Критерии некротизирующего ангиита легких. Классификация некротизирующего ангиита
Добавил пользователь Дмитрий К. Обновлено: 01.02.2026
Для цитирования: СОВРЕМЕННАЯ КЛАССИФИКАЦИЯ И КЛИНИЧЕСКОЕ ТЕЧЕНИЕ АНГИИТОВ (ВАСКУЛИТОВ) КОЖИ. РМЖ. 1997;11:3.
В статье представлена классификация ангиитов и рассмотрены особенности клинического течения различных форм этих заболеваний. Уделено внимание вопросам определения стадии и степени активности процесса, что очень важно для разработки максимально индивидуализированной терапевтической тактики.
Кратко перечислены отклонения лабораторных показателей, наблюдающиеся у больных кожными ангиитами.
The paper presents a classification of angiitides and outlines the clinical presentation of various types of these diseases. Emphasis is laid on the determination of a stage and the progression of a process, which is very important to elaborate the maximum individualized treatment policy.
Laboratory abnormalities observed in patients with skin angiitides are briefly listed.
О.Л. Иванов, доктор мед. наук, проф., заведующий кафедрой кожных
и венерических болезней Московской медицинской академии им. И.М. Сеченова.
Prof. O.L. Ivanov, MD, Head, Department of Skin and Venereal Diseases, I.M. Sechenov Moscow Medical Academy.
С осудистые поражения кожи относятся к нередким и зачастую довольно тяжелым дерматозам, представляющим до сих пор большую проблему в теоретической и практической дерматологии. Основное место в этой обширной группе дерматозов занимают воспалительные поражения сосудов кожи и подкожной клетчатки - так называемые ангииты, или васкулиты, кожи.
Термин "ангиит" (от греч. angion -сосуд) признается в настоящее время этимологически более удачным, чем "васкулит" (от лат. vasculum - сосудик), так как при ангиитах поражаются сосуды различного калибра (от магистральных до капилляров), а не только самые мелкие. По своему содержанию термины "ангиит" и "васкулит" в настоящее время являются синонимами.
Ангиитами (васкулитами) кожи называют дерматозы, в клинической и патоморфологической симптоматике которых первоначальным и ведущим звеном является неспецифическое воспаление стенок дермальных и гиподермальных кровеносных сосудов разного калибра.
В настоящее время насчитывают до 50 нозологических форм, относящихся к группе ангиитов кожи. Значительная часть этих нозологий имеет между собой большое клиническое и патоморфологическое сходство, нередко граничащее с идентичностью, что следует иметь в виду при изучении литературы и постановке больному того или иного диагноза. К сожалению, до сих пор нет единой общепринятой классификации ангиитов кожи, что объясняется недостаточной изученностью этиологии, патогенеза, клинического течения и ряда других вопросов, связанных с этой сложной группой дерматозов. Большинство клиницистов пользуются преимущественно морфологическими классификациями кожных ангиитов, в основу которых обычно берутся клинические изменения кожи, а также глубина расположения (и соответственно калибр) пораженных сосудов.
Приемлемая для практических целей рабочая классификация ангиитов кожи, разработанная в клинике кожных и венерических болезней ММА им. И.М. Сеченова, представлена в таблице.
КЛАССИФИКАЦИЯ АГИИТОВ (ВАСКУЛИТОВ) КОЖИ
пурпура"), экхимозы, геморрагические пузыри
и поверхностных мелких узлов. Возможно сочетание любых элементов
Пурпура Болезнь Шамберга-Майокки
Шамберга. Болезнь Шамберга
некрозы, язвы, рубцы
по периферии и разрешающийся в центре
Представленная классификация включает наиболее часто встречающиеся формы кожных ангиитов. Помимо редких вариантов,не вошедших в данную классификацию,существуют также переходные и смешанные формы ангиитов кожи,сочетающие признаки двух и болееразновидностей ангиитов.
Рис. 2. Хроническая пигментная пурпура.
а - петихиальная сыпь; б - экзематоидный тип.
Рис. 3. Ливедо-ангиит.
| Рис. 4. Узловатая эритема. а - острая; б - подострая (мигрирующая); в - хроническая. | а | б | в |
1. Потекаев Н.С., Иванов О.Л. Ангииты кожи (диагностика, лечение, профилактика). Методические указания. М., 1983, С. 30
2. Иванов О.Л., Современные аспекты, проблемы ангиитов кожи. // Вестник РАМН, 1995, 1, 7-9.
3. Иванов О.Л., Бабаян Р.С., Потекаев Н.С. К вопросу о терминологии и клинике васкулитов (ангиитов) кожи. // Вестн. дерматол. и венерол., 1984, 7, 37-42.
4. Шапошников О.Н., Деменкова Н.В. Сосудистые поражения кожи. Л., Медицина 1974, С. 214.
5. Rhyne T.I., Wilkinson DS. Cutaneous Vasculitis: "angiitis". - In: Textbook of Dermatology. Oxford -London 1979;993-1058.
Критерии некротизирующего ангиита легких. Классификация некротизирующего ангиита
³ФГБНУ «НИИР им. В.А. Насоновой», Москва, Россия
ФГБУ "Научно-исследовательский институт ревматологии им. В.А. Насоновой" РАМН, Москва
ФГНБУ «Центральный научно-исследовательский институт туберкулеза»
ФГБУ "ЦНИИТ" РАМН
Некротизирующий саркоидный гранулематоз
Некротизирующий саркоидный гранулематоз (НСГ) относится к продуктивным васкулитам мелких сосудов с образованием саркоидоподобных гранулем, сопровождающимся ишемическими некрозами разной степени выраженности и давности. Заболевание поражает исключительно легкие. Клинические симптомы заболевания неспецифичны, поэтому выявляются достаточно редко. Основной метод диагностики - морфологическое исследование. Иммунокомплексное воспаление развивается в стенках сосудов с формированием макрофагально-гистиоцитарных гранулем, не содержащих эпителиоидных клеток. Этиология и патогенез НСГ остаются малоизученными. Дифференциальная диагностика в основном приводится с туберкулезом, саркоидозом, гранулематозом с полиангиитом.
