Микроцефалия. Лизэнцефалия у детей
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Определение
Энцефалит - это группа заболеваний, для которых характерно воспаление вещества головного мозга. В настоящее время энцефалитом называют не только инфекционное, но и инфекционно-аллергическое, аллергическое и токсическое поражения мозга.
Причины возникновения энцефалитов
Первичные энцефалиты, к которым относятся клещевой энцефалит, герпетический энцефалит, энцефалит, вызванный вирусом Коксаки (А9, В3, В6), ЕСНО (2, 11, 24) и некоторые другие, возникают в результате проникновения вируса через гематоэнцефалический барьер, по причине чего повреждаются нейроны головного мозга и развивается воспалительный процесс.
Вторичные энцефалиты рассматриваются как осложнение перенесенных инфекционных заболеваний: гриппа, краснухи, кори, ветряной оспы, лептоспироза и др., а также поствакцинальные энцефалиты.
Гриппозный энцефалит вызывают вирусы гриппа А1, А2, А3, В. Патогенетическими механизмами при гриппозной инфекции становятся токсическое поражение головного мозга и недостаточность мозгового кровообращения.
Возбудителем энцефалита при краснухе становится РНК-содержащий вирус семейства Togaviridae.
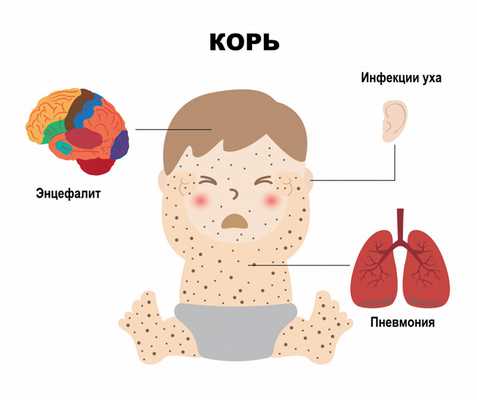
Коревой энцефалит вызывается вирусом кори - это форма поражения головного (иногда и спинного) мозга (энцефаломиелит). Энцефалит считается самым распространенным неврологическим осложнением кори (более 95% всех случаев неврологических осложнений). Он наблюдается с частотой 1:1000 случаев кори, чаще у детей старше 2 лет - как у мальчиков, так и у девочек.
Поствакцинальные энцефалиты могут развиваться после вакцинации, особенно после введения некоторых антирабических прививок. В основе поствакцинальных энцефалитов лежит перекрестная аутоиммунная реакция на антигены вакцины и антигены мозга, морфологически выражающаяся в воспалительном поражении мозговых сосудов и окружающего вещества мозга.
Лептоспирозные энцефалиты вызывается Leptospira interrogans. Источником заражения являются домашние инфицированные животные. Наиболее распространен лептоспироз в южных регионах. В развитии заболевания большое значение имеют аутоиммунные реакции.
В группу подострых склерозирующих лейкоэнцефалитов входят формы хронических и подострых энцефалитов с прогрессирующим тяжелым течением (энцефалит с включениями Даусона, подострый склерозирующий лейкоэнцефалит Ван-Богарта, узелковый панэнцефалит Петте Деринга).
Поскольку различия в их клинической картине и морфологии несущественны, в настоящее время их трактуют как одно заболевание, чаще всего под названием «подострый склерозирующий панэнцефалит».
В развитии болезни большую роль играют персистирующие вирусные инфекции: коревая, энтеровирусная, вирус клещевого энцефалита. У больных подострым склерозирующим панэнцефалитом обнаруживают в крови и ликворе очень высокие титры коревых антител (не отмечаемые даже у больных с острой коревой инфекцией). Кроме того, значимое место занимают аутоиммунные механизмы, а также приобретенный или врожденный дефект иммунной системы.
Классификация заболевания
Классификация энцефалитов отражает факторы, приводящие к развитию заболевания, и связанные с ними клинические проявления и особенности течения.
По срокам возникновения различают:
- первичные энцефалиты - самостоятельные заболевания, вызываемые преимущественно нейротропными вирусами:
- вирусные: полисезонные (герпетический, энтеровирусный, гриппозный, цитомегаловирусный, вирус бешенства и др.); арбовирусные, или трансмиссивные (клещевой, комариный (японский), австралийский долины Муррея, американский Сент-Луис); вызванные неизвестным вирусом (эпидемический);
- микробные и риккетсиозные (при сифилисе, болезни Лайма, сыпном тифе и др.);
- коревые, краснушные, ветряночные;
- поствакцинальные;
- бактериальные и паразитарные (стафилококковый, стрептококковый, туберкулезный, токсоплазменный, хламидийный, малярийный и др.)
Заболевание может протекать в острой, подострой, хронической и рецидивирующей форме и поражать различные отделы головного мозга.
Различают среднее, тяжелое и крайне тяжелое течение энцефалита.
Симптомы энцефалита
Первичные полисезонные энцефалиты
Инкубационный период может варьировать от 2-3 дней до нескольких недель. Наблюдаются продромальные явления в виде сниженного аппетита, вялости, субфебрилитета. Начало заболевания происходит на фоне резкого подъема температуры. Выраженность клинических проявлений и их течение могут быть разными - от легких стертых форм до тяжелых молниеносных, при которых летальный исход наступает на 1-2-е сутки заболевания. Продолжительность острого периода составляет от 3-5 дней до нескольких недель. Тяжелое течение с выраженным и быстро прогрессирующим отеком головного мозга отмечается в основном при энцефалитах герпетической этиологии.На первый план выходит общемозговая симптоматика: рвота, судорожные пароксизмы, вялость, расстройства сознания, возможны дыхательные и сердечно-сосудистые нарушения. Преобладание общемозговых проявлений, нарушения сознания и выраженные менингеальные симптомы (т. е. вовлечение в воспалительный процесс церебральных оболочек) более характерны для детей младшего возраста.
Очаговая симптоматика зависит от локализации патологических изменений в головном мозге - выделяют стволовую, мозжечковую, полушарную формы.
Мозжечковый синдром характеризуется острым расстройством координации, тремором, мышечной гипотонией. Имеет наиболее благоприятное течение с полным регрессом указанной симптоматики и без последующей задержки психомоторного развития даже у маленьких детей.
Стволовой синдром проявляется поражением проходящих в стволе черепных нервов и пирамидных путей. Возникает специфическое нарушение походки и координации движений, вызванное патологической работой вестибулярного аппарата, присутствуют глазодвигательные нарушения (косоглазие, парез взора, двоение в глазах), резкая мышечная гипотония, сменяющаяся затем спастическим парезом рук и ног. Реже развиваются расстройство глотания, сосания, фонации. Возможны нейроэндокринные нарушения, а в тяжелых случаях - сердечные и дыхательные расстройства центрального генеза, которые могут стать причиной летального исхода.
Полушарный синдром чаще сопровождается расстройствами сознания, нарушениями ориентации в пространстве и во времени, вялостью. Возможны эпизоды психомоторного возбуждения, галлюцинаторный синдром. В первые дни возникают эпиприступы, которым, как правило, предшествуют парезы, развивающиеся остро и инсультоподобно.
Типичные симптомы начала герпетического энцефалита - лихорадка, головная боль, нарушение психических функций, эпилептические припадки, мышечная слабость, снижение памяти. Далее присоединяется нарушение поведения, речи и расстройство координации движений.
Вторичные энцефалиты при общих инфекциях
Клиническая картина гриппозного энцефалита развивается чаще в конце заболевания и даже через 1-2 недели после выздоровления: самочувствие больного снова ухудшается, повышается температура тела, возникают общемозговые симптомы (головная боль, рвота, головокружение). Возможно поражение периферической нервной системы в виде невралгии тройничного и большого затылочного нервов, пояснично-крестцового и шейного радикулита, поражения симпатических узлов. В крови определяют лейкоцитоз или лейкопению. Течение благоприятное. Заболевание продолжается от нескольких дней до месяца и заканчивается полным выздоровлением.
Коревой энцефалит развивается остро, чаще на 3-5-й день после появления сыпи. К этому моменту температура тела может уже нормализоваться, но нередко снова поднимается до высоких значений. Выявляют менингеальные симптомы, проявляющиеся головной болью, головокружением, тошнотой, рвотой, нарушением сознания (возбуждением, бредом, галлюцинациями); наблюдают поражение черепно-мозговых нервов; нарушение двигательной функции конечностей и функции тазовых органов. При тяжелом течении летальность достигает 25%, причем тяжесть энцефалита не зависит от течения кори.
![Коревой энцефалит.jpg]()
Энцефалит при ветряной оспе - тяжелое инфекционно-аллергическое заболевание. Его клиническая картина развивается на 3-7-й день после появления высыпаний (редко в более поздние сроки или до периода высыпаний) и проявляется гипертермией, коматозным состоянием, судорогами, менингеальными симптомами (головной болью, головокружением, тошнотой, рвотой, нарушением сознания), нарушением двигательных функций. Рано появляются признаки отека мозга. Течение обычно благоприятное и лишь в единичных случаях - очень тяжелое с риском летального исхода. После выздоровления могут длительно сохраняться парезы, гиперкинезы, судорожные припадки.
Энцефалит при краснухе может развиться через 1-10 (чаще 2-5 дней) после появления сыпи. Известны случаи манифестации острого энцефалита за несколько дней до появления сыпи, а также без высыпаний, что осложняет постановку диагноза. Чаще всего энцефалит развивается у детей в возрасте 3-15 лет, у взрослых случаи энцефалита описываются в качестве казуистики. Редко неврологическая симптоматика сопровождается вторичными высыпаниями.
Лептоспирозные энцефалиты характеризуются острым началом и протекают как респираторно вирусная инфекция. Наблюдается волнообразная лихорадка с болями в мышцах. В последующем в клинической картине могут преобладать симптомы поражения печени и почек, а со 2-3-й недели - поражения нервной системы в виде энцефалита или энцефаломиелита с вовлечением черепных нервов. Течение заболевания, как правило, благоприятное, иногда возможно спонтанное выздоровление.
Подострым склерозирующим лейкоэнцефалитам подвержены в основном дети и подростки до 15 лет, однако иногда болезнь регистрируется и у взрослых. Начало заболевания подострое: появляются симптомы, расцениваемые как неврастенические - рассеянность, раздражительность, утомляемость, плаксивость.
Различают 4 стадии склерозирующего лейкоэнцефалита:
- 1-я стадия продолжается до 6 месяцев: возможны изменения личности, перепады настроения или депрессия, могут присутствовать лихорадка и головная боль;
- 2-я стадия может включать судороги, мышечные спазмы, потерю зрения и слабоумие;
- 3-я стадия характеризуется быстрым прогрессированием деменции, усилением тонуса мышц при ослаблении судорожного синдрома. На этой стадии осложнения могут привести к летальному исходу;
- 4-я стадия характеризуется нарушениями дыхания, частоты сердечных сокращений и кровяного давления, что приводит к коме и смерти.
В 80% случаев заболевание длится 1-3 года, 10% больных живут дольше (до 10 лет), у 10% описано молниеносное течение, когда смерть наступает менее чем через 3 месяца. Специфического лечения нет.
Диагностика энцефалитов
Цель диагностики первичных полисезонных энцефалитов - установление вида возбудителя. Для этого проводится серия серологических исследований. Диагностическое значение имеет многократное нарастание титра антител к определенному вирусу при сравнении результатов исследования сыворотки пациента, взятой в начале заболевания и спустя 2 недели.
Энцефалит, вызванный вирусом простого герпеса, выявляют с помощью определения повышения уровня специфических антител IgM и Ig G в крови.
Синонимы: Анализ крови на антитела к герпесу 1, 2; IgM антитела к вирусу простого герпеса первого и второго типа; Антитела класса M к ВПГ-1, ВПГ-2. Нerpes simplex virus type 1 (HSV-1), Н.
Аномалии развития головного мозга ( Пороки развития головного мозга )
Аномалии развития головного мозга — это результат происходящих во внутриутробном периоде нарушений формирования отдельных церебральных структур или головного мозга в целом. Зачастую имеют неспецифическую клиническую симптоматику: преимущественно эпилептический синдром, задержку психического и умственного развития. Тяжесть клиники напрямую коррелирует со степенью поражения головного мозга. Диагностируются антенатально при проведении акушерского УЗИ, после рождения — при помощи ЭЭГ, нейросонографии и МРТ головного мозга. Лечение симптоматическое: противоэпилептическое, дегидратационное, метаболическое, психокоррегирующее.
МКБ-10
![Аномалии развития головного мозга]()
Общие сведения
Причины
Наиболее весомой причиной сбоев внутриутробного развития является влияние на организм беременной и на плод, различных вредоносных факторов, обладающих тератогенным действием. Возникновение аномалии в результате моногенного наследования встречается лишь в 1% случаев. Наиболее влиятельной причиной пороков головного мозга считается экзогенный фактор. Тератогенным эффектом обладают многие активные химические соединения, радиоактивное загрязнение, отдельные биологические факторы. Немаловажное значение здесь имеет проблема загрязнения среды обитания людей, обуславливающая поступление в организм беременной токсических химических веществ.
Различные эмбриотоксические воздействия могут быть связаны с образом жизни самой беременной: например, с курением, алкоголизмом, наркоманией. Дисметаболические нарушения у беременной, такие как сахарный диабет, гипертиреоз и пр., могут также стать причиной церебральных аномалий плода. Тератогенным действием обладают и многие медикаменты, которые может принимать женщина в ранние сроки беременность, не подозревая о происходящих в ее организме процессах. Мощный тератогенный эффект оказывают инфекции, перенесенные беременной, или внутриутробные инфекции плода. Наиболее опасны цитомегалия, листериоз, краснуха, токсоплазмоз.
Патогенез
Дифференцировка нейробластов (зародышевых нервных клеток) приводит к образованию нейронов, формирующих серое вещество, и глиальных клеток, составляющих белое вещество. Серое вещество отвечает за высшие процессы нервной деятельности. В белом веществе проходят различные проводящие пути, связывающие церебральные структуры в единый функционирующий механизм. Рожденный в срок новорожденный имеет такое же число нейронов, как и взрослый человек. Но развитие его мозга продолжается, особенно интенсивно в первые 3 мес. жизни. Происходит увеличение глиальных клеток, разветвление нейрональных отростков и их миелинизация.
Сбои могут произойти на различных этапах формирования головного мозга. Если они возникают в первые 6 мес. беременности, то способны приводить к снижению числа сформированных нейронов, различным нарушениям в дифференцировке, гипоплазии различных отделов мозга. В более поздние сроки может возникать поражение и гибель нормально сформировавшегося церебрального вещества.
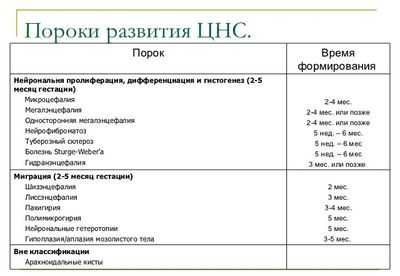
Виды аномалий мозга
Анэнцефалия — отсутствие головного мозга и акрания (отсутствие костей черепа). Место головного мозга занято соединительнотканными разрастаниями и кистозными полостями. Может быть покрыто кожей или обнажено. Патология несовместима с жизнью.
Энцефалоцеле — пролабирование церебральных тканей и оболочек через дефект костей черепа, обусловленный его незаращением. Как правило, формируется по средней линии, но бывает и асимметричным. Небольшое энцефалоцеле может имитировать кефалогематому. В таких случаях определить диагноз помогает рентгенография черепа. Прогноз зависит от размеров и содержимого энцефалоцеле. При небольших размерах выпячивания и наличии в его полости эктопированной нервной ткани эффективно хирургическое удаление энцефалоцеле.
Микроцефалия — уменьшение объема и массы головного мозга, обусловленное задержкой его развития. Встречается с частотой 1 случай на 5 тыс. новорожденных. Сопровождается уменьшенной окружностью головы и диспропорциональным соотношением лицевого/мозгового черепа с преобладанием первого. На долю микроцефалии приходится около 11% всех случаев олигофрении. При выраженной микроцефалии возможна идиотия. Зачастую наблюдается не только ЗПР, но и отставание в физическом развитии.
Макроцефалия — увеличение объема головного мозга и его массы. Гораздо менее распространена, чем микроцефалия. Макроцефалия обычно сочетается с нарушениями архитектоники мозга, очаговой гетеротопией белого вещества. Основное клиническое проявление — умственная отсталость. Может наблюдаться судорожный синдром. Встречается частичная макроцефалия с увеличением лишь одного из полушарий. Как правило, она сопровождается асимметрией мозгового отдела черепа.
Кистозная церебральная дисплазия — характеризуется множественными кистозными полостями головного мозга, обычно соединенными с желудочковой системой. Кисты могут иметь различный размер. Иногда локализуются только в одном полушарии. Множественные кисты головного мозга проявляются эпилепсией, устойчивой к антиконвульсантной терапии. Единичные кисты в зависимости от размера могут иметь субклиническое течение или сопровождаться внутричерепной гипертензией; зачастую отмечается их постепенное рассасывание.
Голопрозэнцефалия — отсутствие разделения полушарий, в результате чего они представлены единой полусферой. Боковые желудочки сформированы в единую полость. Сопровождается грубыми дисплазиями лицевого черепа и соматическими пороками. Отмечается мертворождение или гибель в первые сутки.
Агирия (гладкий мозг, лиссэнцефалия) — отставание развития извилин и тяжелое нарушение архитектоники коры. Клинически проявляется выраженным расстройством психического и моторного развития, парезами и различными формами судорог (в т. ч. синдромом Веста и синдромом Леннокса-Гасто). Обычно заканчивается летальным исходом на первом году жизни.
