КТ при последовательности Пьера Робена (ППР)
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Л.Ф.ВАХИТОВА, Л.К.ФАЗЛЕЕВА,Л.Г.БУЛГАКОВА, О.В. ВАРЛАМОВА
Казанский государственный медицинский университет
Детская республиканская клиническая больница МЗ РТ, г. Казань
Вахитова Лилия Фаукатовна
кандидат медицинских наук, ассистент кафедры госпитальной педиатрии
Ранняя диагностика врожденных аномалий развития в настоящее время остается актуальной из-за увеличения частоты этой патологии и необходимости проведения мероприятий, направленных на профилактику возможных осложнений. В статье описан редкий случай клинически выраженного синдрома Пьера Робена тяжелой степени у новорожденного ребенка.
Ключевые слова: новорожденный, наследственные синдромы, синдром Пьера Робена.
L.F. VAKHITOVA, L.K. PHAZLEEVA, L.G. BULGAKOVA, O.V. VARLAMOVA
Kazan State Medical University
Children’s Republican Clinical Hospital of the Ministry of Health of the Republic of Tatarstan, Kazan
Clinical case of Pierre Robin’s syndrome in a newborn child
Early diagnosis of congenital abnormal development currently remains relevant because of the increasing frequency of this disease and the need for measures aimed at preventing possible complications. This article describes a rare case of clinically apparent severe Pierre Robin’s syndrome in a newborn child.
Key words: newborn, hereditary syndromes, Pierre Robin’s syndrome.
Сведения о нарушении развития нижней челюсти и неба, сочетающемся с нарушением дыхания, появились в медицинской литературе еще в 20-е годы ХІХ века. В 1923 году французским стоматологом Пьером Робеном была установлена связь между гипоплазией нижней челюсти (микрогнатией), расщелиной мягкого неба (рис. 1) и обструкцией дыхательных путей, которая состоит в последовательном появлении симптомов. В дальнейшем патология была названа именем этого ученого, и в зарубежной литературе используется термин «последовательность Пьера Робена» [1, 2]. В настоящее время только в единичных работах приведены эпидемиологические данные по этому синдрому [3, 4]. Согласно результатам ретроспективного исследования, распространенность этой патологии в Дании составляет 1 случай на 14 000 живорожденных детей; по данным американского исследования — 1 случай на 2-30 тысяч. Cоотношение полов — М1: Ж1 [5, 6]. Можно предположить, что данная патология встречается в популяции довольно часто, но выявляемые признаки не рассматриваются в совокупности как проявления единого заболевания. Нарушение эмбрионального развития нижней челюсти происходит как при наличии механической компрессии внутри матки, так и при воздействии инфекции на ранних этапах беременности или нейрогенетических нарушениях. Следует отметить, что последовательность Пьера Робена может быть изолированным синдромом или проявлением генетической патологии. Как часть генетической патологии последовательность Пьера Робена описана почти при 300 синдромах. Наследование может происходить и по аутосомно-доминантному, и по аутосомно-рецессивному типу. Вероятность рождения больного в семье, уже имеющей ребенка с синдромом Пьера Робена, составляет 1-5% [7, 8]. Популяционная частота синдрома — 1 : 12000.
Первичный порок, лежащий в основе синдрома, это резкая гипоплазия нижней челюсти (микрогения). Остальные пороки обусловлены западением языка (глоссоптоз) в связи с уменьшением ротовой полости, что в свою очередь препятствует смыканию небных пластинок. Приблизительно в 25% случаев этот синдром является составной частью синдромов множественных пороков развития, наиболее часто — синдрома Стиклера, а также кампомелического синдрома, синдрома Ханхарта, трисомии 18. У 36% больных аномалад Пьера Робена сочетается с другими пороками развития, в том числе с пороками сердца, аномалиями глаз, ушных раковин, скелета. Также нередко ассоциируется с умственной отсталостью. Клинически проявляется затруднением дыхания и глотания. Изолированные случаи всегда спорадические.
В зависимости от состояния дыхания выделяют три степени тяжести:
1) легкая степень: дыхание не затруднено, незначительные трудности с кормлением, устраняемые консервативным лечением в aмбулаторных условиях;
2) средняя степень: умеренное затруднение дыхания, умеренные трудности с кормлением, требующие лечения в стационаре;
3) тяжелая степень: значительное затруднение дыхания, значительные трудности с кормлением в течение длительного времени, приводящие к необходимости использования дыхательного пособия в виде интраназального зонда в нижние дыхательные пути или трахеостомии [1, 9, 10].
