP | Наилучшая клиническая практика. |

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
Классификация
Первичная (истинная) полицитемия
связана с поражением гемопоэтического ростка, из-за чего наблюдается неадекватно высокий рост количества эритроцитов, лейкоцитов и тромбоцитов;
Вторичная полицитемия является реакцией на изменения в среде
Вторичная к трансфузии:
• отсроченное пережатие пуповины;
• трансфузия от плода к плоду;
• тансфузия от матери к плоду;
• перинатальная асфиксия;
• нахождение ребенка ниже матери сразу после рождения.
Вторичная к внутриутробной гипоксии:
• задержка внутриутробного развития;
• гипертензия вызванная беременностью;
• диабет матери;
• курение матери;
• ВПС матери;
• переношенность.
Причины, связанные с плодом:
• трисомия 13,18,21;
• гипотироидизм, тиротоксикоз;
• врожденная гиперплазия надпочечной железы;
• синдром Беквита-Видемана.
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ И ЛЕЧЕНИЯ
Диагностические критерии
Скрининг на полицитемию новорожденных рекомендуется при следующих ситуациях:
• новорожденный маленький к гестационному возрасту;
• новорожденный от матери с сахарным диабетом;
• новорожденный большой к гестационному возрасту;
• монохориальная двойня, особенно с одним большим ребенком;
• морфологическая картина задержки развития.
Физикальные данные:
Наиболее частые симптомы неонатальной полицитемии:
• изменения цвета кожных покровов: преимущественно периферический вишневый цианоз;
• изменения со стороны центральной нервной системы: ранние признаки: гипотония, сонливость, раздражительность, беспокойство.
Метаболические нарушения:
• гипогликемия;
• желтуха;
• гипокальциемия.
Нарушения со стороны сердечно-легочной системы:
• тахикардия, тахипное, респираторный дистресс;
• цианоз, плетора.
Нарушения со стороны желудочно-кишечного тракта:
• рвота, слабая сосание, некротически энтероколит.
Нарушения со стороны мочевыделительной системы:
• олигоурия.
Гематологические изменения:
• умеренная тромбоцитопения;
• тромбоз.
NB! Примерно в 40% случаев симптомы полицитемии слабо выражены или отсутствуют (бессимптомная гипогликемия).
Лабораторные исследования:
• важным показателем является центральный венозный гематокрит, который при полицитемии превышает 65%;
• биохимические анализы крови всегда обнаруживают гипогликемию, (уровень глюкозы снижается менее 2,2 ммоль/л.); гипокальциемию, (снижение уровня кальция в сыворотке крови до менее 1,74 ммоль/л или снижение уровня ионизированного кальция до менее 0,75 ммоль/л), гипомагниемию (снижение уровня магния менее 0,62 ммоль/л).
NB! Остальные диагностические мероприятия нацелены на выявление причины полицитемии у новорожденных. При подозрении на каждую конкретную нозологию используются свои методы диагностики.
Инструментальные исследования:
• рентгенография грудной клетки: кардиомегалия, отек легких (при развитии дыхательных нарушений, сердца (других частей тела по необходимости);
• ЭхоКГ: повышение резистентности легочных сосудов, снижение сердечного выброса (при подозрении на кардиопатию и ВПС).
Показания для консультации специалистов: консультация кардиолога, невролога, нефролога для обсуждения тактики ведения при обнаружении врожденных пороков сердца, легких, почек.
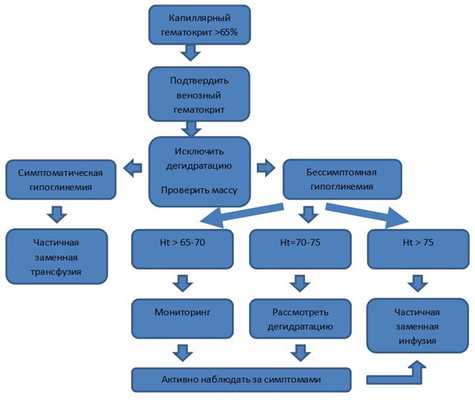
Диагностический алгоритм:
Алгоритм ведения новорожденных с полицитемией
Полицитемия ( Болезнь Вакеза, эритремия, эритроцитоз , Эритремия , Эритроцитоз )
Полицитемия - хронический гемобластоз, в основе которого лежит неограниченная пролиферация всех ростков миелопоэза, преимущественно эритроцитарного. Клинически полицитемия проявляется церебральной симптоматикой (тяжестью в голове, головокружением, шумом в ушах), тромбогеморрагическим синдромом (артериальными и венозными тромбозами, кровотечениями), микроциркуляторными расстройствами (зябкостью конечностей, эритромелалгией, гиперемией кожи и слизистых). Основные диагностические сведения получают при исследовании периферической крови и костного мозга. Для лечения полицитемии применяются кровопускания, эритроцитаферез, химиотерапия.


Общие сведения
Полицитемия (болезнь Вакеза, эритремия, эритроцитоз) - заболевание группы хронических лейкозов, характеризующееся повышенной продукцией эритроцитов, тромбоцитов и лейкоцитов, увеличением ОЦК, спленомегалией. Заболевание является редкой формой лейкемии: ежегодно диагностируется 4-5 новых случаев полицитемии на 1 млн. населения. Эритремия развивается преимущественно у пациентов старшей возрастной группы (50-60 лет), несколько чаще у мужчин. Актуальность полицитемии обусловлена высоким риском развития тромботических и геморрагических осложнений, а также вероятностью трансформации в острый миелобластный лейкоз, эритромиелоз, хронический миелолейкоз.

Причины полицитемии
Развитию полицитемии предшествуют мутационные изменения полипотентной стволовой гемопоэтической клетки, дающей начало всем трем клеточным линиям костного мозга. Наиболее часто выявляется мутация гена тирозинкиназы JAK2 с заменой валина фенилаланином в 617 позиции. Иногда наблюдается семейная заболеваемость эритремией, например, среди евреев, что может свидетельствовать в пользу генетической корреляции.
При полицитемии в костном мозге присутствуют 2 вида клеток-предшественников эритроидного кроветворения: одни из них ведут себя автономно, их пролиферация не регулируется эритропоэтином; другие, как и положено, являются эритропоэтинзависимыми. Считается, что автономная популяция клеток представляет собой не что иное, как мутантный клон - основной субстрат полицитемии.
Патогенез
В патогенезе эритремии ведущая роль принадлежит усиленному эритропоэзу, следствием которого служит абсолютный эритроцитоз, нарушение реологических и свертывающих свойств крови, миелоидная метаплазия селезенки и печени. Высокая вязкость крови обусловливает склонность к сосудистым тромбозам и гипоксическому повреждению тканей, а гиперволемия вызывает повышенное кровенаполнение внутренних органов. В финале полицитемии отмечается истощение кроветворения и миелофиброз.
В гематологии различают 2 формы полицитемии - истинную и относительную. Относительная полицитемия развивается при нормальном уровне эритроцитов и снижении объема плазмы. Данное состояние носит название стрессовой или ложной полицитемии и не рассматривается в рамках данной статьи.
Истинная полицитемия (эритремия) по происхождению может быть первичной и вторичной. Первичная форма является самостоятельным миелопролиферативным заболеванием, в основе которого лежит поражение миелоидного ростка кроветворения.
Вторичная полицитемия обычно развивается при повышении активности эритропоэтина; данное состояние является компенсаторной реакцией на общую гипоксию и может встречаться при хронической легочной патологии, «синих» пороках сердца, опухолях надпочечников, гемоглобинопатиях, при подъеме на высоту или курении и т. д. Истинная полицитемия в своем развитии проходит 3 стадии: начальную, развернутую и терминальную.
I стадия (начальная, малосимптомная) - длится около 5 лет; протекает бессимптомно или с минимально выраженными клиническими проявлениями. Характеризуется умеренной гиперволемией, небольшим эритроцитозом; размеры селезенки в норме.
II стадия (эритремическая, развернутая) подразделяется на две подстадии:
- IА - без миелоидной трансформации селезенки. Отмечается эритроцитоз, тромбоцитоз, иногда - панцитоз; по данным миелограммы - гиперплазия всех ростков кроветворения, выраженный мегакариоцитоз. Длительность развернутой стадии эритремии 10-20 лет.
- IIВ - с наличием миелоидной метаплазии селезенки. Выражены гиперволемия, гепато- и спленомегалия; в периферической крови - панцитоз.
III стадия (анемическая, постэритремическая, терминальная). Характерны анемия, тромбоцитопения, лейкопения, миелоидная трансформация печени и селезенки, вторичный миелофиброз. Возможны исходы полицитемии в другие гемобластозы.
Симптомы полицитемии
Эритремия развивается длительно, постепенно и может быть обнаружена случайно при исследовании крови. Ранние симптомы, такие как тяжесть в голове, шум в ушах, головокружение, ухудшение зрения, зябкость конечностей, расстройство сна и др., часто «списываются» на преклонный возраст или сопутствующие заболевания.
Наиболее характерной чертой полицитемии служит развитие плеторического синдрома, обусловленного панцитозом и увеличением ОЦК. Свидетельством полнокровия служат телеангиэктазии, вишнево-красная окраска кожи (особенно лица, шеи, кистей рук и других открытых участков) и слизистых (губ, языка), гиперемия склер. Типичным диагностическим признаком служит симптом Купермана - окраска твердого нёба остается нормальной, а мягкое нёбо приобретает застойный цианотический оттенок.
Другим отличительным симптомом полицитемии является кожный зуд, усиливающийся после водных процедур и иногда имеющий нестерпимый характер. К числу специфических проявлений полицитемии также относится эритромелалгия - болезненное жжение в кончиках пальцев, которое сопровождается их гиперемией.
В развернутой стадии эритремии могут возникать мучительные мигрени, боли в костях, кардиалгия, артериальная гипертензия. У 80% пациентов обнаруживается умеренная или выраженная спленомегалия; печень увеличивается несколько реже. Многие больные полицитемией замечают повышенную кровоточивость десен, появление синяков на коже, длительные кровотечения после экстракции зубов.
Следствием неэффективного эритропоэза при полицитемии является повышение синтеза мочевой кислоты и нарушение пуринового обмена. Это находит клиническое выражение в развитии так называемого уратового диатеза - подагры, мочекаменной болезни, почечной колики.
Осложнения
Результатом микротромбозов и нарушения трофики кожи и слизистых оболочек служат трофические язвы голени, язвы желудка и двенадцатиперстной кишки. Наиболее часты в клинике полицитемии осложнения в виде сосудистых тромбозов глубоких вен, мезентериальных сосудов, портальных вен, церебральных и коронарных артерий. Тромботические осложнения (ТЭЛА, ишемический инсульт, инфаркт миокарда) выступают ведущими причинами смерти больных полицитемией. Вместе с тем, наряду с тромбообразованием, больные полицитемией склонны к геморрагическому синдрому с развитием спонтанных кровотечений самой различной локализации (десневых, носовых, из вен пищевода, желудочно-кишечных и др.).
Гематологические изменения, характеризующие полицитемию, являются определяющими при проведении диагностики. При исследовании крови выявляется эритроцитоз (до 6,5-7,5х10 12 /л), повышение гемоглобина (до 180-240 г/л), лейкоцитоз (свыше 12х10 9 /л), тромбоцитоз (свыше 400х10 9 /л). Морфология эритроцитов, как правило, не изменена; при повышенной кровоточивости может обнаруживаться микроцитоз. Достоверным подтверждением эритремии служит увеличение массы циркулирующих эритроцитов более 32-36 мл/кг.
Для исследования костного мозга при полицитемии более информативно проведение не стернальной пункции, а трепанобиопсии. При гистологическом исследовании биоптата выявляется панмиелоз (гиперплазия всех ростков кроветворения), в поздних стадиях полицитемии - вторичный миелофиброз.
Для оценки риска развития осложнений эритремии проводятся дополнительные лабораторные тесты (функциональные печеночные пробы, общий анализ мочи) и инструментальные исследования (УЗИ почек, УЗДГ вен конечностей, ЭхоКГ, УЗДГ сосудов головы и шеи, ФГДС и др.). При угрозе тромбогеморрагических и метаболических нарушений необходимы консультации соответствующих узких специалистов: невролога, кардиолога, гастроэнтеролога, уролога.
Лечение полицитемии
С целью нормализации объема ОЦК и снижения риска тромботических осложнений первой мерой являются кровопускания. Эксфузии крови проводятся в объеме 200-500 мл 2-3 раза в неделю с последующим восполнением удаленного объема крови физиологическим раствором или реополиглюкином. Следствием частых кровопусканий может явиться развитие железодефицитной анемии. Кровопускания при полицитемии могут быть успешно заменены эритроцитаферезом, позволяющим извлечь из кровотока только эритроцитарную массу, вернув плазму.
В случае выраженных клинико-гематологических изменений, развития сосудистых и висцеральных осложнений прибегают к миелодепрессивной терапии цитостатиками (бусульфан, митобронитол, циклофосфамид и др.). Иногда проводится терапия радиоактивным фосфором. Для нормализации агрегатного состояния крови назначаются гепарин, ацетилсалициловая кислота, дипиридамол под контролем коагулограммы; при геморрагиях показаны трансфузии тромбоцитов; при уратном диатезе - аллопуринол.
Прогноз
Течение эритремии носит прогрессирующий характер; заболевание не склонно к спонтанным ремиссиям и самопроизвольному излечению. Больные пожизненно вынуждены находиться под наблюдением гематолога, проходить курсы гемоэксфузионной терапии. При полицитемии высок риск тромбоэмболических и геморрагических осложнений. Частота трансформации полицитемии в лейкоз составляет 1% у пациентов, не проходивших химиотерапевтическое лечение, и 11-15% у получавших цитостатическую терапию.
Экстрагенитальная патология в акушерстве: Хронические миелопролиферативные заболевания и беременность
Краткие эпидемиологические данные
Хронические миелопролиферативные заболевания (ХМПЗ) составляют группу Ph-негативных клонально обусловленных хронических лейкозов миелоидного происхождения, сопровождающихся трансформацией полипотентной стволовой кроветворной клетки и характеризующихся пролиферацией одного или нескольких ростков миелопоэза. (2,3) Эти заболевания обычно встречаются во второй половине жизни, средний возраст пациентов -50-60 лет. Эссенциальной тромбоцитемией (ЭТ) несколько чаще болеют женщины, истинная полицитемия (ИП) более характерна для мужского пола. В последнее время имеется тенденция к увеличению частоты ХМПЗ у женщин детородного возраста. В репродуктивном периоде ЭТ встречается чаще, чем остальные ХМПЗ (1).
Классификация
В соответствии с последней классификацией ВОЗ (2001), среди ХМПЗ выделяют 3 нозологические формы: эссенциальную тромбоцитемию, истинную полицитемию и идиопатический миелофиброз (ИМ).
Различают следующие стадии ИП:
Стадия 1 - малосимптомная, продолжительностью до 5 лет и более
Стадия 2А - эритремическая развернутая стадия, без миелоидной метаплазии селезенки, 10-20 лет
Стадия 2В - эритремическая с миелоидной метаплазией селезенки
Стадия 3 - постэритремическая миелоидная метаплазия с миелофиброзом и без него (1)
В развитии ИМ выделяют следующие стадии:
1 .пролиферативная (ранняя/префибротическая)
2. продвинутая (фибротическая/фибротически-склеротическая)
3. трансформация в острый лейкоз (2)
Диагностика
Среди общих симптомов ХМПЗ отмечаются так называемые ослабляющие конституциональные симптомы: субфебрилитет, потеря массы тела, повышенная потливость, а также кожный зуд различной степени выраженности, усиливающийся после водных процедур. Сосудистые осложнения, характеризующиеся многочисленными клиническими проявлениями, являются основной причиной, угрожающей здоровью и жизни пациентов с ХМПЗ. Среди микроциркуляторных сосудистых расстройств преобладают нарушения на уровне головного мозга: мучительные мигрени, головокружения, тошнота и рвота, транзиторные ишемические атаки, инсульты головного мозга, психические расстройства, преходящие нарушения зрения и слуха. Кроме того, микрососудистые осложнения проявляются стенокардией, эритромелалгией, характеризующейся приступами острых жгучих болей в пальцах верхних и нижних конечностей с багровым покраснением кожи и отеком. Тромбозы венозных и артериальных сосудов составляют вторую группу сосудистых расстройств при ХМПЗ и нередко являются причиной летальных исходов (тромбоз глубоких вен нижних конечностей, тромбоэмболия легочной артерии и ее ветвей, инсульты головного мозга, инфаркты миокарда и других органов, тромбозы печеночных и нижней полой вены с развитием синдрома Бадда-Киари). Геморрагические осложнения, спонтанные или спровоцированные даже малыми оперативными вмешательствами, варьируют от незначительных (носовые, десневые кровотечения, экхимозы) до непосредственно угрожающих жизни кровотечений (желудочно-кишечные и другие полостные кровотечения). Спленомегалия, являющаяся характерным симптомом всех ХМПЗ, развивается на разных стадиях заболеваний. Причинами увеличения селезенки являются как депонирование избыточного количества клеток крови при ЭТ, 2А стадии ИП, так и развитие экстрамедуллярного гемопоэза при 2В стадии ИП и ИМ. Нередко спленомегалии сопутствует увеличение печени, хотя встречается и изолированная гепатомегалия. Нарушение обмена мочевой кислоты (гиперурикемия и урикозурия) также является общим признаком всех ХМПЗ. Клинически проявляется почечной коликой, мочекаменной болезнью, подагрой, подагрической полиартралгией и их сочетанием. (1,3)
Стадия гематологических исходов, являющаяся проявлением естественной эволюции ХМПЗ, характеризуется развитием миелофиброза разной степени выраженности или трансформацией в острый лейкоз. Кроме того, возможна взаимная трансформация ХМПЗ, поэтому в настоящее время не является ошибкой смена диагнозов ИП, ЭТ или ИМ. (2)
Неблагоприятные исходы беременности при сочетании с ХМПЗ до появления новых препаратов и разработки современных методов лечения наблюдались в 50-60%. Наиболее частыми осложнениями беременности являются самопроизвольные выкидыши на различных сроках, задержка внутриутробного развития плода (ЗВУР), внутриутробная гибель плода, преждевременные роды, отслойка плаценты, преэклампсия. (5, 6)
Эссенциальная тромбоцитемия у 1/3 больных протекает бессимптомно и обнаруживается только при рутинном исследовании анализа периферической крови. Увеличение селезенки, обычно незначительное, наблюдается в 50-56% случаев, а у 20-50% пациентов наблюдается и гепатомегалия. Первыми проявлениями заболевания у 20-35% пациентов являются кровотечения, а у 25-80% (по различным источникам) - тромбозы. (1)
В начальных стадиях ИП основные проявления заболевания связаны с плеторическим синдромом (гиперпродукция эритроцитов), проявляющимся эритроцианотической окраской кожи лица и видимых слизистых оболочек, особенно мягкого неба, резко контрастирующей с обычной окраской твердого неба (симптом Купермана), чувством жара, повышением температуры конечностей. В то же время некоторые пациенты адаптированы к плеторе и могут не предъявлять никаких жалоб. Примерно у 25% больных в дебюте заболевания развиваются венозные тромбозы, инфаркт миокарда или церебральные нарушения, а в 30-40% случаев отмечены проявления геморрагического синдрома. Кожный зуд наблюдается у каждого второго пациента. Выявляется сплено- и гепатомегалия, а также различные проявления тромбогеморрагического синдрома. В фазе гематологических исходов постэритремический миелофиброз развивается у 10-20% больных, трансформация в острый лейкоз встречается в 20-40% случаев. (1,3)
Увеличение селезенки является основным клиническим симптомом при ИМ и встречается у 97-100% пациентов. ИМ долгое время протекает бессимптомно, и спленомегалия выявляется случайно. Наиболее частой причиной обращения к врачу у пациентов с ИМ является слабость, причиной которой является анемия у половины пациентов, в том числе тяжелой степени у 25%. При значительной спленомегалии пациенты часто жалуются на тяжесть в области живота, ощущение сдавления желудка и кишечника, периодические острые боли, вызываемые инфарктом селезенки и периспленитом, Гепатомегалия встречается более, чем у половины больных на момент диагностики. Эволюция ИМ приводит к развитию острого лейкоза у 5-20% больных. (2)
ЭТ может быть заподозрена при стойком увеличении количества тромбоцитов более 600x10 9 /л. В костном мозге обнаруживается пролиферация большого количества гиперплазированных многодольчатых мегакариоцитов. Костный мозг обычно нормо- или гиперклеточный. Изменений со стороны эритроидного и гранулоцитарного ростков кроветворения не отмечается.
Наличие ИП следует предположить при повышении уровня гемоглобина более 165 г/л у женщин. Как правило, содержание лейкоцитов и тромбоцитов также увеличено и составляет 10-12х10 9 /л и более 400х10 9 /л соответственно. Как правило, отмечается повышение щелочной фосфатазы в нейтрофилах в 80% случаев и витамина В12 в сыворотке. При исследовании костного мозга определяется типичная картина его гиперклеточности с пролиферацией трех ростков кроветворения и нередко гиперплазией мегакариоцитов.
При ИМ в периферической крови обнаруживаются пойкилоцитоз эритроцитов, дакроциты, нормобласты. В префибротической стадии заболевания анемия умеренная или отсутствует, в то время как для поздних стадий заболевания характерна выраженная анемия. При гистологическом исследовании выявляется коллагеновый фиброз, а в поздних стадиях - остеомиелосклероз, приводящие к уменьшению клеточности костного мозга и приводящие к его недостаточности. (2)
В связи со схожестью клинико-морфологических особенностей необходима как внутригрупповая дифференциация, так и с Ph-положительным лейкозом (хроническим миелолейкозом) на основании клинико-лабораторных данных. (2)
Лечение
Программа лечения ХМПЗ во время беременности:
1) всем беременным с тромбоцитозом назначают ацетилсалициловую кислоту в дозе 75 - 100мг;
2) при уровне тромбоцитов более 600x10 9 /л - рекомбинантный интерферон-α (ИФ-α) вводят в дозе 3 млн ME в день (или через день), позволяющей поддерживать число тромбоцитов на уровне 200 - 300x10 9 л;
3) при тромбоцитозе более 400x10 9 л, введение ИФ-α продолжают, если это лечение проводилось еще до беременности и/или существует высокий тромбогенный риск.
4) антикоагулянты прямого действия (низкомолекулярный гепарин) по показаниям при отклонениях в плазменном звене гемостаза. (4)
Для профилактики тромбоэмболических осложнений рекомендовано использование медицинских компрессионных чулок. Чтобы снизить риск развития кровотечения необходимо отменить прием аспирина за 2 недели до родоразрешения. Региональная анестезия не должна использоваться ранее 12 часов от последней профилактической дозы НМГ, в случае применения лечебной дозы НМГ - не ранее, чем через 24 часа. Начинать прием НМГ можно спустя 4 часа после удаления эпидурального катетера. При плановом кесаревом сечении прием профилактической дозы НМГ необходимо прекратить за один день до родоразрешения и возобновить через 3 часа после окончания операции (или через 4 часа после удаления эпидурального катетера). (6)
В послеродовом периоде, опасном по развитию тромбоэмболических осложнений, необходимо продолжение лечения в течение 6 недель. В связи с тем, что рекомбинантный ИФ-α экскретируется с молоком, грудное вскармливание во время лечения противопоказано. (6)
1. Клиническая онкогематология ред. Волковой М.А. М., «Медицина» - 2001-с.263-300.
2. Рукавицын О.А., Поп В.П.// Хронические лейкозы. М., «Бином. Лаборатория знаний» - 2004 - с.44-81.
3. Руководство по гематологии ред. Воробьева А. И. М., «Ньюдиамед» - 2003 -Т.2 - с.16-29.
4. Цветаева Н.В., Хорошко Н.Д., Соколова М.А. и др. Хронические миелопролиферативные заболевания и беременность. // Терапевтический архив. -2006.
5. Barbui Т., Barosi G., Grossi A. et al. Practice guidelines for the therapy of essential thrombocythaemia. A statement from the Italian Society of Hematology, the Italian Society of Experimental Hematology and the Italian Group for Bone Marrow Transplantation. //Haematologica. - 2004 - Feb.,89(2). - p.215-232.
6. Harrison C. Pregnancy and its management in the Philadelphia negative myeloproliferative diseases. // British Journal of Haematology. - 2005 - vol. 129(3) -p.293-306.
Истинная полицитемия (ИП, эритремия, болезнь Вакеза) - эпидемиология, распространенность
Истинная полицитемия (ИП) - хроническое миелопролиферативное заболевание, при котором из-за болезни стволовой (материнской) клетки крови появляется опухоль из ее потомков. У больного увеличивается число всех клеток крови: лейкоцитов, тромбоцитов и особенно эритроцитов. Являясь потомками заболевшей стволовой клетки, все клетки крови выполняют свою обычную работу в целом нормально, но повышение гемоглобина и тромбоцитов опасно, потому что образуются сгустки крови, которые, будто пробки, могут перекрывать (тромбировать) мелкие сосуды. Это вызывает различные осложнения (тромбозы), в том числе инфаркты, инсульты.
Как правило, на начальном этапе заболевшие люди чувствуют себя хорошо, а изменения в анализе крови находят иногда случайно. Симптомы болезни появляются позже. В связи с тем, что крови слишком много, может повышаться артериальное давление, появляется тошнота, недомогание, головные боли. Из-за большого количества гемоглобина лицо пациента становится красным, и окружающие начинают думать, что у заболевшего человека проблемы с алкоголем. У больных полицитемией часто чешется тело, особенно после контакта с водой - после душа, ванны и др. На поздних стадиях могут увеличиваться печень и селезенка, в которых накапливаются опухолевые клетки.
Диагноз
При постановке диагноза «истинной полицитемии» практически всегда находят повышение гемоглобина и мутацию гена «JAK2». Однако важно исключить другие причины, которые приводят к похожим изменениям крови, в первую очередь другие миелопролиферативные заболевания, вредные привычки (например, длительный стаж курения) и профессиональные вредности, а также неопухолевые болезни (ВИЧ, туберкулез, гиперпаратиреоз и другие). Пациента всесторонне обследуют: делают гистологическое, цитологическое и цитогенетическое исследование костного мозга, УЗИ или КТ брюшной полости, другие специальные анализы.
Лечение
В настоящее время лучшим методом лечения истинной полицитемии является старый, но очень действенный метод - кровопускание. Регулярное (примерно 1 раз в 1-2 месяца) удаление 400 мл крови позволяет уменьшить все проявления и предупредить большинство осложнений болезни. Лекарства при полицитемии назначают редко. Если со временем полицитемия перейдет в острый лейкоз (что бывает очень редко), то он лечится по схемам острых лейкозов. Врачи постоянно ищут новые способы лечения с использованием новых средств. Молодых пациентов стараются полностью вылечить путем донорской пересадки материнских стволовых клеток крови.
Читайте также: