Лучевые признаки поражения легких при нейрофиброматозе
Добавил пользователь Alex Обновлено: 28.01.2026
Нейрофиброматоз Реклингхаузена — заболевание, в основе которого лежит развитие опухолей (нейрофибром) из нервной ткани, как в центральной нервной системе, так и на периферических нервах. Симптомы нейрофиброматоза Реклингхаузена зависят от локализации опухолей. Наиболее ранним и типичным признаком заболевания являются пигментные пятна размером более 1,5 см. Диагностика проводится на основании клиники и наследственного характера заболевания. Для выявления опухолей применяют рентгенологические и томографические методы обследования. Специфическое лечение нейрофиброматоза Реклингхаузена не разработано. По показаниям производится удаление опухолей.

Общие сведения
По данным наблюдений, которые проводит клиническая дерматология, чаще всего нейрофиброматоз Реклингхаузена диагностируется в возрасте от 3 до 16 лет. При наличие поражений центральной нервной системы выделяют центральную форму заболевания (НФ2), а при поражении только периферических нервов — периферическую (НФ1). Нейрофиброматоз Реклингхаузена 1 типа встречается 1 раз на 4000 рождений, а НФ2 — гораздо реже — 1 раз на 50000.

Причины нейрофиброматоза Реклингхаузена
Нейрофиброматозы являются наследственными заболеваниями, возникающими в связи с генными нарушениями. Форма Реклингхаузена передается аутосомно-доминантно. Это значит, что вероятность рождения ребенка с патологией у больного родителя составляет 50%. В некоторых случаях нейрофиброматоз Реклингхаузена развивается как результат спонтанной мутации в семье, где случаи этого заболевания ранее не наблюдались.
Симптомы нейрофиброматоза Реклингхаузена
Клиническая картина нейрофиброматоза Реклингхаузена отличается значительным многообразием симптомов. Самым ранним признаком, позволяющим заподозрить заболевание, является наличие у новорожденного или ребенка более 5-ти пигментных пятен диаметром 1,5 см и больше. Обычно они расположены на туловище и шее, но могут быть на лице и конечностях. Как правило, пигментные пятна при нейрофиброматозе имеют молочно-кофейную окраску, но могут встречаться фиолетовые, синие и даже депигментированные пятна.
Периферическая форма характеризуется развитием множественных неврином периферических нервов и нейрофибром в виде подкожных узелков. Опухоли могут локализоваться на туловище, шее, голове, конечностях. Они имеют округлую форму, подвижны и безболезненны при прощупывании. Размер таких образований в среднем 1-2 см, но в отдельных случаях они достигают значительной величины с весом около 2 кг. Кожа над опухолями часто более пигментированна, на верхушке образования может отмечаться рост волос.
Для центральной формы нейрофиброматоза Реклингхаузена характерными являются менингиомы или глиомы, поражающие спинномозговые корешки и черепно-мозговые нервы, реже локализующиеся внутри позвоночного канала или черепа. Симптомы поражения ЦНС при этом зависят от расположения опухоли и степени сдавления спинного или головного мозга. Это могут быть головные боли, потеря чувствительности, нарушения равновесия, признаки поражения черепно-лицевых нервов, нарушения речи. При этом, как правило, нарушения двигательной функции отсутствуют.
Примерно у пятой часто больных отмечается поражение зрительного анализатора, проявляющееся развитием нейрофибром оболочек глаза, глаукомы, экзофтальма, опухолью зрительного нерва. Чаще всего при НФ2 наблюдается двусторонняя потеря слуха, обусловленная развитием неврином обоих слуховых нервов. В некоторых случаях это является единственным проявлением заболевания. Нейрофиброматоз Реклингхаузена может сопровождаться умственной отсталостью, эпилептическими приступами, гинекомастией, преждевременным половым созреванием.
Диагностика нейрофиброматоза Реклингхаузена
Пигментные пятна различного размера встречаются у большинства детей. Однако большое количество таких пятен является поводом для консультации дерматолога. Врач осмотрит не только пигментные образования, но и всего ребенка для выявления других признаков нейрофиброматоза Реклингхаузена. Ранняя постановка диагноза — это возможность проведения своевременного лечения до развития осложнений от разросшихся опухолевых образований.
Для определения локализации опухолей при нейрофиброматозе Реклингхаузена проводится КТ и МРТ позвоночника, МРТ и КТ головного мозга. Диагностика аномалий скелета осуществляется путем проведения рентгенографии позвоночника и черепа. При диагностике НФ2 обязательным является исследование слуха: аудиометрия, тест Вебера, импедансометрия, электрокохлеография, исследование вызванных слуховых потенциалов.
Поскольку нейрофиброматоз Реклингхаузена связан с поражением нервной системы, необходима консультация невролога. При выявлении аномалий скелета пациента должен осмотреть ортопед. Решение вопроса о хирургическом лечении осуществляется нейрохирургом.
Лечение нейрофиброматоза Реклингхаузена
К сожалению, на сегодняшний день медицина не имеет эффективных способов лечения нейрофиброматоза Реклингхаузена. Пациентам проводится симптоматическая терапия. При большом размере периферических опухолей, развитии неврологических нарушений, появлении признаков сдавления мозга проводятся операции по удалению опухолей. Нейрофибромы могут быть удалены с косметической целью. При их озлокачествлении (3-5% случаев) удаление опухоли дополняется курсами химиотерапии и лучевой терапии.
Лечение нарушений слуха проводится с применением слуховых аппаратов. Однако после операции по поводу удаления невриномы слухового нерва слухопротезирование возможно только при помощи кохлеарной имплантации.
Нейрофиброматозы
Нейрофиброматозы - наследственные заболевания, характеризующиеся образованием доброкачественных опухолей в коже, мягких тканях, нервной системе и внутренних органах. Выделяют 6 типов нейрофиброматозов, клинически значимы типы I и II. Общие симптомы включают нейрофибромы на коже, опухоли спинномозговых корешков, слуховых и зрительных нервов, пигментные пятна, костные деформации. Диагностика основана на данных осмотра пациентов, выявлении опухолей с помощью МРТ и КТ спинного и головного мозга, внутренних органов. Лечение симптоматическое - проводится резекция опухолей, рентгенотерапия, химиотерапия.
МКБ-10

Нейрофибромы - доброкачественные опухоли, развивающиеся из оболочек нервных волокон. Чаще всего располагаются в слоях кожи и подкожной клетчатке, иногда поражают головной мозг, нервные волокна, корешки спинного мозга, мягкие ткани, внутренние органы. Нейрофиброматоз - болезнь, при которой образуются многочисленные нейрофибромы. Распространенность разных типов патологии значительно колеблется: заболеваемость 1 типом составляет 1:2 500, 2 типом - 1:50 000. Другие варианты встречаются еще реже, их точная эпидемиология не определена. Гендерной и расовой предрасположенности не выявлено. Дебют клинических проявлений возможен в любом возрасте, зависит от типа болезни.

Причины нейрофиброматозов
Образование множественных нейрофибром детерминировано генетически. При нейрофиброматозе I существует мутация гена НФ1, расположенного на длинном плече 17 хромосомы. Он относится к генам-супрессорам роста опухолевых тканей, большая часть из которых - нейроэктодермального генеза. При дефекте в гене НФ1 нарушается синтез белков, ответственных за клеточную пролиферацию. Мутации носят характер транслокаций, делеций, инверсий, точковых изменений. Больше 80% из них приводят к синтезу нефункциональных белков или к полному отсутствию белковых молекул. Наследование происходит по аутосомно-доминантному механизму с высокой степенью пенетрации: при наличии мутационного гена у одного из родителей вероятность болезни у ребенка составляет 50%, если оба родителя имеют мутацию, риск повышается до 80-90%. Известны случаи спонтанных мутаций.
Причиной нейрофиброматоза II является мутационное изменение гена НФ2, локализованного на 22 хромосоме. Он кодирует производство белка мерлина (шванномина) - супрессора опухолевого роста. Тип наследования - аутосомно-доминантный с небольшой степенью пенетрации. Передача одного мутантного гена зачастую не проявляется, поскольку второй ген обеспечивает синтез достаточного количества белков. Если он повреждается, синтез нормальных фракций мерлина прекращается, пролиферация клеток усиливается, развивается новообразование. При других типах нейрофиброматозов также существуют мутации в генах, обеспечивающих воспроизведение молекул белков-супрессоров роста опухолей.
Патогенез
Общим патогенетическим механизмом развития нейрофиброматозов является наследственно обусловленная недостаточность какого-либо белка, подавляющего процессы опухолевой пролиферации клеток в тканях нейроэктодермального происхождения. При мутации одного гена производство опухолевых супрессоров прекращается наполовину, равновесие роста и гибели клеток смещается в сторону митотического деления. Нормальный аллельный ген частично компенсирует дефицит белка. Тяжесть нейрофиброматоза определяется тем, насколько дефектный ген влияет на активность белка-супрессора - частично или полностью нарушает функциональность, полностью блокирует производство. Кроме этого, выраженность клинических признаков зависит от сохранности противоопухолевого иммунитета.
Во многих органах и тканях пациентов формируются доброкачественные опухолевые образования, состоящие из соединительной ткани и пигментных клеток. На нервных стволах образуются невриномы; на поверхности кожи - пигментированные области, жировые бляшки, расширенные сосуды; на сетчатке глаз - факоматоз. Изменяется строение костей, они остаются недоразвитыми либо чрезмерно утолщаются, искривляется позвоночный столб.
Симптомы нейрофиброматозов
Заболевания проявляются признаками поражения кожи, нервной системы. Классическим клиническим вариантом является нейрофиброматоз типа I, на долю которого приходится 90% случаев болезни. Характерный симптом - гиперпигментация. У больных при рождении или в раннем детстве появляются кожные пятна, цвет которых варьируется от светло-золотистого до коричневого «кофе с молоком». В отдельных случаях пятна имеют фиолетовый или синий оттенок. На радужке глаза обнаруживаются узелки Лиша (пятна пигмента - гамартомы) небольшого размера, белесоватые или светло-бежевые, заметные только при офтальмологическом осмотре. Являются специфическим признаком нейрофиброматоза 1, образуются по мере взросления: у детей до 4 лет распространенность составляет 22%, с 5 до 9 лет - 41%, с 10 до 19 лет - 85%, после 20 лет - 95%.
В период пубертата и позже формируются кожные и плексиформные нейрофибромы, располагающиеся соответственно подкожно (на мелких нервных волокнах, иннервирующих кожу) и на крупных нервах. Они представляют собой небольшие доброкачественные новообразования. Кожные нейрофибромы воспринимаются как косметический дефект, при определенном расположении травмируются. Плексиформные опухоли, локализующиеся по ходу периферических нервов, выявляются на конъюнктиве, веках, в брюшной полости и средостении. Проявляются хронической болью, онемением, судорогами, параличом. Опухоли ЦНС находятся внутри черепа, представлены глиомами зрительных нервных волокон, астроцитомами, эпендимомами, невриномами слухового нерва, менингиомами и нейрофибромами. Клиническая картина определяется размерами новообразований, вовлеченностью мозговых структур в патологический процесс. В детском возрасте диагностируются расстройства психического развития: снижение когнитивных способностей, гиперактивность, редко - деменция.
При тяжелом нейрофиброматозе деформируется костная система. У больных возникает сколиоз, краевые структурные изменения тел позвонков и их отростков, эрозийные поражения краев межпозвоночных отверстий и задних ребер. Характерна атрофия либо, наоборот, гипертрофия трубчатых костей. Кости часто искривлены, на поверхности обнаруживаются периостальные гребни и наслоения. В полостях костей могут образовываться нейрофибромы. Если в процесс вовлекаются кости черепа, появляется внешняя асимметрия, наиболее выраженная при поражении лицевой части и глазниц. Свод черепа может иметь атрофированные участки, дефекты, узуры, иногда отмечается локальное увеличение костного вещества.
При типе 2 формируются высокодифференцированные опухоли, которые, однако, более агрессивны, чем при заболевании 1 типа. Пигментных пятен нет. Образуются невриномы - подвижные и болезненные неоплазии. Нередко они локализуются на слуховом нерве, вызывая потерю слуха. Нейрофиброматоз 3 типа отличается большим количеством нейрофибром, ускоренным развитием нейролемм и глиом зрительного нерва, приводящих к расстройству зрения. Специфический признак - появление нейрофибром на ладонях. При болезни 4 типа симптомы похожи, сохраняется риск поражения зрительных волокон. Для 5 типа характерны пигментные темные пятна, опухоли больших размеров, провоцирующие асимметрию тела. Течение 6 типа сопровождается лишь пигментными пятнами. При 7 типе выявляются нейрофибромы средних размеров, гиперпигментации нет.
Осложнения
В 10% случаев нейрофибромы трансформируются в злокачественные опухоли. В группе высокого риска находятся пациенты с большим катамнестическим стажем, беременные женщины. У 6% детей нарушается умственное развитие: они имеют проблемы при освоении учебных навыков (чтение, письмо, счет), с трудом запоминают новую информацию, долго адаптируются в незнакомых ситуациях. Больные всех возрастов подвержены депрессии, поскольку испытывают дискомфорт, чувство стыда и неловкости из-за обезображенной внешности. Множественные нейрофибромы провоцируют эндокринные расстройства, эпилептические припадки, гипотонию мышц, стеноз почечной и легочной артерии, легочные кисты, интерстициальную пневмонию, гипертрофию клитора, нарушения развития органов ЖКТ.
Диагностика
Подозрение на нейрофиброматоз возникает при множественных подкожных опухолях, пигментных пятнах, спинальной шванноме, ухудшении слуха и зрения. Обследование проводят дерматовенеролог, невролог, офтальмолог, отоларинголог и генетик. Перед инструментальными и лабораторными процедурами осуществляется сбор семейного и личного анамнеза, клинический опрос и осмотр. В ходе генеалогического анализа выявляется передача заболевания в нескольких поколениях, реже определяется первичная спонтанная мутация. На теле пациентов обнаруживаются нейрофибромы, пигментные области (при определенных типах болезни), искривления позвоночника, деформации костей, нарушения зрения, слуха, координации движений. Производится дифференциальная диагностика различных вариантов нейрофиброматозов, исключается синдром Протея, рассеянный липоматоз, синдром Клиппеля-Треноне-Вебера. Для уточнения диагноза назначаются:
- МРТ, КТ. Визуализационные методы исследований позволяют определить наличие нейрофибром в головном мозге, позвоночнике, внутренних органах. Двусторонние невриномы 7 пары черепных нервов являются диагностическим критерием нейрофиброматоза II типа. Часто выявляются глиомы, шванномы, менингиомы. Для I типа свойственно развитие плексиформных и обычных новообразований, глиом.
- Рентгенография костей скелета. Диагностическая процедура выполняется с целью подтверждения и оценки тяжести сколиоза, костных атрофий и гипертрофий, локальных утолщений и эрозийных поражений костных структур. При большинстве типов болезни наблюдается истончение кортикального слоя, ложные суставы, дисплазии крыльев клиновидной кости, дугообразное искривление большеберцовой и малоберцовой костей, кисты длинных костей.
- Офтальмологическое исследование. Нейрофиброматоз типа 1 сопровождается плексиформной нейрофибромой век, меланоцитарными гамартомами радужки, глиомой оптических нервных волокон, астроцитарной гамартомой сетчатки, утолщением роговичных нервов, конъюнктивальной нейрофибромой, ишемическими поражениями венул сетчатки. Патогномоничный признак - пятна светлого оттенка на глазном дне и радужке (гамартомы). При 2 типе диагностируется задняя субкапсулярная катаракта, помутнение хрусталика.