Визуализация структур лица и выявление пороков их развития потенциально возможны уже с 11-12 нед. беременности.
Характерен внешний вид новорожденного — так называемое птичье лицо, что связано с недоразвитием нижней челюсти. В разные сроки, в зависимости от степени тяжести порока, появляется резкое нарушение дыхания, связанное с западением языка, возможны частые апноэ. Из-за нарушения акта глотания во время кормления ребенка может наступить удушье.
Оптимальным и жизненно важным положением малыша с синдромом Робена является положение на боку или на животе с приподнятой (или даже фиксированной в таком положении) головой. Это позволяет избежать обструкции (перекрывания) дыхательных путей смещенным назад корнем языка. Оптимально кормить ребенка в положении на боку, полувертикальном, а иногда практически в вертикальном через назогастральный зонд.
При легкой и среднетяжелой формах заболевания проводят симптоматическую терапию и профилактику возможных осложнений. При аномалии средней тяжести альтернативой хирургического лечения является временная глоссопексия (temporary tongue-lip adhesion) — это подтягивание языка вперед и фиксация его к нижней губе. При тяжелой форме, кроме этого, требуется хирургическое вмешательство, позволяющее удлинить нижнюю челюсть: компрессионно-дистракционный остеосинтез. Метод заключается в том, что на нижнюю челюсть с двух сторон устанавливаются аппараты, проводятся распилы челюсти между лапками аппаратов и фрагменты челюсти плотно прижимаются друг к другу — компрессия. С 5-6-х суток после операции фрагменты челюсти постепенно, со скоростью 1 миллиметр в сутки, разводятся аппаратами на необходимую величину. Процесс удлинения челюсти аппаратами называется дистракцией, а аппарат, соответственно, — компрессионно-дистракционным. Между фрагментами кости образуется новая молодая кость — костная мозоль. В результате дистракции нижняя челюсть перемещается кпереди вместе с языком, который перестает перекрывать верхние дыхательные пути. Самостоятельное дыхание становится возможным в среднем уже на 4-6-е сутки дистракции, которая продолжается до достижения правильного соотношения челюстей. На самостоятельное питание ребенок переводится, как правило, к моменту окончания дистракции или немного позже. Ребенок выписывается с аппаратами на время (3 месяца) превращения костной мозоли в полноценную кость. Через 3 месяца — повторная госпитализация для удаления аппаратов [11, 12].
Важным представляется, что устранение расщелины мягкого неба должно быть проведено до становления речи, поэтому операции производятся в возрасте от полугода до 1,5 года, в зависимости от степени дыхательной обструкции. Проводится операция уранопластики. Часто требуется проведение 3-4 этапов лечения. Перед операцией таким пациентам рекомендуется ношение специального обтуратора, который разделяет ротовую и носовую полость, позволяет малышу правильно дышать, питаться, разговаривать. При оперативном вмешательстве дефект неба восстанавливается при помощи пластического материала, что позволяет получить анатомически правильное строение.
Приводим собственное наблюдение ребенка с синдромом Пьера Робена.
Девочка З., поступила в отделение патологии новорожденных ДРКБ МЗ РТ из роддома на 5-й день жизни с диагнозом: «Синдром Пьера Робена. Атрезия хоан? Неонатальная желтуха. Асфиксия умеренной степени. Синдром задержки фетальной жидкости. Множественные стигмы дизэмбриогенеза. Риск ВУИ» в тяжелом состоянии за счет ДН II-III степени, интоксикации, неврологической симптоматики, микроциркуляторных нарушений.