Пахигирия — укрупнение основных извилин при отсутствии третичных и вторичных. Сопровождается укорочением и выпрямлением борозд, нарушением архитектоники церебральной коры.
Микрополигирия — поверхность коры мозга представлена множеством мелких извилин. Кора имеет до 4-х слоев, тогда как в норме кора насчитывает 6 слоев. Может быть локальной или диффузной. Последняя, полимикрогирия, характеризуется плегией мимических, жевательных и глоточных мышц, эпилепсией с дебютом на 1-ом году жизни, олигофренией.
Гипоплазия/аплазия мозолистого тела. Часто встречается в виде синдрома Айкарди, описанного только у девочек. Характерны миоклонические пароксизмы и сгибательные спазмы, врожденные офтальмические пороки (колобомы, эктазия склеры, микрофтальм), множественные хориоретинальные дистрофические очаги, обнаруживаемые при офтальмоскопии.
Фокальная корковая дисплазия (ФКД) — наличие в коре головного мозга патологических участков с гигантскими нейронами и аномальными астроцитами. Излюбленное расположение — височные и лобные зоны мозга. Отличительной особенностью эпиприступов при ФКД является наличие кратковременных сложных пароксизмов с быстрой генерализацией, сопровождающихся в своей начальной фазе демонстративными двигательными феноменами в виде жестов, топтания на одном месте и т. п.
Гетеротопии — скопления нейронов, на этапе нейронной миграции задержавшихся на пути своего следования к коре. Гетеротопионы могут быть единичными и множественными, иметь узловую и ленточную форму. Их главное отличие от туберозного склероза — отсутствие способности накапливать контраст. Эти аномалии развития головного мозга проявляются эписиндромом и олигофренией, выраженность которых прямо коррелирует с числом и размером гетеротопионов. При одиночной гетеротопии эпиприступы, как правило, дебютируют после 10-летнего возраста.
Диагностика
Тяжелые аномалии развития головного мозга зачастую могут быть диагностированы при визуальном осмотре. В остальных случаях заподозрить церебральную аномалию позволяет ЗПР, гипотония мышц в неонатальном периоде, возникновение судорожного синдрома у детей первого года жизни. Исключить травматический или гипоксический характер поражения головного мозга можно при отсутствии в анамнезе данных о родовой травме новорожденного, гипоксии плода или асфиксии новорожденного. Пренатальная диагностика пороков развития плода осуществляется путем скринингового УЗИ при беременности. УЗИ в I триместре беременности позволяет предупредить рождение ребенка с тяжелой церебральной аномалией.
Одним из методов выявления пороков головного мозга у грудничков является нейросонография через родничок. Намного более точные данные у детей любого возраста и у взрослых получают при помощи МРТ головного мозга. МРТ позволяет определить характер и локализацию аномалии, размеры кист, гетеротопий и других аномальных участков, провести дифференциальную диагностику с гипоксическими, травматическими, опухолевыми, инфекционными поражениями мозга. Диагностика судорожного синдрома и подбор антиконвульсантной терапии осуществляется при помощи ЭЭГ, а также пролонгированного ЭЭГ-видеомониторинга. При наличии семейных случаев церебральных аномалий может быть полезна консультация генетика с проведением генеалогического исследования и ДНК-анализа. С целью выявления сочетанных аномалий проводится обследование соматических органов: УЗИ сердца, УЗИ брюшной полости, рентгенография органов грудной полости, УЗИ почек и пр.
Лечение аномалий мозга
Терапия пороков развития головного мозга преимущественно симптоматическая, осуществляется детским неврологом, неонатологом, педиатром, эпилептологом. При наличии судорожного синдрома проводится антиконвульсантная терапия (карбамазепин, леветирацетам, вальпроаты, нитразепам, ламотриджин и др.). Поскольку эпилепсия у детей, сопровождающая аномалии развития головного мозга, обычно резистентна к противосудорожной монотерапии, назначают комбинацию из 2 препаратов (например, леветирацетам с ламотриджином). При гидроцефалии осуществляют дегидратационную терапию, по показаниям прибегают к шунтирующим операциям. С целью улучшения метаболизма нормально функционирующих мозговых тканей, в какой-то степени компенсирующих имеющийся врожденный дефект, возможно проведение курсового нейрометаболического лечения с назначением глицина, витаминов гр. В и пр. Ноотропные препараты используются в лечении только при отсутствии эписиндрома.
При умеренных и относительно легких церебральных аномалиях рекомендована нейропсихологическая коррекция, занятия ребенка с психологом, комплексное психологическое сопровождение ребенка, детская арт-терапия, обучение детей старшего возраста в специализированных школах. Указанные методики помогают привить навыки самообслуживания, уменьшить степень выраженности олигофрении и по возможности социально адаптировать детей с церебральными пороками.
Прогноз и профилактика
Прогноз во многом определяется тяжестью церебральной аномалии. Неблагоприятным симптомом выступает ранее начало эпилепсии и ее резистентность к осуществляемой терапии. Осложняет прогноз наличие сочетанной врожденной соматической патологии. Эффективной мерой профилактики служит исключение эмбриотоксических и тератогенных влияний на женщину в период беременности. При планировании беременности будущим родителям следует избавиться от вредных привычек, пройти генетическое консультирование, обследование на наличие хронических инфекций.
Микроцефалия
Микроцефалия - недоразвитие черепа и головного мозга, сопровождающее умственной отсталостью и неврологическими отклонениями. Микроцефалия характеризуется малыми размерами черепа, ранним смыканием черепных швов и закрытием родничка, судорожным синдромом, задержкой моторного развития, интеллектуальным дефектом, недоразвитием или отсутствием речи. Диагностика микроцефалии основывается на данных антропометрии, краниографии, КТ и МРТ головного мозга, ЭЭГ, НСГ; возможно пренатальное выявления микроцефалии у плода. При микроцефалии проводится симптоматическое лечение и реабилитационные мероприятия, направленные на социализацию ребенка.
![Микроцефалия]()
Микроцефалия (микрокефалия) - тяжелый порок развития ЦНС, в основе которого лежит уменьшение массы головного мозга и уменьшение окружности черепа более чем на два-три сигмальных отклонения по сравнению со средними половозрастными показателями. Различные формы микроцефалии встречаются с частотой 1 случай на 10000 детей, в равных соотношениях среди мальчиков и девочек. Микроцефалия является причиной олигофрении в 10% наблюдений. При рождении окружность головы у ребенка с микроцефалией, как правило, не превышает 25-27 см (при норме - 35-37 см), а масса головного мозга - 250 г (в норме около 400 г).
Причины микроцефалии
С учетом времени и причин возникновения в педиатрии и детской неврологии выделяют первичную (наследственную, истинную) и вторичную (синдромальную и эмбриопатическую) микроцефалию. Первичная микроцефалия является компонентом наследственных болезней с аутосомно-рецессивным и рецессивным, сцепленным с полом типами наследования (синдром Джакомини, синдром Пейна). На долю истинной микроцефалии приходится 7-34% от всех форм патологии.
Вторичная микроцефалия отмечается при хромосомных аберрациях, наследственных энзимопатиях (фенилкетонурии), патологии беременности и родов. Синдромальная микроцефалия встречается более чем при 125 хромосомных аномалиях, наиболее частыми из которых являются болезнь Дауна (трисомия по 21 хромосоме), синдром Эдвардса (трисомия по 18 хромосоме), синдром Патау (трисомия по 13 хромосоме), синдром «кошачьего крика» (моносомия 5р) и др.
Вторичная эмбриопатическая микроцефалия обусловлена воздействием на плод тератогенных факторов и может являться следствием внутриутробных инфекций (краснухи, цитомегаловирусного энцефалита, герпеса, токсоплазмоза) и интоксикаций (алкогольной, наркотической, профессиональной), радиационного влияния, гипоксии, внутричерепных родовых травм, метаболических нарушений, гормональных заболеваний матери (сахарного диабета, тиреотоксикоза).
Микроцефалия у детей часто сочетается с другими аномалиями: расщелинами губы и неба («заячьей губой» и «волчьей пастью»), несовершенным остеогенезом, врожденной катарактой, пигментным ретинитом, первичной кардиомиопатией, лимфедемой, врожденными пороками сердца и легких, гипоплазией почек, что в значительной мере отягощает прогноз.
Патоморфологическое исследование головного мозга при микроцефалии выявляет уменьшение его массы свыше 25% от нормы, недоразвитие больших полушарий, особенно лобных отделов. При микроцефалии могут иметь место явления микро- или макрогирии (аномально узких либо широких извилин), лиссенцефалии или агирии (сглаженности либо отсутствия извилин), порэнцефалии (наличия патологических кистозных полостей в ткани мозга); агенезия мозолистого тела, расширение ликворных пространств, умеренная гидроцефалия, нарушения миелинизации.
Симптомы микроцефалии
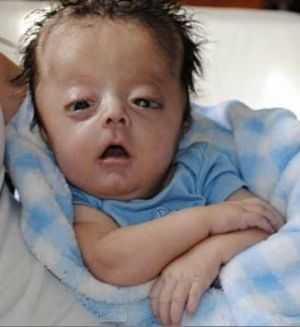
Объем черепа у ребенка с микроцефалией уменьшен уже при рождении, в дальнейшем его развитие заметно отстает от возрастной нормы. Отмечается преобладание лицевого черепа над мозговым. Типичный внешний вид больного с микроцефалией характеризуется узким и скошенным лбом, выступающими надбровными дугами, большими ушами. Большой родничок и черепные швы закрываются уже в первые месяцы жизни. В дальнейшем больные с микроцефалией обычно отстают в массе и росте (вплоть до карликовости), имеют диспропорциональное телосложение, узкое высокое (готическое) небо, большие редкие зубы.
Неврологические нарушения при микроцефалии могут включать мышечную дистонию, спастические парезы, атаксию, судороги, косоглазие. Часто дети с микроцефалией могут страдать эпилепсией и детским церебральным параличом. Дети с микроцефалией поздно начинают держать головку, сидеть, ползать, ходить. Отмечается грубая задержка речевого развития, нечеткость артикуляции, резкая ограниченность словарного запаса, нарушение понимания обращенной речи.
Степень интеллектуальных нарушений у ребенка с микроцефалией может варьировать от дебильности до идиотии. При нерезко выраженной умственной отсталости больные с микроцефалией могут быть обучаемы, способны к самообслуживанию и выполнению несложных поручений. Однако в большинстве случаев дети с микроцефалией требуют ухода, контроля и надзора со стороны взрослых.
По особенностям темперамента дети с микроцефалией могут быть отнесены к торпидной или эретической группе. В первом случае детям свойственна малоподвижность, вялость, безучастность к окружающему, пассивно-подражательная деятельность; во втором случае - гиперактивность, суетливость, подвижность, неустойчивое внимание. Эмоциональная сфера у больных с микроцефалией остается относительно сохранной: дети приветливы, добродушны; реже - эмоционально неустойчивы и склонны к аффективным вспышкам.
Диагностика микроцефалии
Пренатальная диагностика микроцефалии основана на сравнении биометрических параметров плода, получаемых в процессе динамического ультразвукового наблюдения. Однако чувствительность акушерского УЗИ в диагностике микроцефалии составляет всего 67%, а сам порок выявляется только после 27-30 недель беременности. Поэтому при подозрении на микроцефалию, связанную с хромосомной или генетической патологией, УЗИ-скрининг всегда должен дополняться инвазивной пренатальной диагностикой (биопсией хориона, амниоцентезом или кордоцентезом) и кариотипированием плода.
После рождения диагноз микроцефалии подтверждается на основании визуального осмотра новорожденного: уменьшения окружности головы более чем на 2SD-3SD от средней нормы, диспропорции лицевой и мозговой частей черепа. Дети с микроцефалией должны быть проконсультированы генетиком на предмет выявления наследственных заболеваний.