- Аудиологическое исследование. При опухолевом поражении слуховых нервов и жалобах на нарастающую глухоту (тугоухость) выполняется электрокохлеография и импедансометрия. Результаты указывают на снижение остроты слуха, наличие слуховой нейропатии, определяют причину и локализацию нарушения.
Лечение нейрофиброматозов
В настоящее время терапия данной группы заболеваний заключается в симптоматической помощи больным. Пациенты регулярно проходят обследования, нацеленные на контроль формирования и увеличения опухолей. При наличии нейрофибром, провоцирующих боль, расположенных в местах повышенного риска травмирования, сдавливающих или смещающих жизненно важные органы, проводится их хирургическое удаление. Применяются классические методики резекции неоплазий и участков нервов, криодеструкция, лазерная хирургия. При множественных новообразованиях назначается лучевая терапия, химиотерапия. Больным с поражением опорно-двигательного аппарата показаны реабилитационные мероприятия (физиолечение, ЛФК).
Активно разрабатываются способы этиологического лечения нейрофиброматозов. На стадии клинических испытаний находится терапия ингибиторами RAS (белков-активаторов роста опухолей) у лиц с нейрофиброматозом первого типа. Этап теоретических разработок проходят методы генной инженерии. Усилия ученых-генетиков направлены на создание и внедрение в организм больных нормального НФ1 гена, отвечающего за синтез нейрофибромина, на расшифровку и введение гена ФН2, обеспечивающего транскрипцию белка шванномина. В некоторых медицинских центрах предпринимаются попытки применения патогенетической терапии, в основе которой лежит комплексное использование стабилизаторов мембран тучных клеток, антипролиферативных препаратов и ферментов, корректирующих метаболические процессы.
Прогноз и профилактика
Нейрофиброматозы являются прогностически благоприятными заболеваниями - малигнизация опухолей происходит редко, в большинстве случаев больные остаются трудоспособными и социально адаптированными. При правильных и регулярных реабилитационных мероприятиях нарушения со стороны костной системы и задержка умственного развития успешно корректируются. Поскольку заболевание является наследственным, профилактика возможна на этапе планирования беременности, парам из групп риска (с отягощенным семейным анамнезом) рекомендуется медико-генетическое консультирование с определением вероятности рождения больного ребенка.
1. Нейрофиброматоз: этиология, патогенез, лечение/ Скаварская Е.А.// Международный журнал педиатрии, акушерства и гинекологии - 2014 - Т.5, №2.
2. Клинико-диагностические аспекты нейрофиброматоза/ Попова А.А.// Университетская медицина Урала - 2016 - №2.
3. Нейрофиброматоз первого типа (болезнь Реклингхаузена)/ Н.А. Шнайдер, А.И. Горелов// Сибирское медицинское обозрение - 2007.
Постлучевой пневмонит ( Радиационный пневмонит )
Постлучевой пневмонит - повреждение лёгочной ткани, развивающееся под действием высоких доз ионизирующей радиации. Проявляется одышкой, сухим или продуктивным кашлем, плевральными болями, сопровождается повышением температуры тела. Диагноз выставляется на основании анамнестических данных и клинических симптомов, подтверждается результатами спирометрии, рентгенологического исследования, компьютерной томографии и МРТ органов грудной клетки. Лечение пневмонита проводится с помощью фармакотерапии кортикостероидными гормонами, антибиотиками, антикоагулянтами; оксигенотерапии, физиотерапии.

Постлучевой пневмонит (лучевой пульмонит, радиационная пневмония) относится к локальным радиационным поражениям лёгочной паренхимы. Ионизирующим излучением повреждается участок альвеолярной ткани, отсюда другое название - лучевой альвеолит. Ярко выраженные клинические проявления встречаются у 15-60% получающих радиотерапию по поводу рака легких и молочной железы. В 3-4% случаев постлучевой пневмонит приобретает крайне тяжёлое течение и заканчивается летально. У некоторых больных протекает латентно, характерные изменения выявляются только специальными методами исследования.
Причины
Пусковым механизмом служит превышение толерантной дозы радиоактивного воздействия на лёгочную ткань. При лучевой терапии онкологических заболеваний грудной полости, рака молочной железы доза радиации составляет в среднем 70-80 Гр. В фокус облучения попадает зона лёгкого с предельной переносимостью 35-40 Гр, вследствие чего развиваются радиационные поражения. Частота возникновения, течение патологического процесса напрямую зависят от величины суммарной очаговой дозы. Большое значение имеют следующие факторы:
- Возраст пациента. Согласно исследованиям в области онкологии и пульмонологии, больные старше 70 лет страдают от осложнений радиотерапии в 1,5 раза чаще, чем 40-60-летние. У детей толерантность респираторных органов к лучевой нагрузке в 2,5 раза ниже, чем у взрослых.
- Локализация очага. Установлена прямая зависимость частоты радиационно-индуцированного пульмонита от расположения очага, на который производится воздействие. Чем ближе по отношению к средостению находится фокус облучения, тем реже и позднее выявляются проблемы. Больше страдают периферические участки лёгких.
- Комплексное и комбинированное лечение. Сочетание радиотерапии с хирургическим лечением и (или) химиотерапией увеличивает количество случаев постлучевого пневмонита, провоцирует его раннее развитие, утяжеляет течение. Противоопухолевые препараты способны самостоятельно негативно воздействовать на дыхательную систему, вызывать пневмопатии.