Ребенок от VIII беременности, четвертых родов. I беременность — роды в 1991 г., здоровый ребенок. II беременность — роды в 1995 г., ребенок с синдромом Пьера Робена, умер в возрасте четырех мес. в результате автомобильной катастрофы. III беременность — роды в 1996 г., здоровый ребенок. IV-я беременность — выкидыш. V, VI, VII беременности — м/аборты. Настоящая беременность протекала: I триместр — угроза прерывания беременности; II триместр — на сроке 22 нед. выявлен порок развития по УЗИ. III триместр — кольпит на 40-41-й нед. Маме 40 лет, папе 44 года. Наследственность отягощена: у отца — микрогения. Роды IV, на сроке 40-41 нед., через естественные родовые пути, дистация плечиков. Оценка по шкале Апгар: 6-7-8 баллов. Проведена ИВЛ мешком Амбу — 3’. Масса при рождении 3550 г, рост 50 см, окр. гол 34 см, окр. гр. кл. 32 см. МУМТ на 5-й день — 9%. Состояние ребенка в роддоме тяжелое. Ребенок находился в вынужденном положении на боку, на животе. Кормление через зонд по 8 мл смесью НАН-1. Движения неактивные. Гипотония, гипорефлексия. Кожные покровы субиктеричные на розовом фоне. Носовое дыхание затруднено, дыхание шумное. Аускультативно дыхание жесткое, проводные хрипы. Тоны сердца ритмичные. Живот мягкий. Стул самостоятельный. Диурез адекватный. В роддоме проведено лечение: амписид, глюкозо-солевые растворы, аминовен для парентерального питания, фототерапия.
При поступлении в отделение патологии ДРКБ состояние оценено как тяжелое за счет интоксикации, дыхательной недостаточности II-III, неврологических расстройств, метаболических и микроциркуляторных нарушений. На осмотр реагировала беспокойством, закатывала глаза. Плач громкий. Двигательная активность снижена. Возбудимость повышена. Мышечный тонус диффузно снижен, гипорефлексия. Стигмы дизэмбриогенеза: микрогения, готическое небо, расщелина твердого неба, аномальная форма ушных раковин, короткая шея, широкая переносица, эпикант, арахнодактилия. Тургор мягких тканей удовлетворительный. Кожные покровы бледно-розовые, отмечается мраморность, периоральный, периорбитальный цианоз, акроцианоз в покое. Пупочная ранка сухая, чистая. Голова округлой формы. БР 1,5×1,7 см, на уровне костей черепа. Швы закрыты. Разведение в тазобедренных суставах полное. Носовое дыхание затруднено. Грудная клетка правильной формы. Отмечается втяжение межреберий и нижней апертуры грудной клетки. Дыхание ритмичное. Аускультативно выслушиваются обильные проводные хрипы, дыхание ослаблено по всем легочным полям. Тоны сердца ритмичные, приглушены. Выслушивается негрубый систолический шум на верхушке. Живот увеличен в объеме, мягкий, печень +2,5 см (край эластической консистенции, острый, ровный), селезенка — не пальпируется. Физиологические отправления — без патологии.
Девочка находилась в палате интенсивной терапии в положении на боку, так как резко беспокоилась в положении на животе. Получала необходимое лечение. Через 2 дня после госпитализации состояние ребенка резко ухудшилось из-за обструкции дыхательных путей, нарастания дыхательной недостаточности, микроциркуляторных нарушений. Ребенок был переведен в отделение реанимации новорожденных. Из-за невозможности интубации трахеи проведена операция «нижняя трахеотомия», вставлена трахеостомическая трубка (рис. 1).
Ребенок с синдромом Пьера Робена (собственное наблюдение)
Диагностированы аспирационная пневмония, острое течение, ДН II-III ст., церебральная ишемия средней тяжести, постгеморрагическая анемия средней тяжести. Проводилось лечение: антибактериальная, противогрибковая терапия, гемостатические, ноотропные препараты, инфузионная терапия. Далее, несмотря на проводимую деэскалационную антибактериальную терапию, состояние ребенка оставалось тяжелым за счет интоксикации, дыхательной недостаточности, респираторных нарушений. Образовался порочный круг: трахеостома поддерживает воспалительный процесс в легких и невозможность проведения остеосинтеза и удаления трахеостомы. Девочка находится под наблюдением детских хирургов. В плане после купирования воспаления проведение оперативного лечения.
1. Bronshtein M., Blazer S., Zalel Y. et al. Ultrasonographic diagnosis of glossoptosis in fetuses with Pierre Robin sequence in early and mid pregnancy // Am. J. Obstet. Gynecol. — 2005 Oct. — Vol. 193, № 4. — P. 1561-1564.
2. Bixler D., Christian J.C. Pierre Robin syndrome occurring in two unrelated sibshops. — Birth Defects, 1971. — Vol. VII, № 7. — P. 67-71.
3. Chiriac A., Dawson A., Krapp M. et al. Pierre-Robin syndrome: a case report // Arch. Gynecol. Obstet. — 2007. — Jul 6.