Для определения степени и прогноза микроцефалии важно проведение полного инструментального неврологического обследования: нейросонографии, ЭЭГ, ЭхоЭГ, КТ и МРТ головного мозга. Рентгенография черепа позволяет дифференцировать микроцефалию от краниосиностоза.
Лечение микроцефалии
Патогенетического лечения микроцефалии не существует, поэтому медицинская помощь в основном сводится к симптоматической поддержке больных. На регулярной основе показано проведение медикаментозных курсов, улучшающих обменные процессы в мозговой ткани (пирацетам, пиритинол, витаминные комплексы), по показаниям - противосудорожные и седативные препараты. В рамках реабилитационных мероприятий детям с микроцефалией необходимы занятия лечебной физкультурой, массаж, трудотерапия.
Дети с микроцефалией нуждаются в диспансерном наблюдении педиатра и детского невролога, ежемесячной антропометрии. Воспитание и обучение детей с микроцефалией проводится специалистами дефектологами; коррекция системного недоразвития речи - логопедами. Реабилитационные мероприятия направлены на максимальную адаптацию и социализацию детей с микроцефалией.
Прогноз и профилактика микроцефалии
Прогноз в отношении продолжительности жизни и социализации вариабелен. Некоторые дети способны к обучению в коррекционной школе, овладению элементарными навыками самообслуживания. В целом прогноз при микроцефалии неблагоприятен: продолжительность жизни таких пациентов снижена, большинство из них пожизненно находятся в специнтернатах для умственно-отсталых.
Профилактика микроцефалии у детей предусматривает тщательное планирование беременности, обследование на инфекции (TORCH-комплекс, ПЦР), антенатальную охрану плода. Раннее внутриутробное выявление микроцефалии является основанием для решения вопроса об искусственном прерывании беременности. Медико-генетическое консультирование семей, имеющих детей с микроцефалией, необходимо для оценки потенциального риска при последующих беременностях.
Микроцефалия у детей: симптомы, лечение и причины заболевания
Микроцефалия у детей - тяжёлое состояние, при котором наблюдается уменьшение черепной коробки и объёмов её внутреннего содержимого. Заболевание часто протекает с умственной отсталостью, неврологическими проблемами, способно спровоцировать осложнения в виде олигофрении, привести к сокращению жизненных лет больного. Патологию ЦНС диагностируют у 1 из 10000 младенцев.
![Микроцефалия у детей]()
Симптомы микроцефалии у детей
Код нарушения по МКБ-10 - Q02 Микроцефальный синдром диагностируют у малышей обоих полов. Признаки аномалии обнаруживают у грудничков сразу после появления на свет.
Основными симптомами имеющегося порока являются уменьшенный размер черепа, малый вес головного мозга:
Клиническая картина микроцефалии у ребенка дополняется другими признаками:
- недостаточной телесной массой;
- наличием зауженного лба, выступающих надбровных дуг;
- недоразвитием нёба;
- непропорциональностью телосложения;
- низким ростом, карликовостью.
У пациентов-микроцефалов рост лицевого черепа проходит намного быстрее, чем развитие мозговой части головы. Как выглядит больной ребёнок, смотрите на фото.
![Дети микроцефалы]()
![Микроцефалия у ребёнка]()
У некоторых новорожденных патология протекает с косоглазием, судорожным синдромом, нарушениями координации движений, парезами. По мере взросления у пациента возникают проблемы с речью, наблюдается отставание в плане моторного развития, неспособность к элементарному самообслуживанию.
Важно! Микроцефалия часто комбинируется с заболеваниями, тяжело поддающимися лечению - ДЦП, эпилепсией, аутизмом, идиотией, дебильностью.
Причины развития первичной и вторичной микроцефалии
Первичная форма микроцефалии развивается у малышей, родившихся от родителей с дефектными генами. Этот вид нарушения диагностируют у 35% больных. Признаки аномалии фиксируют уже в раннем периоде внутриутробного развития.
Вторичная микроцефалия не связана с генетикой матери или отца.
![Беременная девушка]()
Эта форма заболевания ассоциируется со следующими факторами:
- инфекциями, передающимися от беременной женщины эмбриону (токсоплазмозом, краснухой, корью);
- употреблением на этапе вынашивания спиртного, наркотических веществ;
- лечением медикаментами, запрещёнными при беременности (противоопухолевыми и другими токсичными препаратами);
- неправильным питанием будущей матери (наибольший вред наносит недостаток белковой пищи);
- кислородным голоданием малыша, находящегося в утробе;
- радиационным излучением, вызывающим мутации в организме плода.
Диагностика патологии у ребёнка и плода
Диагностика детей с микроцефалией находятся в сфере компетенции невролога. Медицинские обследования, необходимые для выявления патологии, периодически назначают женщинам в период вынашивания. В большинстве случаев аномалию обнаруживают на плановом УЗИ.
![УЗИ]()
После рождения ребёнка потребуется изучение анамнеза отца и матери. Новорожденного с маленьким мозгом обследуют с целью подтверждения или опровержения аномалии. Начальная диагностика состоит в измерении массы и длины тела пациента, окружности головы и грудины. Полученные результаты сверяются с показателями центильных таблиц.
Грудничкам назначают ряд информативных процедур, позволяющих определить важные особенности болезни:
- анализы крови;
- биопсию хориона;
- нейросонографию;
- генетическое исследование методом ПЦР;
- УЗИ мозга;
- электроэнцефалографию;
- МРТ, КТ;
- эхоэнцефалографию.
Врождённая или приобретённая микроцефалия принадлежит к неизлечимым заболеваниям. Детям с такой патологией назначают симптоматическое лечение, обучают навыкам самообслуживания. Проводится работа, направленная на развитие интеллектуальных способностей пациентов, социальную адаптацию. Улучшению состояния микроцефалов способствуют физиотерапевтические процедуры, трудотерапия.
![Дефектолог]()
Воспитание и обучение детей-микроцефалов осуществляются специалистами в области дефектологии. Коррекцией речевых дефектов занимаются логопеды.
Пациенты с микроцефалией периодически проходят процедуру антропометрии, состоящую в измерении объёмов головы.
Лекарства
Детям, имеющим малый размер черепной коробки, показаны регулярные курсы с препаратами из следующего списка:
- ноотропами, улучшающими обмен веществ в тканях мозга, повышающими умственные способности (Пирацетамом, Энцефаболом);
- лекарствами, применяемыми при возникновении судорожных приступов (Реланиумом, магния сульфатом);
- антигипоксантами, имеющими нейропротекторные и микроциркуляторные свойства, предотвращающими кислородное голодание (Актовегином, Кавинтоном);
- дегидратационными средствами, снижающими риск развития мозгового отёка (Фуросемида, Маннитола).
При повышенной нервной возбудимости применяют седативные средства растительного происхождения (отвар пустырника, настой валерианового корня). Снятию агрессивности, тревожности способствуют нейролептики, транквилизаторы (Сонапакс, Диазепам). Для нормального функционирования ЦНС показаны витамины группы В.
Физиотерапия
При приобретённой или врождённой микроцефалии назначают физиотерапевтические процедуры:
- массаж;
- магнитотерапию;
- гальванизацию;
- лечебные ванны;
- ЛФК.
Физиотерапия поможет улучшить кровообращение и общее состояние нервной системы, сократить интенсивность двигательных нарушений, повысить тонус мышц. Процедуры обязательно сочетаются с медикаментозным курсом.
Прогноз при микроцефалии
Для большинства пациентов с микроцефальным синдромом прогноз неблагоприятен. Продолжительность жизни часто не превышает 15 лет. Некоторые больные живут до наступления 30-летия. К летальному исходу приводят нарушения в работе внутренних органов и систем, возникающие из-за неполноценного функционирования головного мозга.
![Ребёнок микроцефал на руках у девушки]()
Если комплексное лечение микроцефалии оказалось эффективным, у ребёнка улучшаются память и внимание, расширяется лексикон, повышается уровень работоспособности, нормализуется эмоциональное состояние, совершенствуется двигательная активность.
При выявлении патологии на ранних сроках - рекомендуют прерывание беременности.
Чтобы снизить риск развития болезни у ребёнка, будущим родителям рекомендуется придерживаться здорового образа жизни, укреплять иммунитет. На этапе подготовки к беременности необходимо проходить детальное медицинское обследование и корректное лечение обнаруженных TORCH-инфекций.
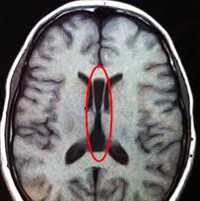
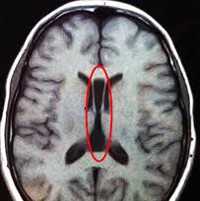
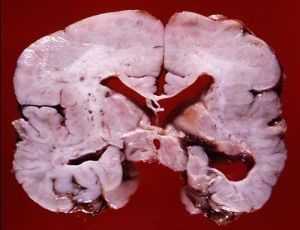
Мальформации головного мозга: полимикрогирия, агирия и пахигирия
![пахигирия разрез мозга]()
Полимикрогирия, агирия и пахигирия относятся к врожденным порокам развития головного мозга, также называются мальформациями головного мозга.
Данные аномалии возникают в результате нарушенного процесса формирования головного мозга или отдельных его структур во время пренатального периода, и представляют собой аномальные изменения в строении структур головного мозга.
Не существует точных данных о распространенности врожденных пороков головного мозга, однако наиболее распространенной считается полимикрогирия.
![Пороки развития ЦНС]()
О каждой мальформации подробней:
![синдром Уокера-Варбурга]()
- Полимикрогирия - редко встречающаяся патология развития коры головного мозга, при которой на поверхности больших полушарий формируется значительное количество мелких и недоразвитых извилин. Часто сочетается с другими генетическими патологиями и аномалиями развития коры головного мозга. Наиболее часто патология развивается в области сильвиевой борозды (60% случаев заболевания), однако может возникать в любой части коры головного мозга. Патология бывает диффузной, мультифокальной и очаговой. Также ее можно разделить на одностороннюю (40% случаев) и двустороннюю (60% случаев), симметричную и ассиметричную.
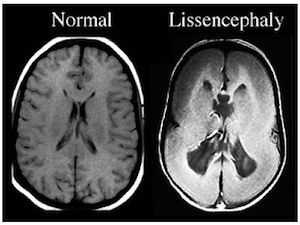
- Агирия (лиссэнцефалия) - аномалия формирования коры головного мозга, при которой извилины недоразвиты, слабо выражены, либо отсутствуют, архитектоника коры нарушена. Внешний вид головного мозга ребенка идентичен внешнему виду мозга плода на 3-4 месяце пренатального периода. Агирия является тяжелой формой лиссэнцефалии. Может быть как идиопатическим заболеванием, так и сопровождать другие патологии (синдром Миллера-Дикера, синдром Нормана-Робертса, синдром Уокера-Варбурга, врожденную мышечную дистрофию Фукуямы).
- Пахигирия является редкой аномалией развития центральной нервной системы, при которой в коре больших полушарий головного мозга формируется небольшое количество широких и плоских извилин. При пахигирии основные извилины укрупнены, а вторичные и третичные отсутствуют, борозды укорочены и выпрямлены, архитектоника церебральной коры нарушена. Данную патологию рассматривают как «неполную», более легкую форму лиссэнцефалии.
Провоцирующие факторы
Основная причина развития полимикрогирии, пахигирии и агирии в неправильном протекании процесса нейрональной миграции, причиной чего являются:
- генетические нарушения;
- вирусные инфекции;
- недостаточное кровоснабжение головного мозга ребенка в пренатальный период (2 триместр беременности).
Полимикрогирия может быть вызвана цитомегаловирусной инфекцией, недостаточным насыщением плаценты кислородом, а мутации происходят в генах COL18A1 (21q22.3), PAX6 (11p13), K1AA1279 (10q22.1), TUBB2B (6p25), EOMES (3p21.3-p21.2), RAB3GAP1 (2q21.3), SRPX2 (Xq21.33-q23).
Агирия может возникнуть из-за вирусных инфекций, которые проникают в матку или в плод в период первого триместра, недостаточного кровоснабжения мозга плода в первые месяцы беременности. Среди генетических факторов можно назвать мутации в гене RELN (7q22), генах X хромосомы, 17 хромосомы.
При пахигирии возникают мутации в генах LIS1 (17p13.3) и DCX (Xq22.3-q23).
Характерная симптоматика
Для полимикрогирии характерны такие симптомы:
- тяжелые задержки в развитии;
- гипоплазия головного мозга;
- нарушения в лицевых мышцах, а также мышцах отвечающих за глотание, жевания, движения языком;
- умственные отклонения разной степени тяжести;
- квадрипарез и гемипарез конечностей;
- двигательные нарушения;
- осложнения дыхания;
- судороги;
- эпилепсия;
- псевдобульбарный синдром;
- артрогрипоз;
- церебральный паралич.
Пахигирия проявляет себя в возрасте до 2 лет. Основными ее признаками являются:
![Микроцефалия]()
- задержка физического, двигательного развития;
- эпилепсия;
- гипотония (пониженный мышечный тонус);
- умственная отсталость;
- инфантильные спазмы;
- микроцефалия;
- низкий контроль мышц;
- трудности при глотании, проблемы с приемом пищи.
Также аномалия может проявлять себя следующими признаками:
- микроцефалия (пропорционально небольшой размер головы);
- отставание в росте, в речевом и двигательном развитии;
- общее вялое состояние ребенка;
- большое расстояние между глазами, диспропорционально маленькие глаза, тонкая верхняя губа, высокий лоб, короткий нос, низко расположенные уши;
- эпилепсия, судорожный синдром;
- отставание в умственном развитии;
- повышенный мышечный тонус, из-за отсутствия тормозящего контроля центральной нервной системы над нейронами передних рогов спинного мозга;
- увеличенное расстояние между легкими, почками;
- стремительно развивающееся слабоумие;
- отсутствие контроля над органами малого таза (энурез, недержание кала);
- мышечная дистрофия.
Фото мозга в разрезе и на МРТ ребенка с диагнозом агирия
Постановка диагноза
Для диагностики мальформаций головного мозга применяют такие исследования:
- компьютерная томография (определяет нарушения в сером и белом веществе головного мозга);
- генетические анализы;
- магнитно резонансная томография (помогает определить тип заболевания);
- электроэнцефалография (помогает определить характер электрической активности мозга).
![мальформация и нормальный мозг]()
При полимикрогирии кора головного мозга аномально тонкая, увеличены субарахноидальные пространства и желудочки мозга. Поражения могут располагаться с одной или двух сторон, могут быть локальными или обширными. Кора может выглядеть аномально толстой из-за большого количества мелких извилин, которые сливаются и образуют толстые, глубокие массы.
Пахигирию определяют по результатам МРТ, на которых видны утолщения коры головного мозга, небольшие извилины, и недоразвитая сильвиевая борозда.
В пренатальный период определить мальформации мозга возможно при помощи УЗИ плода. При подозрениях на аномалии в развитии направляют на дополнительные исследования (МРТ, генетический анализ генов-кандидатов).
Самым ранним сроком, на котором можно определить данные патологии, является двадцатая неделя беременности.
Медицина практически бессильна
Эффективные методы лечения мальформаций головного мозга отсутствуют в современной медицине. Терапия направлена на облегчение симптомов, и поддержание функционирования организма на приемлемом уровне.
При полимикрогирии и пахигирии рекомендованы такие методы облегчения состояния пациентов:
- физиотерапия, в том числе водные процедуры;
- логопедические методы;
- ношение ортезов;
- трудотерапия (при нарушениях моторных функций);
- прием противосудорожных препаратов;
- хирургические методы.
![Сирдалуд]()
При агирии неэффективными считаются классические методы терапии: прием ноотропных препаратов, массаж, физиотерапия.
Наибольшую эффективность показывает лечение противосудорожными препаратами и миорелаксантами центрального действия, которые позволяют снизить тонус в скелетных мышцах.
Часто эпилепсия, сопровождающая патологии развития головного мозга, оказывается резистентной к терапии, и тогда рекомендуется использовать комбинацию из двух препаратов.
Проводятся исследования по лечению данных аномалия при помощи пересадки стволовых клеток, трансплантации факторов роста.
Результаты показали возможность стабилизировать состояние пациентов, однако выздоровления добиться не удалось. Данные методы имеют достаточно высокий уровень риска и имеют плохой прогноз, поэтому применение стволовых клеток не имеет широкого распространения.
Прогноз и продолжительность жизни
Пациенты с полимикрогирией, имеющие легкие формы патологии, могут дожить до взрослого возраста. При тяжелых формах летальный исход наступает в раннем и молодом возрасте по причине пневмонии или судорог.
Для пациентов с пахигирией не существует единого прогноза. Он может варьироваться в каждом конкретном случае и зависит от тяжести патологии головного мозга, и интенсивности симптомов. Но обычно дети с пахигирией живут не дольше 20 лет.
Недоразвитие мозга у детей при агирии необратимое и прогноз неблагоприятный. Большая часть пациентов не доживает до возраста 5 лет. У всех больных присутствует полная инвалидизация.
Из-за угнетенных глотательных рефлексов часто требуется зондовое питание. Летальный исход может наступить по причине заболеваний, возникающих на фоне основной патологии: парез кишечника, сердечно-сосудистая недостаточность, дыхательная недостаточность, атрофия дыхательной мускулатуры, гипостатическая пневмония.
Читайте также:
- КТ, МРТ при карциноме слезной железы
- Агенезия мозолистого тела. Отсутствие (агенезия) черепных нервов
- Классификация эпилепсии и эпилептических припадков у детей
- Серологическая диагностика золотистого стафилококка. Типирование бактериофагами золотистого стафилококка. Определение чувствительности золотистого стафилококка к антибиотикам. Лечение и профилактика стафилококковых инфекций.
- Прогрессирующая колбочковая дистрофия