Ионизирующее излучение повреждает клетки альвеолярного эндотелия. Нарушается продукция сурфактанта, альвеолы спадаются. Одновременно поражается внутренняя оболочка капилляров, что приводит к тромбообразованию, частичной закупорке и повышению проницаемости сосудов. Страдает газообмен. Изменения по типу экссудативного альвеолита возникают в первые дни. В течение 1-3 месяцев повреждённый эндотелий частично восстанавливается, происходит реканализация ряда сосудов. Активируются фибробласты, погибшие клетки заменяются соединительной тканью.
Макроскопически определяются полнокровные участки лёгочной паренхимы плотноэластической консистенции, фибринозный выпот в плевральной полости. При микроскопическом исследовании выявляется утолщение альвеолярной мембраны, повреждения капиллярной сети с признаками тромбоза, стаза и полнокровия. В области межальвеолярных перегородок формируются зоны фиброза.
Классификация
Острый постлучевой пневмонит является ранним радиационным повреждениям органов дыхания. Возникает на фоне облучения или в течение первых трёх месяцев после него. Стабильные изменения, обнаруженные в более поздние сроки, расцениваются как пневмофиброз. Американскими онкологами разработана классификация заболевания в зависимости от степени тяжести:
- I степень. Болевые ощущения в груди отсутствуют или минимальные. Кашель редкий. Одышка появляется при значительной физической нагрузке. Определяется снижение жизненной ёмкости лёгких (ЖЕЛ) на 10-25% от должного значения. Имеются косвенные рентгенологические признаки болезни.
- II степень. Больного беспокоит периодическая терпимая торакалгия, приступы кашля. При ходьбе ощущается нехватка воздуха. ЖЕЛ находится в пределах 50-75% от нормальной величины. На рентгенограмме видны очаговые тени.
- III степень. Боль становится интенсивной, кашель - постоянным. Одышка возникает при малейшей нагрузке. ЖЕЛ составляет 25-50 % от нормы. Размеры инфильтрации на снимке соответствуют облучённому участку.
- IV степень. Стойкий болевой синдром, непрекращающийся кашель требуют назначения наркотических анальгетиков. Дыхание затруднено в покое. Наблюдается значительное (более чем на 75%) снижение ЖЕЛ. При рентгенографии выявляется одностороннее субтотальное или тотальное затенение. Пациент нуждается в респираторной поддержке.
Симптомы постлучевого пневмонита
Заболевание начинается остро. Ведущим симптомом является кашель. Его интенсивность варьирует от редкого покашливания до постоянных мучительных приступов. Может отделяться светлая слизистая мокрота, которая при присоединении вторичной инфекции становится гнойной жёлто-зелёной. Иногда возникает кровохарканье. Одышка вызывается нагрузкой, в тяжёлых случаях беспокоит при разговоре, в состоянии покоя. Боль в груди может отсутствовать или появляться эпизодически; при наличии фибринозного плеврита становится постоянной и интенсивной. Температурная реакция колеблется от стойкого субфебрилитета до гипертермии.
Классической клинической картине иногда предшествует общая лучевая реакция. Пациент становится излишне раздражительным, предъявляет жалобы на слабость, головную боль, головокружение, бессонницу. Развивается миокардиодистрофия, возникают перебои в сердечной деятельности. Нарушения работы пищеварительного тракта проявляются тошнотой, рвотой, диареей. Резко снижается аппетит, наблюдается извращение вкуса, избыточное отделение слюны.
Небольшой по объёму постлучевой пневмонит часто распознаётся несвоевременно. Обнаруживается на стадии пневмофиброза с бронхоэктазами, плевродиафрагмальными спайками, смещением средостения. Из-за массивного склеротического процесса возникают необратимые дыхательные нарушения. Формируется хроническое лёгочное сердце, значительно ухудшается качество жизни. При обширных изменениях развивается тяжёлая острая дыхательная недостаточность, требующая перевода на искусственную вентиляцию лёгких. Может наступить летальный исход.
Чёткая связь респираторных нарушений с проводимой радиотерапией позволяет пульмонологу заподозрить постлучевой пневмонит. При сборе анамнеза уточняются дозы, количество и давность сеансов облучения, сочетание с химиотерапией или операцией. Особое внимание уделяется локализации и размерам очага. Следует учитывать наличие общей реакции. Основные диагностические мероприятия включают:
- Физикальное исследование. Определяются зоны притупления перкуторного звука на стороне поражения. Аускультативно выслушиваются непостоянные средне- и мелкопузырчатые хрипы, шум трения плевры.
- Лабораторные анализы. Такие проявления лучевой реакции, как угнетение гемопоэза, иммунные нарушения, выявляются лабораторными методами. В клиническом анализе крови наблюдается снижение уровня лейкоцитов, тромбоцитов, эозинофилия.
- Рентгенография и КТ лёгких. На рентгенограмме заметны усиление и деформация лёгочного рисунка, фокусы инфильтрации в проекции зоны облучения, плевральный выпот. Одностороннее поражение является патогномоничным признаком заболевания. КТ позволяет раньше выявить и более точно определить границы повреждения.
КТ органов грудной клетки. Снижение пневматизации в периферических отделах правого легкого после лучевой терапии по поводу рака молочной железы.
Дополнительно выполняется исследование газового состава крови и функции внешнего дыхания. В неясных случаях для уточнения диагноза используется МРТ. Постлучевой пневмонит следует дифференцировать с метастатическим обсеменением, бактериальной пневмонией, а также с туберкулёзом и микозами, которые могут быть спровоцированы вторичным иммунодефицитом.
Лечение постлучевого пневмонита
При подтверждении радиационной пневмонии назначается консервативная терапия. Применяются фармакологические средства, физиотерапевтические методы, лечебная физкультура. Основные мероприятия направлены на снижение смертности, восстановление респираторных функций организма, минимизацию последствий. Для достижения терапевтического эффекта комплексно используются следующие группы препаратов:
- Системные кортикостероиды. Назначаются с целью стимуляции продукции сурфактанта. Уменьшают воспалительную реакцию лёгкого, сглаживают симптомы. Положительный ответ наступает быстро, обычно на 2-4 день. Если за этот промежуток времени состояние пациента не улучшилось, дальнейшее применение кортикоидных гормонов не имеет смысла.
- Антикоагулянты. Препятствуют образованию тромбов в капиллярах малого круга кровообращения. Снижают риск развития смертельно опасного осложнения - тромбоэмболии лёгочной артерии.
- Антибиотики. Применяются для лечения бактериальных осложнений в условиях снижения иммунитета. Назначаются с учётом чувствительности микрофлоры мокроты к антибактериальным средствам. При отсутствии данных предпочтение отдаётся препаратам с широким спектром действия.
Дополнительно используются отхаркивающие средства, ангиопротекторы, аскорбиновая кислота. При острой респираторной недостаточности показаны наркотические противокашлевые препараты, кислородная поддержка. Из физиотерапевтических методов рекомендуются ингаляции с димексидом, переменные магнитные поля. В восстановительном периоде широко применяются массаж и дыхательная гимнастика.
Профилактика и прогноз
Для предотвращения нежелательных реакций при проведении лучевой терапии пациенту рекомендуется сбалансированное калорийное питание, прогулки на свежем воздухе. Необходимо контролировать показатели крови, общее состояние больного. При появлении тромбопении, лейкопении, кровохарканья лучевую терапию следует прекратить. Рациональный индивидуальный подход, использование методов фигурных полей облучения позволяют значительно снизить частоту возникновения индуцированных радиацией осложнений. При радикальной радиотерапии рака лёгкого в профилактических целях рекомендуется одновременное переливание облучённой аутокрови, использование энтеросорбентов.
Прогноз во многом определяется течением основного заболевания, площадью повреждения лёгкого. Адекватно пролеченные небольшие пульмониты нередко подвергаются обратному развитию. Формирование фиброза продолжается от 6 месяцев до 2 лет, после чего изменения носят стабильный характер, и нарушения становятся необратимыми. Реабилитационные мероприятия способны влиять на процесс фиброзирования. Острый респираторный дистресс-синдром в ряде случаев заканчивается смертью.
1. Постлучевой пневмонит в практике пульмонолога/ Семищева Н. Л., Карапетян Е. И., Мальцева Т. А., Авдеева Н. В., Одиреев А. Н.// Бюллетень физиологии и патологии дыхания. - 2014.
2. Лучевые пневмониты у больных раком легкого/ Курсова Л.В., Иванова И.Н., Мардынский Ю.С., Золотков А.Г., Рагулин Ю.А.// Сибирский онкологический журнал. - 2010 - №2 (38).
3. Клинические рекомендации по оказанию медицинской помощи пострадавшим от воздействия ионизирующего излучения в чрезвычайных ситуациях. - 2013.
4. Поздние лучевые повреждения органов грудной клетки/ Пасов В.В., Зубова Н.Д., Иволгин Е.М., Курпешева А.К.// Сибирский онкологический журнал. - 2009 - №6 (36).
Нейрофиброматоз

«Нейрофиброматоз относят к группе факоматозов, он возникает из-за генетических нарушений и поражает всю нервную систему: ее клетки не имеют возможности нормально расти, когда у человека диагностируют нейрофиброматоз. Новообразования появляется в головном и спинном мозге из соединительной ткани. Считаю, что в TomoClinic созданы современные условия для лечения таких онкологических заболеваний».
Что такое нейрофиброматоз?
Болезнь носит генетический характер, передается аутосомно-доминантным типом наследования. Ген картирован в 17 хромосоме. Риск передачи патологии по наследству от одного из родителей составляет 50%. Болезнь в одинаковом соотношении поражает и мужчин, и женщин. Диагностируется в детском и подростковом возрасте.
Признаки нейрофиброматоза имеют множественный характер, что затрудняет своевременное диагностирование. Важным симптомом для определения нейрофиброматоза является возникновение узелков Лиша. Узелки Лиша — белесые пятна на радужке глаза, которые визуализируются во время офтальмологического осмотра при помощи специального инструмента. Патология не сопровождается болевыми ощущениями и не видна без применения специальных приборов.
Типы нейрофиброматоза
- Нейрофиброматоз 1 типа. Также называется «нейрофиброматоз реклингхаузена». Классический тип болезни, встречается раз на 3-4 тысячи новорожденных. Причиной является повреждением гена «нф1» в 17 хромосоме. «нф1» принадлежит к ряду генов, отвечающих за подавление опухолей. Этот тип болезни в одинаковом соотношении поражает и мужчин, и женщин. Нейрофиброматоз 1 типа сопровождается пигментными пятнами на коже, узелками Лиша на радужке глаза, костными аномалиями. Этот тип заболевания является наиболее распространенным, встречается в 90% всех случаев.
- Нейрофиброматоз 2 типа. Проявляется как и 1 тип, но причиной развития болезни является аномалия гена в 22 хромосоме. Патология сопровождается одиночным развитием неврином — подвижных и болезненных опухолей. Двусторонняя невринома слухового нерва может привести к потере слуха. При нейрофиброматозе 2 типа пигментные пятна на коже не диагностируются. Доброкачественные опухоли имеют высокий риск трансформации в злокачественные новообразования. Нейрофиброматоз 2 типа диагностируется в 10 раз реже патологии первого типа.
- Нейрофиброматоз 3 типа. Характеризуется большим количеством нейрофибром, которые провоцируют ускоренное развитие глиомы зрительного нерва и нейролеммы. Болезнь не диагностируется у детей, поражает молодых людей в возрасте 30 лет. Отличительной чертой нейрофиброматоза третьего типа является появление нейрофибром на ладонях.
- Нейрофиброматоз 4-7 типа. Для болезни 4 типа характерно множественное поражение кожи нейрофибромами. Присутствует высокий риск развития глиомы зрительного нерва и нейролеммы. При 5 типе патологии характерно появление пигментных пятен и нейрофибром. Опухоли достигают крупных размеров, отягощают кожу, вызывают заметную асимметрию. Шестой тип нейрофиброматоза классифицируется лишь визуальными проявлениями пигментных пятен на поверхности кожи. Для седьмого типа нейрофиброматоза характерно развитие только нейрофибром, гиперпигментация кожи не наблюдается.
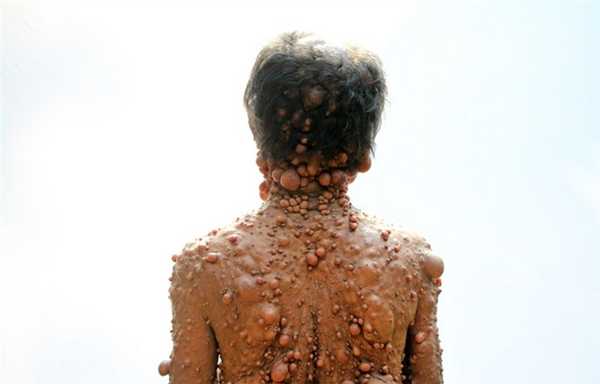
Причины нейрофиброматоза
Основной причиной развития нейрофиброматоза у грудника является генетическая предрасположенность.
Если поврежденный ген присутствует только у одного из родителей, вероятность болезни составляет не менее 50%. При присутствии гена у обоих родителей вероятность развития болезнь у новорожденного составляет свыше 80%.
На тип и тяжесть симптомов при нейрофиброматозе влияет степень экспрессии гена. Несмотря на наследственный характер болезни известны случаи, когда заболевание развивалось из-за спонтанной мутации гена во время зачатия.
Симптомы нейрофиброматоза
Болезнь не имеет четких симптомов, признаки проявляются по-своему в каждом индивидуальном случае. Опасность нейрофиброматоза заключается в разносторонности симптомов, которые, поражая различные системы организма, размывают клиническую картину. Специалисты отмечают, что нарушения зрения характерны для нейрофиброматоза 1 типа, а патологии слуха — преимущественно для нейрофиброматоза 2 типа.
Основные признаки, которые характерны для нейрофиброматоза:
- Гиперпигментация. Визуальным симптомом нейрофиброматоза является возникновение и распространение пигментных пятен на коже. Пятна светло-коричневого оттенка возникают при рождении или в первые годы жизни. При наличии более 6 пигментных пятен в нехарактерных местах (подмышки, паховая область, шея) возникает подозрение на болезнь. Пигментные пятна являются первичным признаком заболевания.
- Опухоли на коже. Нейрофибромы (опухоли) появляются на теле пациента в пубертатный период. С возрастом или в период вынашивания плода нейрофибромы могут трансформироваться в злокачественные опухоли.
- Нервная система. Болезнь поражает центральную нервную систему, что провоцирует заминку в развитии ребенка. Дети с нейрофиброматозом склонны к расстройству восприятия, страдают от гиперактивности. Примерно у 6% детей диагностируется умственная отсталость. При нейрофиброматозе кожи у детей может развиваться опухоль зрительного нерва с последующей потерей зрения.
- Зрение. При офтальмологическом осмотре с помощью специального инструмента диагностируются пигментные пятна на радужке. Пятна имеют светло-бежевый окрас, они менее выражены, чем на коже. Поражение глаз при нейрофиброматозе пигментными пятнами не несет особой опасности для пациента, но является тревожным сигналом для дальнейшего обследования.
- Опорно-двигательная система. Нейрофиброматоз может быть причиной деформации костей и суставов пациента. Многие патологии костей являются врожденными дефектами, но патологии позвоночника проявляются в пубертатный период.
- Эндокринная система. Нейрофиброматоз может провоцировать развитие опухолей щитовидной железы.
- Сердечно-сосудистая система. У пациентов с диагнозом нейрофиброматоз развивается артериальная гипертензия, стеноз.
Диагностика нейрофиброматоза
Поводом для диагностики нейрофиброматоза 1 типа является наличие более 6 пятен светло-коричневого цвета на коже. Они возникают в период внутриутробного развития и в первые пару месяцев жизни. Характерным признаком заболевания являются пигментные пятна, расположенные в области паха, подмышек и на конечностях.
Серьезным поводом для обследования является наличие подкожных нейрофибром, спинальной шванномы и помутнение хрусталика. Прежде чем приступить к обследованию на диагностической аппаратуре, врач проводит сбор анамнеза, осматривает видимые признаки (пигментные пятна на коже, опухоли). Обязательным является неврологический осмотр для оценки координации движений.
В ходе обследования применяются методы диагностики:
- МРТ или КТ. Магнитно-резонансная и компьютерная томографии используются для визуализации позвоночника, внутренних органов, головного мозга. Эти методы точно определяют размер и локализацию опухолей.
- Рентген. Рентгенография позвоночника проводится с целью определения степени сколиоза.
- Электрокохлеография. Метод исследования, который используется для диагностики слуховой нейропатии, нарушений остроты слуха.
- Импедансометрия. Исследование позволяет определить место и причину нарушений в слуховой системе пациента.
- Офтальмоскопия. Является способом обследования глазного дна, позволяющим оценить сетчатку и зрительный нерв.
Помимо аппаратных исследований одним из основных методов диагностики является генетическое обследование и генеалогический анализ.

Нейрофиброматозы - это группа наследственных заболеваний, которые сопровождаются образованием доброкачественных опухолевых образований в области мягких тканей, кожи, внутренних органов и нервной системы. В клинической практике выделено 6 типов заболевания, но значимыми являются 1 и 2.
Причины нейрофиброматозов
Патология является генетически детерминированной и передаётся по аутосомно-доминантному механизму. При этом пенетрация является высокой. В редких случаях возможны случайные мутации.
В отличие от 1 типа при втором степень пенетрации ниже, несмотря на аутосомно-доминантный характер наследования. В случае отсутствия мутации у второго родителя заболевание может не проявиться.
Патогенез
К общему патологическому механизму развития любого типа нейрофиброматоза относят недостаточное содержание в организме белка, который будет подавлять опухолевую пролиферацию в клетках, имеющих нейроэктодермальное происхождение. Тяжесть течения заболевания будет зависеть от степени повреждения гена, отвечающего за активность белка - супрессора. Это может быть полное или частичное нарушение функционирования.
Также следует учитывать, что клиническая картина зависит от того, насколько выражен противоопухолевый иммунитет.
Симптомы нейрофиброматозов

К наиболее характерным проявлениям относят поражение нервных клеток и кожных покровов. При первом типе с рождения на поверхности кожных покровов появляются очаги гиперпигментации с светло-коричневым или оттенком «кофе с молоком», реже они могут быть фиолетовыми или синими.
В ходе офтальмологического осмотра удаётся выявить гемартромы на радужке глаза.
В пубертатном периоде под кожей или по ходу крупных нервов происходит формирование кожных и плексиформных фибром. Постепенно присоединяется хроническая боль, онемение, судороги и паралич.
На фотке тяжелого течения происходит деформация скелета с атрофией или гипертрофией трубчатых костей.
При втором типе удаётся обнаружить высокодифференцированные опухоли, которые не сопровождаются развитием болевого синдрома и являются подвижными.
При третьем типе формируется большое количество нейрофибром с глиомами зрительного нерва. Для четвёртого типа характерна схожая симптоматика с высоким риском повреждения зрительных волокон.
5 и 6 тип проявляется очагами гиперпигментации, которые могут иметь большую площадь.
Диагностика
Постановка диагноза начинается с осмотра пациента и выявления у него подкожных опухолей, искривления позвоночника, костных деформаций и пигментных пятен. В ходе беседы определяется ухудшение слуха и зрения.
Врачу важно собрать анамнез и провести генеалогический анализ для того, чтобы выяснить, является ли заболевание генетически-обусловленным или же является результатом спонтанной мутации.
Из дополнительных методов назначают:
- Рентгенографию. Общедоступный способ позволяет выявить сколиоз, атрофические или гипертрофические процессы в костях, локальные утолщения и эрозии. Для заболевания характерна атрофия кортикального слоя костей, появление ложных суставов.
- Магнитно-резонансную или компьютерную томографии. Исследование применяют для оценки состояния головного мозга, позвоночника и внутренних органов. При нейрофиброматозов определяются различные по размеру невриномы, глиомы, шванномы и менингиомы.
- Офтальмологическое исследование. В ходе обследования можно выявить плексиформную нейрофиброму век, меланоцитарные гамартомы радужки, астроцитарные гамартомы сетчатки и утолщение нервов роговицы.
- Аудиометрию. Для нейрофиброматоза характерно снижение остроты слуха со слуховой нейропатией.
Лечение
Терапия нейрофиброматозов является симптоматической. Необходимо исключить трение и сдавление образовавшихся опухолей. Кроме того, выполняют иссечение неоплазий или участков нервов с помощью различных хирургических методов.
В случае множественных образований показано лучевое воздействие и химиотерапия.
Читайте также:
- Сбор полового анамнеза при мужском бесплодии. Оценка репродукции
- Лечение вестибулярного неврита. Течение (прогноз) вестибулярного неврита
- Эрозия зубов: причины, симптомы и лечение
- Высвобождение энергии из глюкозы через пентозофосфатный цикл. Превращение глюкозы в жиры
- Жизнеспособность сперматозоидов. Продолжительность жизни сперматозоидов