4. Evans A.K, Rahbar R., Rogers G.F. et al. Robin sequence: a retrospective review of 115 patients // Int. J. Pediatr. Otorhinolaryngol. — 2006 Jun. — Vol. 70, № 6. — P. 973-980.
5. Козлова С.И. Демикова Н.С. Семанова Е. Блинникова О.Е. Наследственные синдромы и медико-генетическое консультирование. — М.: Практика, 1996. — С. 122-123.
6. Наследственные синдромы и медико-генетическое консультирование: справочник / Козлова С.И., Семанова Е., Демикова Н.С., Блинникова О.Е. — Л.: Медицина, 1987. — С. 185.
7. Jakobsen L.P., Knudsen M.A., Lespinasse J. et al. The genetic basis of the Pierre Robin Sequence // Cleft Palate Craniofac J. — 2006 Mar. — Vol. 43, № 2. — P. 155-159.
8. Kirschner R.E., Low D.W., Randall P. et al. Surgical airway management in Pierre Robin sequence: is there a role for tongue-lip adhesion? // Cleft Palate Craniofac J. — 2003 Jan. — Vol. 40, № 1. — P. 13-18.
9. Printzlau A., Andersen M. Pierre Robin sequence in Denmark: a retrospective population-based epidemiological study // Cleft Palate Craniofac J. — 2004 Jan. — Vol. 41, № 1. — P. 47-52.
10. Tolarova M., Senders C. Medicine, Pierre Robin Malformation. — Last updated June 2006.
11. Wagener S., Rayatt S.S., Tatman A.J. et al. Management of infants with Pierre Robin sequence // Cleft Palate Craniofac J. — 2003 Mar. — Vol. 40, № 2. — P. 180-185.
12. Кириллова Л.Г., Ткачук Л.И., Шевченко А.А., Силаева Л.Ю., Лисица В.В., Мироняк Л.А. Синдром Пьера Робена у детей // Международный неврологический журнал. — 2010. — № 3.
5. Kozlova S.I. Demikova N.S. Semanova E. Blinnikova O.E. Nasledstvennye sindromy i mediko-geneticheskoe konsul’tirovanie. — M.: Praktika, 1996. — S. 122-123.
6. Nasledstvennye sindromy i mediko-geneticheskoe konsul’tirovanie: spravochnik / Kozlova S.I., Semanova E., Demikova N.S., Blinnikova O.E. — L.: Medicina, 1987. — S. 185.
12. Kirillova L.G., Tkachuk L.I., Shevchenko A.A., Silaeva L.Ju., Lisica V.V., Mironjak L.A. Sindrom P’era Robena u detej // Mezhdunarodnyj nevrologicheskij zhurnal. — 2010. — № 3.
Синдром Пьера Робена ( Аномалия Робена , Последовательность Робена )
Синдром Пьера Робена - это генетически обусловленная аномалия, характеризующаяся гипоплазией нижней челюсти, небной расщелиной и глоссоптозом. Комбинация пороков ЧЛО вызывает затруднения при вскармливании, обструктивное апноэ, диффузный цианоз, риск аспирации пищи и асфиксии. Патология диагностируется по данным рентгенографии и КТ челюсти, фиброларингоскопии, полисомнографии, генодиагностики. Консервативная тактика включает позиционную терапию, СРАР, ортодонтическое лечение. Методы хирургической коррекции порока представлены глоссопексией, пластикой неба, компрессионно-дистракционным остеосинтезом.
МКБ-10



Общие сведения
Синдром (последовательность, аномалия) Робена включает триаду признаков: микрогнатию, глоссоптоз, расщепление твердого и мягкого неба. Врожденная анатомическая деформация описана в 1923 г. французским зубным врачом Пьером Робеном. По разным оценкам, дети с данной аномалией рождаются с популяционной частотой 1:8500-30000, независимо от пола. Ведущей проблемой младенцев с синдромом Робена является обструкция верхних дыхательных путей и критическое нарушение дыхания вследствие недоразвития нижней челюсти и смещения мягких тканей вглубь полости рта.

Причины
Последовательность Пьера Робена может встречаться как изолированный синдром (20-40%) или как часть других генетических патологий. В первом случае челюстно-лицевая аномалия формируется под влиянием неблагоприятных условий внутриутробного развития, в числе которых может быть:
- механическое сдавление плода в полости матки, ограничивающее рост челюсти (рубцами, амниотическими тяжами, опухолями или другим плодом при многоплодной беременности);
- внутриутробные инфекции;
- маловодие;
- дефицит фолиевой кислоты в организме беременной;
- тератогенные воздействия на плод, вызванные вредными экологическими факторами, радиацией, химическими веществами, приемом ЛС.