Некротизирующий саркоидный гранулематоз (НСГ) - продуктивный васкулит мелких артерий и вен с образованием массивных скоплений саркоидоподобных гранулем, сопровождающийся ишемическим некрозом разной степени выраженности и давности. Заболевание поражает исключительно легкие. Этиология и патогенез НСГ остаются малоизученными из-за редкого выявления этого заболевания, отсутствия патогномоничных лабораторных, лучевых и функциональных критериев. Согласно мировому опыту, основным методом диагностики НСГ остается морфологическое исследование 3.
Термин «некротизирующий саркоидный гранулематоз» был впервые предложен в 1973 г. А. Liebov [5]. Автор описал 11 наблюдений пациентов, в биоптатах легких которых гистологически был выявлен продуктивный васкулит. Васкулит сопровождался наличием саркоидоподобных гранулем с образованием конгломератов и некротическими изменениями. При этом отсутствовала внутригрудная лимфаденопатия, характерная для саркоидоза. У этих пациентов наступала регрессия процесса спонтанно или после непродолжительной терапии.
Эпидемиология
Это заболевание встречается редко. Достоверных данных о его распространенности до настоящего времени в доступной нам литературе не отмечено.
Патологический процесс чаще поражает женщин, чем мужчин, в соотношении 2,2:1. Возраст пациентов значительно варьирует, в среднем 49 лет (от 11 до 75 лет) [6, 7].
Лучевая диагностика
Рентгенологический метод исследования выявляет преимущественно односторонние очаговые диссеминаты в виде узелков или узелковых инфильтратов более 1 см в диаметре [1]. Инфильтраты располагаются субплеврально или периваскулярно, как правило в нижних, реже средних отделах легкого, сочетаясь с мелкоочаговой диссеминацией. Описано и двустороннее поражение легких с формированием полостей как в узелках, так и в инфильтрате [6]. Первоначально заболевание может начинаться с развития мелкоочаговой диссеминации легких, которая впоследствии трансформируется в узелковые образования. При вовлечении в патологический процесс плевры развивается экссудативный плеврит с накоплением экссудата до 200 мл, который обычно носит эпизодический характер [8, 9]. Позитронно-эмиссионная томография выявляет накопление флюородеоксиглюкозы в участках консолидации легочной ткани [1, 6, 8, 9].
Клинические проявления
В 20% случаев пациенты не предъявляют каких-либо определенных жалоб, хотя у всех имеются рентгенологические изменения в легких. Респираторные симптомы НСГ неспецифичны: кашель, поверхностное дыхание, боль в грудной клетке. Реже больные жалуются на лихорадку и слабость. Легочные функциональные тесты могут показывать различные отклонения: снижение форсированного жизненного объема легких и артериальную гипоксемию. Результаты лабораторных анализов обычно указывают на ускорение СОЭ до 40-85 мм/ч. В сыворотке крови повышено содержание JgM, JgG. Внелегочные проявления (в частности, почечные) и поражение верхних дыхательных путей крайне редки. Лишь у 10% больных встречается прикорневая лимфаденопатия. Периферические лимфатические узлы не увеличены. В ряде наблюдений сообщается об изменениях в паратрахеальных и внутригрудных лимфатических узлах, характер которых неясен [1, 5, 6]. В таких случаях необходимо проводить дифференциальную диагностику с саркоидозом, туберкулезом и др.
Морфологическая характеристика
Макроскопически ткань легкого в области патологического участка имеет мелкобугристый характер. На разрезе определяются множественные мелкие просовидные, нечетко контурирующие белесовато-сероватые уплотнения от 1 до 3-4 см в диаметре, имеющие округлую, овальную или вытянутую форму. На разрезе поверхность уплотнений зернистая, гомогенная, светло-серого цвета, местами с участками, напоминающими казеозный некроз в фазе организации. Перифокально можно увидеть более мелкие полупрозрачные высыпания размером до 0,1 см. Плевра в области поражения несколько утолщена, белесоватого оттенка (рис. 1).
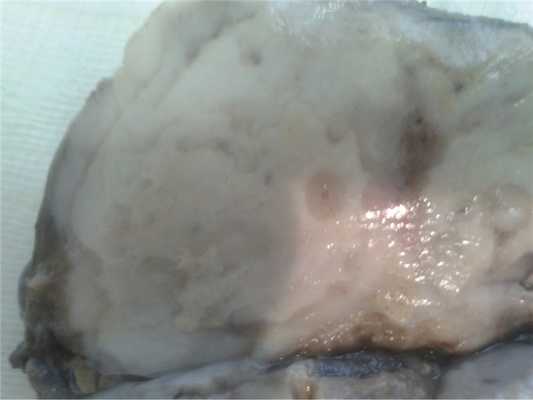
Рис. 1. Макроскопический препарат. НСГ. Легкое: крупноочаговый узел зернистого вида.
Микроскопически НСГ характеризуется наличием васкулитов мелких ветвей легочной артерии, реже - вен с наличием в стенках сосудов одиночных или сливающихся в конгломераты гранулем (рис. 2, а, б).
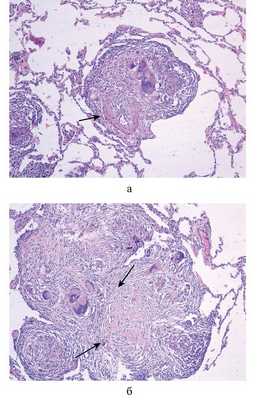
Рис. 2. Гистологические препараты. НСГ. Гранулематозный васкулит легочной артериолы. а - одиночная гранулема в среднем слое гиалинизированной стенки сосуда, ×120 (сосуд обозначен стрелкой); б - гиалиноз в конгломерате гранулем области спавшихся сосудов, ×240 (обозначен стрелками). Окраска гематоксилином и эозином.
Гранулемы и/или конгломераты располагаются в среднем слое сосуда, растягивая эластический каркас и адвентицию (см. рис. 2, а). Гранулемы имеют «оплавленный» вид. Растущие гранулемы сдавливают просвет сосуда вплоть до полного его спадения (см. рис. 2, б). В сосудах, где просвет частично сохранен, можно наблюдать умеренную пролиферацию эндотелия, утолщение базальной мембраны, отек и гиалиноз стенок (см. рис. 2, б). Гранулемы при НСГ в основном состоят из гистиоцитов, плазматических, многоядерных гигантских клеток типа инородных тел и Пирогова-Лангханса, лимфоцитов, немногочисленных лейкоцитов, преимущественно эозинофильных (рис. 3, а, б). Гистиоциты имеют округлое или вытянутое ядро с плотным хроматином и маленьким ядрышком. Ядра расположены в основном центрально, реже - эксцентрично. Гистиоциты отличаются от эпителиоидных клеток округлой формой, меньшими размерами, гиперхромным ядром, более интенсивным окрашиванием цитоплазмы с наличием в отдельных клетках фагосом.
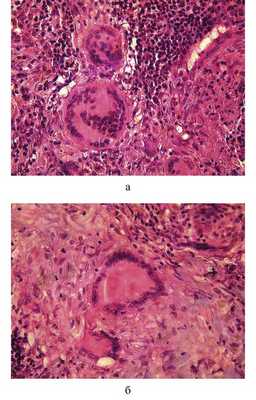
Рис. 3. Гистологические препараты. НСГ. Клеточный состав гранулем. а - преобладание лимфоцитов и гистиоцитов, ×480; б - многоядерный гигантский макрофаг, ×600. Окраска гематоксилином и эозином.
Макрофаги располагаются как в центре, где могут достигать крупных размеров (более 150 мкм), так и по периферии гранулем. В макрофагах определяются светлые фагоцитарные вакуоли, иногда фрагменты лимфоцитов. В ряде случаев центральная часть гранулемы может содержать небольшую зону фибриноидного некроза.
Формирование конгломератов при НСГ носит волнообразный характер и происходит при наличии выраженных признаков васкулита. В связи с тем что точную локализацию гранулем при НСГ определить не всегда возможно, некоторые авторы допускают их расположение не только в стенках сосудов, но и в более отдаленных участках легочной ткани. Помимо конгломератных гранулем, в легочной ткани определяются разновеликие очаги гиалиноза. Они имеют преимущественно гомогенную структуру, не содержат клеточных элементов воспаления и ядерного материала. В центре очага при окраске орсеином определяются стенки спавшихся сосудов, небольшие скопления гемосидерина. Очаг гиалиноза имеет волнистый контур за счет постоянно сливающихся между собой новообразованных гранулем, расположенных в стенках сосудов (рис. 4).
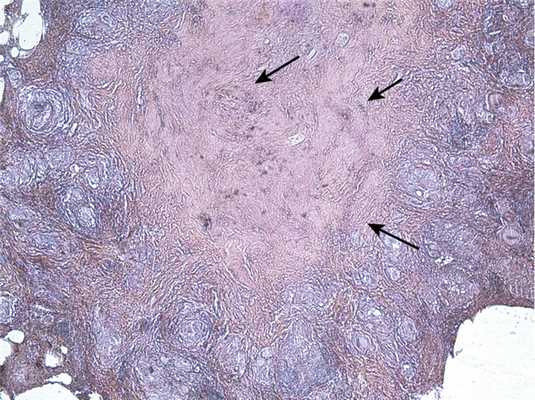
Рис. 4. Гистологический препарат. Очаг НСГ в легочной ткани. Стенки спавшихся сосудов в очаге гиалиноза после окраски орсеином (обозначены стрелками). Окраска пикрофуксином по ван Гизону + орсеин, ×150.
Конгломератные гранулемы и очаги гиалиноза при НСГ могут располагаться субплеврально: без поражения плевры или вовлекая ее в воспалительный процесс. В случаях поражения плевры она неравномерно утолщена за счет сливающихся гранулем. В центре таких гранулем чаще, чем в легочной ткани, встречаются очаги фибриноидного некроза (рис. 5).
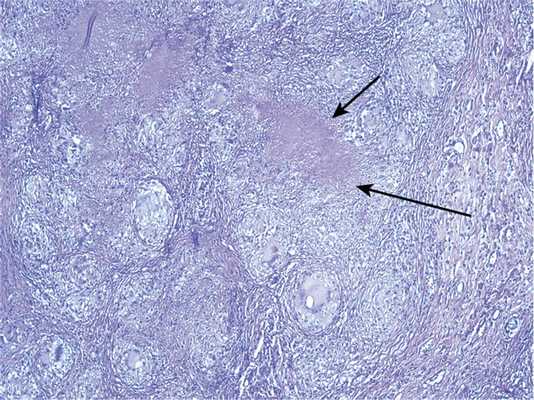
Рис. 5. Гистологический препарат. НСГ легкого с реакцией в плевре: конгломерат из макрофагально-гистиоцитарных гранулем с очагом фибриноидного некроза (обозначены стрелками). Окраска гематоксилином и эозином, ×250.
Характерными фоновыми изменениями легочной паренхимы при НСГ являются полнокровие сосудов с эритродиапедезом и участками кровоизлияния различного масштаба. В просвете альвеол видны эритроциты и сидерофаги. За пределами очагов легочная ткань преимущественно сохраняет свою гистоархитектонику без признаков альвеолита и интерстициального фиброза.
Прикорневые лимфатические узлы при НСГ могут быть увеличены. При этом гистологически лимфоидная ткань частично или полностью замещена гранулематозным воспалением с преобладанием гистиоцитов. Разновеликие гранулемы сливаются между собой, могут иметь зоны фибриноидного некроза и признаки гиалинизации разной степени выраженности.
Лечение и прогноз
Некротизирующий саркоидный гранулематоз отличается доброкачественным течением с благоприятным прогнозом и самоизлечением в ряде случаев [1, 6, 9]. Пациенты с таким диагнозом хорошо лечатся кортикостероидами. В отдельных случаях применяют цитостатики [2]. Летальных исходов при НСГ не описано.
ФГУ "НИИ пульмонологии" ФМБА России
Гистологическая дифференциальная диагностика гранулематозных болезней легких (часть I)
Журнал: Архив патологии. 2019;81(1): 65‑70
Гранулематозные болезни — это гетерогенная группа заболеваний различной этиологии, проявляющихся разнообразными клиническими синдромами и вариантами тканевых изменений, неоднородной чувствительностью к терапии и преобладанием общего доминирующего гистологического признака — наличием гранулем, определяющих клинико-морфологическую сущность каждой болезни. Гранулема является хронической воспалительной реакцией, в которой принимают участие клетки макрофагальной системы и другие клетки воспаления. После воздействия антигена происходит активация T-лимфоцитов, макрофагов, эпителиоидных клеток и гигантских многоядерных клеток, приводящая к образованию гранулемы. Гранулема включает также внеклеточный матрикс, продуцируемый фибробластами, позволяющий отграничить и изолировать антиген. Гранулематозные заболевания классифицируются по этиологии на инфекционные и неинфекционные. Однако, согласно последним исследованиям, патогенные микроорганизмы могут быть причиной развития гранулем при заболеваниях, ранее считавшихся неинфекционными. В ряде случаев классифицировать гранулематозный процесс как инфекционный и неинфекционный весьма трудно. Цель настоящего исследования — привлечь внимание читателей к разнообразию гранулематозных заболеваний, описать ключевые моменты патолого-анатомических проявлений различных болезней неинфекционной природы, а также определить подход к дифференциальной диагностике гранулематозов.
Название «гранулема» происходит от латинского слова granulum, означающее «зерно», греческое окончание (-oma) используется для обозначения узелкового образования. Таким образом, гранулема представляет собой узелковое очерченное образование. При микроскопической оценке термин используется для обозначения компактных агрегатов клеток. Микроскопически гранулемы могут состоять из гистиоцитов и/или эпителиоидных клеток, гигантских многоядерных клеток и других клеток воспаления (лимфоциты, нейтрофилы, эозинофилы). Эпителиоидные и гигантские многоядерные клетки являются производными моноцитов/макрофагов, первые являются дифференцированными секреторными клетками, тогда как последние специализированы на функции фагоцитоза [1]. Гигантские многоядерные клетки образуются путем слияния или неполного клеточного деления, что было доказано в экспериментальных исследованиях [2], причем раньше образуются гигантские клетки инородных тел, затем - клетки Пирогова—Лангханса. Лимфоциты располагаются преимущественно по периферии гранулемы и представляют популяцию T-клеток, тогда как B-лимфоциты разрозненно лежат вне гранулемы. В зависимости от заболевания T-лимфоциты представлены преимущественно T-хелперами 1 и 2 или же цитотоксическими T-супрессорами.
Гранулематозные болезни — гетерогенная группа заболеваний различной этиологии, проявляющихся разнообразными клиническими синдромами и вариантами тканевых изменений, неоднородной чувствительностью к терапии, при этом преобладает общий доминирующий гистологический признак — наличие гранулем, определяющих клинико-морфологическую сущность каждой болезни [3]. Настоящая публикация преследует цель привлечь внимание читателей к разнообразию гранулематозных заболеваний легких, описать ключевые моменты патолого-анатомических проявлений различных заболеваний инфекционной и неинфекционной природы, а также определить подход к дифференциальной диагностике гранулематозов.
При гранулематозных заболеваниях неинфекционной природы, как правило, развиваются ненекротические гранулемы, за некоторыми исключениями, о которых будет сказано ниже.
Саркоидоз
Поражение легких при саркоидозе описано в 90% наблюдений. Морфологическим субстратом саркоидоза является эпителиоидно-клеточная гранулема — компактное скопление мононуклеарных фагоцитов — макрофагов и эпителиоидных клеток. Каждая саркоидная гранулема имеет следующие стадии развития: 1) ранняя или лимфогистиоцитарная гранулема иногда с примесью нейтрофилов (рис. 1); Рис. 1. Ранняя фаза саркоидной гранулемы: периваскулярная лимфогистиоцитарная нечетко очерченная гранулема с единичной многоядерной клеткой. Здесь и на рис. 2—8: окраска гематоксилином и эозином, ×200. 2) гранулема со скоплением эпителиоидных клеток в центре и макрофагов по периферии; 3) эпителиоидно-лимфоцитарная гранулема; 4) гранулема с гигантскими многоядерными клетками (сначала клетки «инородных тел», а в последующем клетки Пирогова—Лангханса); 5) гранулема с ранними признаками некроза (пикноз ядер) или апоптоза (появление апоптотических телец) эпителиоидных или гигантских клеток в центре; 6) гранулема с центральным фибриноидным, гранулярным, ишемическим некрозом, как правило, в виде мелких фокусов; 7) гранулема с парциальным фиброзом (или гиалинозом, при окраске серебром выявляют ретикулиновые волокна); 8) гиалинизированная гранулема.
Процесс организации гранулем начинается с периферии, что придает им четко очерченный, «штампованный» вид (рис. 2). Рис. 2. Множественные четко очерченные эпителиоидно-клеточные саркоидные гранулемы в виде конгломератов, ×40. Зачастую при саркоидозе в одном и том же материале можно встретить гранулемы разного «возраста», от ранних, рыхлых, нечетко оформленных гистиоцитарных до четко очерченных («штампованных») эпителиоидно-клеточных, нередко гранулемы располагаются в виде конгломератов (рис. 3) Рис. 3. Типичная «штампованная» саркоидная гранулема, ×100. [4].
Значительное число лимфоцитов в ткани легких при саркоидозе представлено преимущественно Т-клетками. Для дифференциальной диагностики саркоидоза полезно оценивать бронхоальвеолярный смыв, в котором при этом заболевании среди лимфоцитов преобладают Т-хелперы (CD4+), соотношение CD4/CD8 составляет 3,5—10:1 (в норме 1,8:1). Гигантские клетки в составе гранулем могут содержать в цитоплазме включения, такие как астероидные тельца, тельца Шауманна или кристаллоидные структуры. Эти включения характерны для саркоидоза, однако не являются патогномоничными, так как могут присутствовать и при других гранулематозных заболеваниях [5].
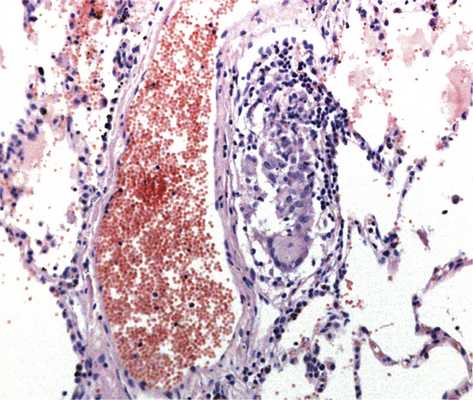
При исследовании биоптатов бронхов и легких при гранулематозных заболеваниях, как правило, обнаруживают диссеминированное поражение с васкулитом, периваскулитом, перибронхитом; гранулемы чаще всего локализуются в межальвеолярных перегородках. Гранулематозное поражение бронхов и бронхиол при саркоидозе встречается часто и описано у 15—55% пациентов. Нередко гранулемы располагаются в стенке сосуда, частота гранулематозного васкулита может достигать 69%. В этих наблюдениях следует дифференцировать саркоидоз с некротизирующим саркоидным гранулематозом, который ряд авторов относят к узловой форме саркоидоза [6]. Для последнего характерно развитие некрозов.
В целом для саркоидоза характерными являются наличие четко очерченных, «штампованных гранулем», располагающихся по ходу лимфатических сосудов, концентрический фиброз вокруг гранулем, гиалиноз внутри и между гранулемами, гранулематозный васкулит, отсутствие хронического интерстициального воспаления вне гранулематозного поражения и фокусов организующейся пневмонии.
Помимо саркоидоза, встречается так называемая неспецифическая саркоидная реакция в виде эпителиоидно-клеточного гранулематоза. Она обычно наблюдается в регионарных лимфатических узлах, но может встречаться и в ткани легких при псевдоопухолях, злокачественных новообразованиях, паразитарных заболеваниях, экзогенном аллергическом альвеолите, туберкулезе. Гистологически саркоидная реакция отличается ограниченностью и топической связью с указанными патологическими процессами.
Гиперсенситивный пневмонит
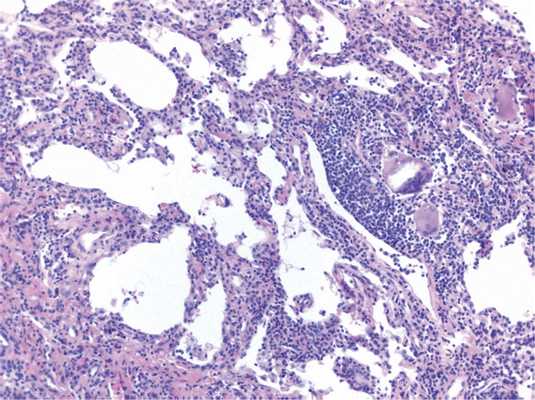
В основе патогенеза гиперсенситивного пневмонита лежит развитие иммунологической реакции легочной ткани по III (иммунокомплексному) и IV типу при ингаляции аллергена. Этиологическим фактором болезни обычно являются термофильные бактерии, грибы, животные протеины. Другие бактерии и их продукты, амебы и некоторые химические вещества значительно реже являются причиной развития заболевания. При гиперсенситивном пневмоните гранулемы нечетко организованы, более «рыхлые», состоят из гистиоцитов, лимфоцитов, многоядерных клеток, иногда — эозинофилов (рис. 4) Рис. 4. Гиперсенситивный пневмонит: нечетко очерченная гранулема, состоящая из гигантских многоядерных клеток с игольчатыми структурами, окруженных лимфоцитами, лимфоцитарная инфильтрация межальвеолярных перегородок, ×100. [7], характерна перибронхиолярная локализация гранулем. Для диагностики гиперсенситивного пневмонита следует выявить триаду признаков: неспецифическую интерстициальную пневмонию в перибронхиолярных зонах, ненекротизирующие гистиоцитарные (гигантоклеточные гранулемы), фокусы облитерирующего бронхиолита. При повышении числа лимфоцитов в бронхоальвеолярном смыве, в отличие от саркоидоза, среди них преобладают цитотоксические Т-клетки и Т-супрессоры (CD8+), соотношение CD4/CD8
Хроническая аллергическая болезнь, вызванная металлами
Хронический бериллиоз представляет собой аллергический гранулематоз. Гранулемы сходны с таковыми при саркоидозе, могут быть несколько крупнее. Как и при саркоидозе, гранулемы располагаются перилимфатически, со сходным клеточным составом, включая субпопуляции T-лимфоцитов, характерно поражение лимфатических узлов. Такой же вариант гранулематоза может развиваться в результате действия циркония. Диагностику следует основывать на данных анамнеза и тесте трансформации лимфоцитов в ответ на действие того или иного металла [9].
Особую сложность представляет, на наш взгляд, дифференциальная диагностика некротических гранулематозных процессов. Инфекционные заболевания, как одну из наиболее частых причин некротических гранулематозных процессов, следует дифференцировать прежде всего с полиангиитом с гранулематозом (ранее — гранулематоз Вегенера), аспирационной пневмонией, реже — с узелковой формой ревматоидного артрита, некротизирующим саркоидным гранулематозом (НСГ), инфарктом легкого и лимфоматоидным гранулематозом [10]. Несмотря на специфические гистологические изменения при этих заболеваниях, существует тем не менее перекрест, и лишь комбинация множества гистологических особенностей позволяет сформулировать окончательный диагноз. Инфекционная некротическая гранулема обычно имеет ровные контуры, как правило, эозинофильный некроз, окруженный валом гистиоцитов и гигантских многоядерных клеток. Напротив, при полиангиите с гранулематозом зоны некроза обычно с неровными контурами, напоминающими географическую карту, с большим количеством клеточного детрита, что придает некрозу «грязный» вид (рис. 5). Рис. 5. Гранулематоз Вегенера: некроз с клеточным детритом в виде «географической карты», ×40. Зоны некроза также окружены гистиоцитарным валом, однако гигантские клетки обычно немногочисленны и лежат разрозненно, не формируя компактные гранулемы. Характерной чертой полиангиита с гранулематозом является наличие некротического васкулита с фибриноидным некрозом мышечной оболочки (рис. 6), Рис. 6. Гранулематоз Вегенера: фибриноидный некроз стенки сосуда с лимфолейкоцитарной инфильтрацией, ×100. встречаются такие сосуды в зоне воспаления, поражение стенки часто эксцентрично, при этом заболевании поражаются ветви легочных артерий и вен. Возможно также развитие капиллярита, что сопровождается внутриальвеолярными кровоизлияниями. Некротический васкулит часто можно встретить в зоне воспаления и некроза и при инфекционном процессе, поэтому для подтверждения диагноза полиангиита с гранулематозом необходимо внимательно оценить сосуды вне зон некроза, желательно при этом использовать дополнительные окраски для выявления эластической ткани (по Верхофф, Ван-Гизону и т. д.). Кроме того, для полиангиита с гранулематозом в отличие от инфекционного гранулематоза нехарактерно вовлечение в процесс лимфатических узлов [11].
Некротический гранулематоз встречается также при аллергическом ангиите с гранулематозом (ранее — болезнь Черджа—Стросса) в сочетании с некротическим васкулитом и эозинофильной пневмонией. Гранулемы при аллергическом ангиите с гранулематозом хорошо оформленные, с центральным некрозом, содержащим множество эозинофилов, имеет место эозинофильная инфильтрация стенок сосудов и бронхиол, некротический васкулит с наличием эозинофилов и гигантских многоядерных клеток. Однако классическая триада изменений в виде гранулематоза, некротического васкулита и эозинофильной пневмонии в легких встречается нечасто, поэтому для установления диагноза необходимы дополнительные клинические и лабораторные данные.
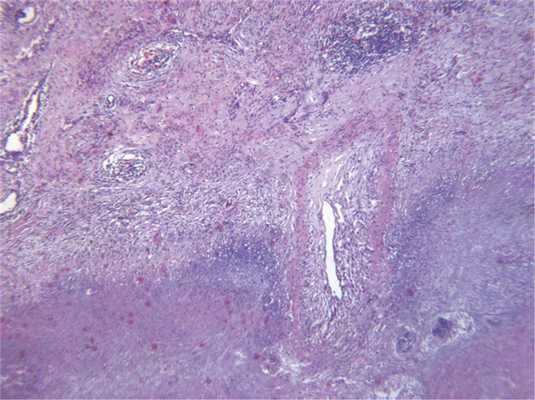
Некротические гранулемы могут формироваться в легком и при ревматоидном артрите, однако диагноз в этом случае следует формулировать с осторожностью. Прежде всего следует учитывать клинические данные, поскольку узелковые формы ревматоидного артрита развиваются только в активной фазе заболевания, у серопозитивных пациентов с наличием выраженного суставного синдрома. При этом заболевании некроз, как правило, окрашен эозинофильно, клеточный детрит располагается обычно между некрозом и окружающим его гистиоцитарным валом, может сочетаться с васкулитом, однако некротический васкулит нехарактерен (рис. 7) Рис. 7. Ревматоидный артрит: обширная зона некроза с гистиоцитарным валом по периферии, лимфоидная инфильтрация стенок сосудов с обтурацией просветов, ×40. [12]. Описанные гистологические изменения практически неотличимы от таковых инфекционного гранулематоза, кроме того, описаны редкие наблюдения сочетания ревматоидного артрита и туберкулеза, поэтому необходимо исключить наличие инфекции.
При дифференциальной диагностике васкулитов с развитием гранулематозной реакции, описанных выше, следует учитывать специфические серологические признаки того или иного заболевания.
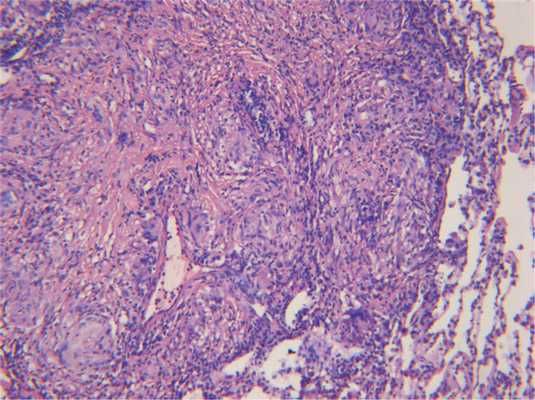
Инфекционный гранулематоз следует также дифференцировать с НСГ. Последний имеет также некоторые сходства с полиангиитом с гранулематозом. При НСГ характерно развитие интерстициального некроза, который чаще эозинофильно окрашен, однако иногда может содержать клеточный детрит. Однако некроз при НСГ сочетается с имеющимися ненекротизирующими гранулемами саркоидного типа, состоящими преимущественно из эпителиоидных и гигантских многоядерных клеток с небольшим числом лимфоцитов, эти гранулемы имеют тенденцию к слиянию, часто располагаются вблизи кровеносных сосудов или в их стенке, не приводя, однако, к развитию васкулита (рис. 8) Рис. 8. Некротизирующий саркоидный гранулематоз: гранулематозный васкулит, ×100. [6]. Для диагностики НСГ необходимо наличие триады признаков: гранулемы саркоидного типа, гранулематозного васкулита и некротического воспаления.
Поскольку некроз при инфекционном гранулематозе может быть коагуляционного типа, его также следует дифференцировать с инфарктом легкого. В стадии организации инфаркты могут быть окружены фибробластами и клетками воспаления, напоминая гранулематозное воспаление. В резецированных кусочках легкого при инфаркте, как правило, можно обнаружить тромбы в ветвях легочной артерии, явившиеся причиной развития инфаркта [13].
Наличие гигантских многоядерных клеток «инородных тел» и выраженной воспалительной реакции при аспирационной пневмонии в ряде случаев может напоминать гранулематозный процесс, для диагностики этого заболевания необходима тщательная оценка гистологических срезов с целью обнаружения различных инородных остатков пищи (растительных клеток, фрагментов поперечнополосатых мышц и др.).

Дифференциально-диагностические признаки основных неинфекционных заболеваний легких приведены в таблице. Ключевые диагностические признаки основных неинфекционных заболеваний легких [14]
Таким образом, для проведения дифференциальной диагностики гранулематозных заболеваний необходимо оценить форму (очерченность, «зрелось») гранулемы, ее клеточный состав, наличие/отсутствие некроза, цвет некроза, клеточную реакцию по периферии некроза, состояние ткани легкого вне зоны гранулематозного воспаления, а также другие специфические признаки. Дифференциальная диагностика гранулематозных процессов инфекционной природы будет рассмотрена в части II.
Авторы заявляют об отсутствии конфликта интересов.
Сведения об авторах
Микроскопический полиангиит
Микроскопический полиангиит - генерализованный некротизирующий васкулит, протекающий с преимущественным поражением мелких сосудов (капилляров, артериол, венул) легких, почек и кожи. При микроскопическом полиангиите может отмечаться кашель, одышка, боли в грудной клетке, легочное кровотечение, быстропрогрессирующий гломерулонефрит, почечная недостаточность, кожные петехиальные высыпания, артралгии. Лабораторным подтверждением микроскопического полиангиита служит обнаружение антител к цитоплазме нейтрофилов. Лечение микроскопического полиангиита включает иммуносупрессивную терапию глюкокортикоидами и цитостатиками, плазмаферез; при развитии почечной недостаточности - гемодиализ или трансплантацию почки.
Общие сведения
Микроскопический полиангиит (микроскопический полиартериит) - заболевание из группы системных васкулитов, характеризующееся поражением сосудов мелкого калибра с явлениями некротизирующего гломерулонефрита и легочного капиллярита. В связи с особенностями морфологических, клинических и иммунологических нарушений в 1948 г. микроскопический полиангиит был выделен в отдельную нозологию. Распространенность микроскопического полиангиита составляет 0,36 случаев на 100 тыс. населения. В ревматологии заболевание регистрируется в 10 раз чаще, чем узелковый периартериит и в 2 раза чаще, чем гранулематоз Вегенера. Микроскопический полиангиит практически с одинаковой частотой диагностируется у мужчин и женщин; чаще развивается у лиц старше 50-60 лет.
Причины
Этиология микроскопического полиангиита неизвестна. В настоящее время ведется изучение иммуногенетических аспектов патологии, исследуется пусковая роль вирусной инфекции в развитии заболевания. Вместе с тем, достоверно известно, что основной механизм патогенеза некротизирующего васкулита связан с образованием антител к цитоплазме нейтрофилов (ANCA), вызывающих повреждение эндотелия сосудов. Аутоантитела обладают специфичностью в отношении миелопероксидазы (p-ANCA) и сериновой протеиназы (c-ANCA), реже - других ферментов нейтрофилов. В активную стадию заболевания антинейтрофильные цитоплазматические антитела выявляются у всех пациентов.
Патоморфологические изменения при микроскопическом полиангиите представлены некротизирующим васкулитом мелких сосудов (артериол, венул, капилляров) без явлений гранулематозного воспаления. Наиболее характерно поражение сосудов почек (некротизируюший гломерулонефрит), легких (геморрагический альвеолит, капиллярит) и кожи (лейкоцитокластический венулит). Изредка поражаются артерии среднего и крупного калибра.
Симптомы микроскопического полиангиита
Дебют микроскопического полиангиита характеризуется появлением неспецифических гриппоподобных симптомов: субфебрильной температуры тела, мигрирующих миалгий и артралгий, общей слабости, недомогания, ночной потливости. Примерно у трети больных развивается поражение верхних дыхательных путей (язвенно-некротический или атрофический ринит, синусит, средний отит) и зрительной системы (конъюнктивит, кератит, эписклерит, увеит). Эти изменения обратимы и подвергаются обратному развитию на фоне терапии иммунодепрессантами.
Возможно возникновение стойких артритов мелких и крупных суставов, синовитов пястно-фаланговых и межфаланговых суставов, периферической полинейропатии. Обычно уже в начальной стадии микроскопического полиангиита обнаруживается поражение кожи в виде сосудистой пурпуры, эритемы, ливедо, узелковых или буллезных высыпаний, реже - язвенных изменений и асептических некрозов мягких тканей.
Ведущими проявлениями, определяющими течение и прогноз микроскопического полиангиита, служат легочный и почечный синдромы. Геморрагический альвеолит развивается более чем у половины пациентов. Клинические проявления легочного синдрома включают кашель, прогрессирующую одышку, боль в грудной клетке, нарастающую дыхательную недостаточность. Типично развитие кровохарканья, а иногда - профузного легочного кровотечения, которое является ведущей причиной смерти пациентов с микроскопическим полиангиитом.
Поражение почек по типу некротического гломерулонефрита обнаруживается практически во всех случаях заболевания. Патологический симптомокомплекс характеризуется стойкой протеинурией, гематурией, нефротическим синдромом, мягкой или умеренной артериальной гипертензией. Течение гломерулонефрита при микроскопическом полиангиите быстропрогрессирующее, злокачественное, рано приводящее к развитию острой почечной недостаточности. К нечасто встречающимся висцеральным проявлениям микроскопического полиангиита относятся бронхообструктивный синдром, ишемический энтероколит, протекающий с болями в животе и кишечным кровотечением.
Различают несколько вариантов клинического течения микроскопического полиангиита:
- молниеносный - летальный исход наступает в течение нескольких недель вследствие легочного кровотечения или ОПН;
- подострый - характерны тяжелый нефротический синдром или быстропрогрессирующий гломерулонефрит;
- непрерывно рецидивирующий - обострения наступают каждые 6-12 месяцев; в клиническом течении преобладает неспецифическая симптоматика, гломерулонефрит, кровохарканье;
- латентный - доминируют суставной синдром, гематурия, кровохарканье.
Диагностика
В начальной фазе микроскопического полиангиита пациенты довольно часто обследуются у узких специалистов: отоларинголога, офтальмолога, дерматолога, пульмонолога, фтизиатра, нефролога и др. Однако после исключения изолированных синдромов, диагностикой и лечением заболевания занимается ревматолог.
Специфическим иммунологическим маркером микроскопического полиангиита служит обнаружение в сыворотке крови антител к цитоплазме нейтрофилов. С целью оценки степени активности патологического процесса исследуется общий анализ мочи и крови, фибриноген, С-реактивный белок, электролиты, креатинин, железо сыворотки крови. В рамках морфологической верификации патологии может потребоваться проведение биопсии кожи, слизистой оболочки верхних дыхательных путей, легкого, почки. Рентгенография и КТ легких выявляют двустороннюю очаговую инфильтрацию, при длительном течении микроскопического полиангиита - легочный фиброз. Широко используется ультразвуковая и радиоизотопная диагностика (УЗИ почек, сцинтиграфия легких и др.).
Лечение микроскопического полиангиита
Лечение микроскопического полиангиита преследует цели купирования обострений, достижения и поддержания ремиссии. В острой стадии лечение проводится в стационаре. На этом этапе назначается иммуносупрессивная терапия глюкокортикоидами и циклофосфамидом. Возможен вариант проведения пульс-терапии метилпреднизолоном в сочетании с плазмаферезом. В комплексном лечении быстропрогрессирующих, тяжелых форм микроскопического полиангиита используются и другие методы экстракорпоральной гемокоррекции (криоаферез, каскадная фильтрация плазмы, лимфоцитаферез).
Поддерживающая иммуносупрессивная терапия продолжается даже после достижения полной клинико-лабораторной ремиссии. При нарастании почечной недостаточности проводится гемодиализ; в терминальной стадии ХПН решается вопрос о возможности трансплантации почки.
Прогноз и профилактика
Заболевание имеет относительно неблагоприятный прогноз. На фоне лекарственной терапии 5-летняя выживаемость составляет 65%. Плохими прогностическими факторами в отношении общей выживаемости служат кровохарканье, высокий уровень креатинина крови, олигурия, высокая протеинурия, пожилой возраст. Основными причинами смерти выступают легочное кровотечение, острая дыхательная или почечная недостаточность, инфекционные осложнения. Пациенты с микроскопическим полиангиитом нуждаются в длительной терапии иммунодепрессантами и пожизненном наблюдении ревматолога. Профилактика заболевания не разработана.
Полиморфный дермальный ангиит
Полиморфный дермальный ангиит (болезнь Гужеро-Рюитера) — хронический рецидивирующий дерматоз, обусловленный неспецифическим воспалительным процессом в стенках кожных сосудов. Характеризуется выраженным разнообразием клинических проявлений: бляшки, узелки, кожные кровоизлияния, волдыри, пузыри, участки отека кожи, гнойнички, поверхностные некрозы, язвы, рубцы. Основной метод диагностики полиморфного дермального ангиита — дерматоскопия. Лечение может включать назначение противовоспалительных и антигистаминных препаратов, антибиотиков, глюкокортикоидов, витаминов, препаратов кальция, хлорохина. Тяжелые формы заболевания требуют применения экстракорпоральных методов очищения крови.
В настоящее время дерматология придерживается иммунокомплексной теории развития полиморфного дермального ангиита, согласно которой поражения сосудов обусловлены действием на их стенку циркулирующих иммунных комплексов (ЦИК). В роли антигенов при этом могут выступать локальные или общие инфекции (стафилококковые, стрептококковые, ангина, грипп, ОРВИ, краснуха, корь и др.). При полиморфном дермальном ангиите в местах поражения ЦИК происходит утолщение и выбухание сосудистой стенки, в ее толще могут образовываться узелки. В дальнейшем она пропитывается фибрином, проницаемость ее увеличивается, что приводит к появлению кровоизлияний. Происходит некроз отдельных участков стенки сосуда и компенсаторное разрастание других.
Симптомы полиморфного дермального ангиита
Полиморфный дермальный ангиит, как правило, протекает с поражением кожи голеней. Однако может наблюдаться и другая локализация высыпаний. В некоторых случаях появлению кожных элементов предшествует общая симптоматика: утомляемость, повышение температуры, слабость, артралгии, головная боль. Появившись на коже, высыпания держаться до нескольких месяцев. После их разрешения и заживления создается впечатление полного выздоровления пациента, но при этом всегда существует риск развития рецидива.
По преобладанию тех или иных элементов сыпи выделяют несколько разновидностей полиморфного дермального ангиита. Уртикарный вариант полиморфного дермального ангиита проявляется появлением на любых участках кожи различного размера волдырей, что напоминает клиническую картину крапивницы. Но, в отличие от последней, высыпания сопровождаются не зудом, а своеобразным дискомфортом и болезненностью.
Геморрагический вариант — самый распространенный тип полиморфного дермального ангиита. Он характеризуется появлением очаговых внутрикожных кровоизлияний и может напоминать другие геморрагические пурпуры (например, при системных васкулитах или геморрагической лихорадке). Сюда же относят болезнь Шенлейн-Геноха - геморрагический вариант полиморфного дермального ангиита с общей симптоматикой, болями в животе и кровянистым стулом.
Папулонодулярный вариант встречается относительно редко. Его проявлением являются уплощенные узелки и крупные (больше грецкого ореха) узлы, не сопровождающиеся никаким дискомфортом, но болезненные при прощупывании. Папулонекротический вариант полиморфного дермального ангиита представлен небольшими воспалительными узелками, в центре которых со временем происходят некротические изменения. Образуется черная корочка, под которой обнаруживаются кровоточащие язвочки. Заживление происходит с образованием рубчиков. Характерна локализация элементов на разгибательной поверхности в области суставов конечностей. Эту форму заболевания необходимо дифференцировать с папулонекротическим туберкулезем кожи.
Пустулезно-язвенный вариант полиморфного дермального ангиита начинается с возникновения на коже любой части тела пустул и пузырьков, напоминающих проявления фолликулита или акне. Затем на месте высыпаний формируются глубокие язвы, склонные к увеличению в размерах и заживающие с образованием грубых рубцов.
Некротически-язвенный вариант полиморфного дермального ангиита характеризуется образованием тромбов в пораженных сосудах, что вызывает некроз целых участков кожи с образованием черного струпа. Имеет тяжелое течение с молниеносным началом и быстрым ухудшением состояния пациента, нередко приводящим к летальному исходу. Если пациент не погибает, то некротические участки трансформируются в длительно заживающие и плохо поддающиеся лечению язвы, оставляющие после себя большие рубцы. Полиморфный вариант отличается сочетанным проявлением сразу нескольких вариантов полиморфного дермального ангиита.
Диагностика и лечение полиморфного дермального ангиита
Диагностику проводит дерматолог. Основным методом исследования при этом является дерматоскопия, в ходе которой выявляется набухание и воспаление стенок проходящих в коже сосудов, пропитывание их фибрином, некротические изменения, тромбозы, выход клеток крови за пределы сосуда и участки разрастания эндотелия.
В лечении применяют противовоспалительные препараты (фенилбутазон, индометацин), антигистаминные средства (дезлоратадин, лоратадин, хлоропирамин), препараты кальция, витамины В15, Р и С. При выявлении связи полиморфного дермального ангиита с инфекцией проводится антибиотикотерапия. В тяжелых случаях необходимо применение глюкокортикостероидной терапии и методов экстракорпоральной гемокоррекции: гемосорбции, плазмафереза, криоафереза. При рецидивирующем течении назначают длительный курс хлорохина.
В местном лечении полиморфного дермального ангиита используют повязки и мази с кортикостероидами, при язвах и некротических поражениях — мазь Вишневского, метилуроциловую мазь, примочки с химотрипсином.
Читайте также:
- Статьи по болезням полости рта и глотки
- Слои головного мозга при облучении. Изменения нервных волокон при лучевой болезни
- Диагностика и лечение стеноза наружного слухового прохода. Операция Швартце, Руттина
- Лимфоцитарный васкулит (пигментированный пурпурозный дерматоз, болезнь Шамберга) у ребенка
- Анатомия главных бронхов. Кровоснабжение и иннервация главных бронхов