Кроме этого, аномалия Пьера Робена встречается в структуре генетических синдромов с множественными пороками развития. Всего насчитывается порядка 300 таких заболеваний, среди которых синдромы Стиклера, Хангарта, Эдвардса, кампомелическая дисплазия и др. Более чем в 30% случаев синдром Пьера Робена сочетается с ВПС, скелетными аномалиями, дефектами развития глаз и ушных раковин.
Спорадические случаи возникают в результате генетических мутаций de novo. Синдромальная последовательность наследуется по тому же принципу, что и патология, с которой она ассоциирована (сообщается об аутосомно-рецессивном, аутосомно-доминантном, Х-сцепленном варианте). Согласно исследованиям, генетические мутации затрагивают области хромосом 2 (2q24.1-33.3), 4 (4q32-qter), 11 (11q21-q23.1) и 17 (17q21-q24.3).
Патогенез
Синдром Пьера Робена принято называть «последовательностью», поскольку его признаки представляют собой цепочку взаимовлияющих друг на друга событий. Нарушение развития нижней челюсти у плода происходит в период между 7-й и 11-й неделями гестации под воздействием генетических мутаций или экзогенных факторов. Гипоплазия челюсти способствует неправильному положению языка между небными пластинками, которое препятствует их своевременному закрытию и вызывает образование расщелины.
Нижняя ретрогнатия и маленький объем ротовой полости создают условия для смещения языка в сторону задней стенки глотки и возникновения глоссоптоза. Оттянутый назад язык вызывает у новорожденного серьезные затруднения дыхания, вследствие чего часто возникает инспираторная обструкция ВДП.
Классификация
В зависимости от выраженности нарушения глотания и дыхания выделяют три степени тяжести синдрома Пьера Робена:
- легкая - дыхание не нарушено, имеются небольшие затруднения при кормлении младенца, которые устраняются консервативными методами в домашних условиях;
- средняя - трудности дыхания и кормления выражены умеренно, ребенку требуется стационарная помощь, зондовое питание;
- тяжелая - выраженные затруднения дыхания и самостоятельного питания, требующие постановки трахеостомы и желудочного зонда.
Симптомы
Новорожденные с синдромом Пьера Робена имеют характерное «птичье» лицо, обусловленное гипоплазией нижней челюсти (микрогнатия) и ее смещением назад (ретрогнатия). Объем полости рта полости уменьшен, язык оттянут вглубь (глоссоптоз). Как правило, у пациентов обнаруживается U-образная субмукозная расщелина мягкого неба, также встречается готическое небо.
Смещение языка кзади вызывает проблемы с сосанием в раннем возрасте, из-за чего у ребенка отмечается задержка роста и набора веса, отставание в моторном развитии. Тяжесть дыхательных нарушений варьируется от храпа до опасных эпизодов обструктивного апноэ. В легких случаях дыхательные расстройства возникают только в положении на спине и при беспокойстве ребенка, проходят самостоятельно. В более серьезных ситуациях приступы остановки дыхания длительные (до 20 сек. и более), приводящие к развитию гипоксии и гиперкапнии.
Тяжелые эпизоды апноэ сопровождаются акроцианозом или диффузным цианозом. Постоянная задействованность вспомогательных дыхательных мышц способствует формированию воронкообразной деформации грудной клетки.
Синдромальная последовательность Пьера Робена, как правило, сочетается с врожденными аномалиями других органов: тугоухостью и недоразвитием слухового аппарата, патологией зрительного анализатора (близорукость, катаракта), скелетными деформациями (полидактилия, врожденные ампутации), сердечными пороками. Примерно у половины пациентов обнаруживаются дефекты ЦНС: микроцефалия, гидроцефалия, эпилепсия, задержка психоречевого развития, олигофрения.
Осложнения
Практически все дети с синдромом Пьера Робена страдают речевыми дефектами (открытая ринолалия), нарушением прикуса, рецидивирующими ушными инфекциями. На фоне пролонгированных приступов обструктивного апноэ постепенно формируется гипоксическая энцефалопатия, легочное сердце. В некоторых случаях ребенок может погибнуть от асфиксии. Дисфагические расстройства могут повлечь за собой аспирацию слюны, пищи, желудочного содержимого при срыгивании, что чревато развитием аспирационной пневмонии. Смертность детей составляет порядка 16%, а при наличии сочетанных пороков достигает 41%.
Диагностика
В настоящее время синдром Пьера Робена диагностируется пренатально с помощью скринингового УЗИ в 11-12 недель беременности. После рождения диагноз подтверждается данными клинической картины и инструментальных исследований. Необходимая диагностика:
- Рентгеновские методы. Для первичной оценки анатомии челюстно-лицевой зоны выполняется рентгенография нижней челюсти, гортани и трахеи. В рамках предоперационного обследования обязательно проведение КТ челюстей, лицевого черепа, мягких тканей шеи. Рентгеновская визуализация помогает определить степень микро- и ретрогнатии, обструкции дыхательных путей, рассчитать параметры дистракции.
- Эндоскопия гортани. С помощью фиброларингоскопии уточняется состояние носоглотки, гортани и начальных отделов трахеи. Оценивается уровень и степень обструкции ВДП, обнаруживаются другие аномалии ЛОР-органов.
- Полисомнография. Считается «золотым стандартом» определения тяжести синдрома обструктивного апноэ. С ее помощью выясняется частота и длительность эпизодов остановки дыхания, подбирается оптимальная терапия.
- Другие методы. Регулярно проводится исследование газового состава крови, пульсоксиметрия. Перед планируемой операцией выполняется фотофиксация внешнего вида ребенка в фас и профиль, регистрация прикуса. При синдромальных формах аномалии Робена показана генодиагностика.
Лечение синдрома Пьера Робена
Лечебная тактика при аномалии Робена зависит от выраженности респираторных и дисфагических расстройств. Она может заключаться в проведении позиционной терапии, вспомогательной терапии, хирургического лечения. На различных этапах жизни ребенку требуется сопровождение врача-отоларинголога, челюстно-лицевого хирурга, ортодонта, логопеда.
Консервативное лечение
При легкой степени дыхательной обструкции рекомендуется постуральная терапия. Ребенка выкладывают на живот или на бок с приподнятой или зафиксированной головой, обеспечивающей выдвижение вперед языка и нижней челюсти. Персистирующая обструкция может купироваться с помощью назофарингеального воздуховода. Одним из методов с подтвержденной эффективностью является СРАР-терапия, позволяющая осуществлять неинвазивную масочную вентиляцию под постоянным положительным давлением.
При невозможности самостоятельного питания кормление ребенка производится через назогастральный зонд. Тяжелые степени синдрома Пьера Робена требуют установки трахеостомы или интубации трахеи с подключением ребенка к аппарату ИВЛ.
Консервативные методы позволяют купировать симптомы, но ни один из них не устраняет имеющиеся врожденные аномалии. Поэтому такое лечение должно проводиться очень длительно, до тех пор, пока размеры дыхательных путей ребенка не позволят ему дышать самостоятельно без риска обструкции. После нормализации жизненных функций проводится ортодонтическое лечение, логопедическая коррекция.
Хирургическое лечение
Устранение небной расщелины проводится поэтапно: велофарингопластика выполняется в 6-8 мес., уранопластика - в 12-18 мес. В качестве временной меры при подготовке к радикальному лечению может осуществляться глоссопексия - фиксация языка к нижней губе, альвеолярному отростку. Ранее применявшиеся методы вытяжения нижней челюсти, транспозиции жевательных мышц не являются достаточно эффективными.
Радикальным хирургическим вмешательством, позволяющим удлинить нижнюю челюсть, является компрессионно-дистракционный остеосинтез. Операция заключается в проведении двусторонней остеотомии нижней челюсти с фиксацией на костных фрагментах специального аппарата. Активация устройства обеспечивает постепенное перемещение нижней челюсти вместе с комплексом мягких тканей дна полости рта кпереди. Процесс дистракции занимает около 3-х месяцев, в результате чего становится возможным сначала самостоятельное дыхание, а затем - питание.
Прогноз и профилактика
Изолированный синдром Робена имеет более благоприятный прогноз, чем синдромальные формы, ассоциированные с множественными пороками и умственной отсталостью. Также существенное значение имеет своевременность и радикальность лечения. Вместе с тем, на данный момент смертность от асфиксии, других осложнений и сопутствующих аномалий остается достаточно высокой.
Для снижения риска рождения ребенка с синдром Пьера Робена рекомендуется не допускать воздействия тератогенов, инфицирования, дефицита микронутриентов в период гестации. Беременным необходимо вовремя проходить генетические и ультразвуковые пренатальные скрининги.
1. Синдром Пьера Робена у детей/ Кириллова Л.Г., Ткачук Л.И., Шевченко А.А., Силаева Л.Ю., Лисица В.В., Мироняк Л.А.// Международный неврологический журнал. - 2010. - №3 (33).
3. Хирургическое лечение новорожденных и грудных детей с синдромом Пьера Робена/ Дубин С.А., Комелягин Д.Ю., Злыгарева Н.В., Строгонов И.А.,Р огинский В.В., Полуэктов М.Г.// Российский вестник детской хирургии, анестезиологии и реаниматологии. - 2011.
4. Клинический случай синдрома Пьера Робена у новорожденного ребенка/ Вахитова Л.Ф., Фазлеева Л.К., Булгакова Л.Г., Варламова О.В.// Практическая медицина. - 2013.
Синдром Пьера-Робена
Синдром (секвенция, последовательность, аномалад) Пьера Робена - это врожденный порок развития, который характеризуется резким недоразвитием нижней челюсти, расщелиной твердого и мягкого неба, глоссоптозом. Вследствие резкого уменьшения просвета дыхательных путей, возникающего в ответ на уменьшение размера нижней челюсти и смещения тканей дна полости рта кзади, язык занимает приподнятое положение с тенденцией к опрокидыванию. Анатомическая деформация верхних дыхательных путей приводит к возникновению у детей синдрома обструктивного апноэ во сне, синдрома внезапной смерти, поражению сердца и его проводящей системы, возникновению системной гипертензии малого круга.
В большом количестве случаев дыхательные нарушения у детей с синдромом Пьера Робена могут возникать исключительно во время ночного сна, вызывая синдром обструктивного апноэ сна (СОАС). Синдром обструктивного апноэ во сне и рецидивирующие воспалительные явления в нижних дыхательных путях вызывают задержку умственного и физического развития ребенка, а так же патологические изменения его личности и характера.
Для диагностики синдрома обструктивного апноэ сна, в настоящее время, применяется исследование - полисомнография. Полисомнография - это регистрация большого количества показатетелей жизнедеятельности организма во время ночного сна. В настоящее время - это единственное объективное исследование, которое может определить наличие и оценить степень тяжести СОАС.
Лечение новорожденных детей, родившихся с этим заболеванием зависитот клинических проявлений дыхательных нарушений и от результатов проведенного полисомнографического исследования. В случае имеющегося у ребенка СОАС легкой или средней степени тяжести лечение проводится с применением силиконовых вестибулярных пластинок. В случае если у ребенка имеется СОАС тяжелой степени тяжести, ребеку показана операция по удлиннению нижней челюсти
В последнее время, для лечения детей с синдромом Пьера Робена, в России и за рубежом активно внедряется в практику метод компрессионно-дистракционного остеосинтеза. Данный метод имеет явные преимущества по сравнению со всеми остальными. Они заключаются в том, что этот метод малотравматичен, физиологичен, имеет стойкий результат. (Дубин С. А. 2005, Рогинский В. В. Комелягин Д. Ю. 2006г., Топольницкий 0. З. 2005).
КТ при последовательности Пьера Робена (ППР)
Синдром Пьера Робена — это врожденный порок развития, проявляющийся тремя основными признаками: расщелина нёба, недоразвитие нижней челюсти, «западение» языка.
У новорожденных и грудных детей этот синдром часто сопровождается нарушением двух важнейших функций:
1. Дыхания: на вдохе ребенок хрипит с втяжениями грудной клетки. Иногда ребенок не может дышать вообще и тогда в роддоме ребенку в трахею устанавливают интубационную трубку для обеспечения дыхания. Общее название такого нарушения дыхания — синдром обструктивного апноэ, который является жизнеугрожающим состоянием и может быть причиной синдрома внезапной смерти.
2. Глотания. При этом питание может осуществляться только через желудочный зонд.
Важно отметить, что синдром обструктивного апноэ может наблюдаться и у детей более старших возрастов при заболеваниях, которые сопровождаются недоразвитием костей лицевого скелета: синдромы Франческетти (Тричера-Коллинза), I-II жаберных дуг (гемифациальная микросомия, синдром Гольденхара), Апера, Крузона и др.; артрозах и анкилозах височно-нижнечелюстных суставов, сопровождающихся недоразвитием нижней челюсти.
С такими нарушениями новорожденные дети прямо из родильного отделения переводятся в отделение реанимации, где проводится длительное выхаживание ребенка.
Между тем, длительное пребывание ребенка на трубке и зондовом питании чревато развитием пневмонии, ослаблением глотательного рефлекса. А срок выхаживания может быть неопределенно долгим, пока ребенок не вырастет и просвет верхних дыхательных путей позволит ему самостоятельно дышать и питаться (обычно к 5-6 месяцам).
При синдроме Пьера Робена всему виной недоразвитие (укорочение) нижней челюсти, из-за чего язык смещен назад и перекрывает просвет дыхательных путей (рис. 1). Если удлинить нижнюю челюсть, то язык переместится за ней вперед и откроет дыхательные пути (рис. 2).
Рис. 1. При недоразвитии нижней челюсти язык смещен кзади и перекрывает просвет верхних дыхательных путей. Рис. 2. В результате дистракции нижняя челюсть перемещается кпереди вместе с языком, который перестает перекрывать верхние дыхательные пути.
Существует метод, позволяющий удлинить нижнюю челюсть — компрессионно-дистракционный остеосинтез.
Метод заключается в том, что на нижнюю челюсть с 2-х сторон устанавливаются аппараты, проводятся распилы челюсти между лапками аппаратов и фрагменты челюсти плотно прижимаются друг к другу — компрессия (рис. 3). С 5-6 суток после операции фрагменты челюсти постепенно, со скоростью 1 миллиметр в сутки, разводятся аппаратами на необходимую величину. Процесс удлинения челюсти аппаратами называется дистракцией, а аппарат соответственно — компрессионно-дистракционным. Между фрагментами кости образуется новая молодая кость — костная мозоль (рис. 4).
Рис. 3. Способ установки аппарата. Красным показана плоскость распила. Рис. 4. Результат удлинения нижней челюсти. Красный участок — костная мозоль между фрагментами нижней челюсти.
Операция проводится под интубационным наркозом. Такое оперативное лечение у новорожденных и младенцев проводится по жизненным показаниям.
Самостоятельное дыхание становится возможным в среднем уже на 4-6 сутки дистракции, которая продолжается до достижения правильного соотношения челюстей. На самостоятельное питание ребенок переводится, как правило, к моменту окончания дистракции или немного позже.
Ребенок выписывается с аппаратами на время (3 месяца) превращения костной мозоли в полноценную кость. Через 3 месяца — повторная госпитализация для удаления аппаратов, которое также проводится под наркозом.
Поскольку страховой полис и финансирование больницы не покрывают стоимость аппаратов, их необходимо приобретать отдельно. Их стоимость — 250-300 € за аппарат отечественного предприятия КОНМЕТ и 1200-1500 € за аппарат немецкого предприятия MARTIN. Все остальное лечение (в том числе и операция) проводятся бесплатно.
Клинический пример
В отделение реанимации из родильного дома поступил ребенок 2-х недель жизни с диагнозом синдром Пьера Робена, синдром обструктивного апноэ тяжелой степени. Питание может осуществляться только через зонд. У ребенка имеется дефицит веса. В связи с выраженными нарушениями дыхания и питания ребенку проведен компрессионно-дистракционный остеосинтез нижней челюсти.
После окончания дистракции ребенок выписан на самостоятельном дыхании и питании.
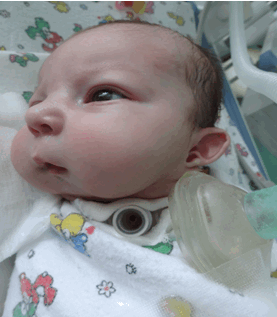
Ребенок 2 недель с синдромом Пьера Робена.
Выраженное недоразвитие нижней челюсти. У ребенка дефицит веса.
Через нос установлен желудочный зонд. Проявления синдрома обструктивного апноэ: задержка дыхания на вдохе с втяжением грудной клетки и грубыми хрипами. Внешний вид ребенка с установленным аппаратом.
Внешний вид ребенка после удаления аппаратов. Внешний вид ребенка после удаления аппаратов.
C возраста 1-1,5 года таким детям проводится уранопластика — операция по устранению расщелины неба. Лечение (в том числе и операция) проводится бесплатно.
Читайте также: