Нервная система при муколипидозах и сиалидозах
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Что такое Псевдогурлеровская полидистрофия (муколипидоз тип III) -
Псевдогурлеровская полидистрофия (муколипидоз тип III) - вариантная форма болезни I-клеток; отличается лишь клинической картиной и течением, а биохимические нарушения сходны.
Симптомы Псевдогурлеровской полидистрофии (муколипидоза типа III):
Проявляется в возрасте 2-4 лет тугоподвижностью межфаланговых, локтевых, тазобедренных и коленных суставов, в связи с чем обычно предполагают ювенильный ревматоидный артрит. Однако симптомы воспаления отсутствуют. Рост замедляется в пубертатном периоде, возможны незначительная задержка психомоторного развития, психические расстройства. Отмечаются легкое огрубление черт лица, помутнение роговицы (в 7-8 лет), признаки множественного дизостоза на рентгенограммах, прогрессирующая деструкция тазобедренных суставов.
К каким докторам следует обращаться если у Вас Псевдогурлеровская полидистрофия (муколипидоз тип III):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Псевдогурлеровской полидистрофии (муколипидоза типа III), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Дифференциальная диагностика мукополисахаридозов
Валентина Мохор, «МВ». 25.01.2022 Фото носит иллюстративный характер. Из открытых источников.
О мукополисахаридозах (МПС), редких наследственных заболеваниях обмена веществ из группы лизосомных болезней накопления, в последние годы говорят часто. Еще недавно схожесть симптоматики МПС с другими заболеваниями и недостаточная лабораторная диагностика часто приводили к ошибочным диагнозам и неадекватному лечению. Вместе с тем ранняя диагностика мукополисахаридозов и правильная терапия во многом определяют прогноз заболевания и перспективы пациента.
Основа патогенеза МПС — накопление в органах и тканях патологического субстрата, а именно гликозаминогликанов (ГАГ), или мукополисахаридов, которые являются базовым компонентом соединительной ткани. Избыточное накопление ГАГ приводит к дисфункции клеток, органов и тканей и вызывает разнообразные клинические проявления. МПС относится к мультисистемным заболеваниям, поэтому дифференциальная диагностика данной нозологии актуальна для врачей любых специальностей, особенно педиатрического профиля.

Опытом правильной диагностики МПС (на примере синдрома Хантера как наиболее распространенного типа мукополисахаридоза) поделилась старший научный сотрудник лаборатории редких наследственных болезней у детей Центра редких болезней ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России, кандидат мед. наук Татьяна Подклетнова.
Объективная лабораторная диагностика
Основа диагностики — клинические проявления заболевания. Все типы МПС имеют общие внешние клинические признаки, которые могут и должны вызвать у врача на амбулаторном приеме настороженность в отношении МПС: гаргоилизм, костные изменения, органомегалия и т. д.
Лабораторная диагностика включает три этапа.
1. Количественная и качественная оценка экскреции ГАГ в моче. Неспецифический тест, который с высокой степенью вероятности позволяет заподозрить у пациента МПС, не дает, однако, четкой информации о типе заболевания.
При легких формах заболевания избыточное количество ГАГ в моче может не определяться, следовательно, тест будет ложно-отрицательным. На основании только этого лабораторного анализа ставить окончательный диагноз нельзя!
2. Энзимодиагностика. Измерение активности фермента в лейкоцитах крови. При МПС любого типа снижается уровень определенного фермента в лейкоцитах или плазме крови. В частности, при МПС 2-го типа снижается уровень идуронат-2-сульфатазы в плазме (норма: 250-620 nM/mg/h); МПС 2-го типа: 0-20 nM/mg/h); при МПС 1-го типа — α-L-идуронидазы в периферической крови (норма: 61-176 nM/mg/h; МПС 1-го типа: 0-18 nM/mg/h) и т. д. Однако существует ряд заболеваний, при которых уровень ферментов в крови также может снижаться, например, множественная сульфатазная недостаточность. Необходим третий этап.
3. Молекулярно-генетическая диагностика. Наличие мутаций в том или ином гене дает окончательный ответ и возможность установить объективный диагноз. Так, МПС 1-го типа обусловлен мутацией в гене IDUA, МПС 2-го типа — мутацией в гене IDS и т. д.
Существуют и другие методы диагностики, которые обязательно должны быть предложены семьям, планирующим рождение ребенка, но у которых уже есть случаи МПС среди близких родственников. Речь о пренатальной и предимплантационной диагностике.
Пренатальная диагностика проводится между 10-й и 12-й неделями уже наступившей беременности. Выполняется определение ферментной активности в ворсинах хориона, проводится молекулярно-генетическая диагностика плода. Надежность метода достигает 99 %. У родителей есть возможность при выявлении патологии прервать беременность на малом сроке. Или же принять осознанное решение о рождении ребенка с генетическим заболеванием. В этом случае ранняя пренатальная диагностика дает возможность начать адекватное лечение на самом раннем этапе, что, естественно, влечет за собой совершенно иные, более благоприятные прогнозы относительно качества и продолжительности жизни ребенка.
Предимплантационная диагностика проводится перед искусственным оплодотворением и позволяет избежать рождения ребенка с данной генетической патологией.
Шаг 1. Дифдиагностика с другими видами мукополисахаридозов
В современной классификации существуют 7 основных типов мукополисахаридозов. Наиболее часто (помимо МПС 2-го типа) встречаются МПС 1-го, 3-го, 4-го, 6-го типов.
Особенности МПС 1-го типа (синдром Гурлера, синдром Гурлера — Шейе, синдром Шейе):
- гипертрофия десен, редкие мелкие зубы, макроглоссия;
- частые респираторные инфекции, отиты, бронхиты;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки, глаукома;
- низкий рост, кифосколиоз, деформация грудной клетки, врожденный вывих бедра, контрактуры крупных и мелких суставов;
- синдром запястного канала;
- цервикальный стеноз;
- шумное носовое дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- задержка психомоторного развития, регресс навыков;
- экскреция гепарансульфата и дерматансульфата с мочой.
МПС 1-го типа имеет аутосомно-рецессивный тип наследования, т. е. заболевание одинаково свойственно лицам мужского и женского пола, в то время как МПС 2-го типа — мужское заболевание, девочки болеют крайне редко. Помутнение роговицы при МПС 2-го типа практически не встречается либо очень «нежное», визуально не видное. При МПС-1 помутнение роговицы видно визуально. Костные деформации и проявления при МПС-1 превалируют, ярче выражены, быстрее прогрессируют.
Особенности МПС 3-го типа (синдром Санфилиппо):
- болеют лица мужского и женского пола;
- ведущим является поражение ЦНС: интеллектуальный дефицит, нарушения поведения и сна, двигательные нарушения, эпилепсия;
- высокий рост, кифосколиоз, деформация грудной клетки, контрактуры крупных и мелких суставов менее выраженные;
- частые респираторные инфекции, отиты, бронхиты;
- тугоухость;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция гепарансульфата с мочой.
Наиболее яркое отличие МПС 3-го типа — более выраженная неврологическая симптоматика по сравнению с другими типами МПС, для которых характерен интеллектуальный дефицит.
Например, при МПС-1 и МПС-2 иногда бывает и так, что ребенок изначально развивается в соответствии с возрастом, лишь затем появляются симптомы задержки и происходит регресс. В случае МПС-3 ребенок с раннего детства имеет прогрессирующую задержку развития, нарушения поведения, к которым позже присоединяются другие неврологические проявления: двигательные нарушения, судороги. Что касается соматических проявлений, то и здесь есть отличия: дети с МПС 3-го типа имеют достаточно высокий рост (другим типам свойственна низкорослость); при этом типе мукополисахаридоза не бывает цервикального стеноза и синдрома запястного канала.
Особенности МПС 4-го типа (синдром Моркио):
- изменения черт лица по типу гаргоилизма отсутствуют;
- частые респираторные инфекции, отиты, бронхиты;
- сохранный интеллект;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки;
- низкий рост, кифосколиоз, килевидная деформация грудной клетки, грубые деформации крупных и мелких суставов при отсутствии контрактур;
- цервикальный стеноз;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция кератансульфата с мочой.
Примечательно, что детям с МПС 4-го типа не присущ гаргоилизм. Кроме того, особенностью этого типа МПС являются специфические костные деформации: килевидная деформация грудной клетки, грубая деформация позвоночника, вальгусная деформация конечностей. МПС-4 отличает и абсолютно сохранный интеллект пациента (в отличие от МПС-1 или МПС-2, при которых есть пациенты как с высоким уровнем интеллекта, так и с достаточной грубой задержкой интеллектуального развития).
При ранней постановке диагноза и правильном ведении болезни пациенты с синдромом Моркио могут быть успешно адаптированы в обществе.
Особенности МПС 6-го типа(синдром Марото — Лами):
- гипертрофия десен, редкие, мелкие зубы, макроглоссия;
- частые респираторные инфекции, отиты, бронхиты;
- сохранный интеллект;
- тугоухость;
- помутнение роговицы, пигментная дегенерация сетчатки;
- низкий рост, кифосколиоз, деформация грудной клетки, врожденный вывих бедра, контрактуры крупных и мелких суставов;
- синдром запястного канала;
- цервикальный стеноз;
- шумное дыхание, гипертрофия аденоидов и миндалин, синдром обструктивного апноэ сна;
- недостаточность митрального и аортального клапанов, кардиомиопатия;
- экскреция дерматансульфата с мочой.
Этот тип МПС имеет всю симптоматику, характерную для других типов мукополисахаридоза. Из-за видимого помутнения роговицы, грубых костных деформаций, выраженных контрактур суставов МПС 6-го типа похож на тяжелые формы МПС-1 и МПС-4.
Важно отметить, что при данном типе МПС, как и при МПС-4, пациенты имеют сохранный интеллект и при ранней диагностике заболевания, своевременном начале ферментозаместительной терапии могут быть социализированы в обществе.
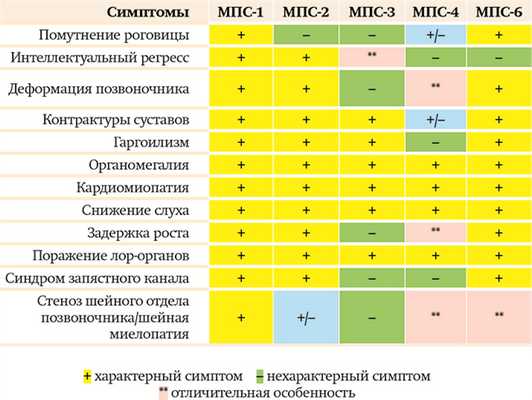
Таблица может помочь врачу на амбулаторном приеме предположить МПС, чтобы потом подтвердить/ опровергнуть диагноз лабораторными исследованиями
Шаг 2. Исключение других болезней накопления
Ряд болезней накопления имеет схожие с МПС внешние клинические признаки. Например, симптоматика альфа-маннозидоза включает:
- грубые черты лица;
- изменения скелета по типу множественного дизостоза;
- поясничный лордоз;
- диффузную мышечную гипотонию;
- шумное носовое дыхание;
- умеренную гепатоспленомегалию;
- нейросенсорную тугоухость;
- пупочную, паховую грыжи;
- умственную отсталость различной степени;
- частые респираторные инфекции, бронхиты, отиты.
Но для альфа-маннозидоза ведущим клиническим признаком является высокая степень нейросенсорной тугоухости, наличие признаков периферической полинейропатии (для МПС-1, МПС-2, МПС-6 характерен только синдром запястного канала), гуморальные и клеточные иммунодефициты. У пациентов с МПС, несмотря на то, что они склонны к частым респираторным инфекциям, иммунодефициты отсутствуют. Но главное отличие в результатах лабораторных исследований. При альфа-маннозидозе отмечается снижение активности альфа-маннозидазы в лейкоцитах. Накопление маннозосодержащих олигосахаридов. ГАГ в моче отсутствует. По результатам ДНК-диагностики заболевание обусловлено мутациями в гене MAN2B1.
Муколипидоз 2-го и 3-го типа также имеет симптомы, которые «роднят» его с разными типами МПС. Муколипидоз 2-го типа имеет более тяжелое течение и клинические проявления практически с первых месяцев жизни, быстрое прогрессирование. Муколипидоз 3-го типа мягче, отличается более поздним проявлением симптоматики и чуть меньшей скоростью прогрессирования. При этом вне зависимости от типа муколипидозу свойственны общие с мукополисахаридозом симптомы:
- грубый гаргоилизм;
- низкорослость;
- помутнение роговицы;
- тугоподвижность суставов;
- интеллектуальный дефицит;
- паховые и пупочные грыжи;
- органомегалия и др.
Отличить одно заболевание от другого помогает лабораторная диагностика. При муколипидозе 2-го типа отмечается повышение уровня лизосомных ферментов (N-ацетилгексозаминидазы) в плазме и низкий уровень в лейкоцитах крови. При муколипидозе 3-го типа — резкое снижение активности N-ацетилглюкозамин-1-фосфотрансферазы в лейкоцитах и культуре кожных фибробластов (ККФ). В отличие от МПС муколипидоз обусловлен мутациями в гене GNPTAB.
Фукозидоз — очень редкая патология, которая имеет общие с МПС симптомы: огрубление черт лица, низкорослость, органомегалию, грыжи, нейросенсорную тугоухость, контрактуры крупных и мелких суставов, помутнение роговицы и т. д.
Как и другие болезни накопления, фукозидоз может протекать с разной скоростью прогрессирования. При тяжелой форме болезни клинические проявления возникают уже на первом году жизни, быстро прогрессируют, приводя к тяжелой инвалидизации и раннему летальному исходу. При мягком течении клинические симптомы появляются в конце первого десятилетия, медленнее прогрессируют.
Главной отличительной особенностью фукозидоза являются специфические кожные проявления — ангиокератома, ангидроз. Наличие ангиокератомы вкупе с характерной для МПС симптоматикой дает основания предполагать фукозидоз. Окончательное слово за лабораторной диагностикой. При фукозидозе отмечается резкое снижение активности α-L-фукозидазы и ККФ, накопление гликолипидов и гликопротеинов в клетках. Молекулярно-генетическая диагностика указывает на мутации в гене FUCA1.
GM1-ганглиозидоз , в отличие от предыдущего заболевания, широко распространен среди болезней накопления. Несмотря на некоторые схожие с МПС симптомы (гаргоилизм, низкорослость, гиперплазия десен, костные деформации, органомегалия, тугоподвижность суставов и т. д.), GM1-ганглиозидоз имеет яркие отличительные особенности. В частности, ему свойственны макулодистрофия по типу «вишневой косточки», поражение почек, быстро прогрессирующее поражение ЦНС, эпилепсия, грубая задержка психомоторного развития, ангиокератома. По результатам лабораторной диагностики отмечается резкое снижение активности β-галактозидазы в лейкоцитах и ККФ. Заболевание обусловлено мутациями в гене GLB1. Болезнь практически всегда имеет тяжелое течение и неблагоприятный исход в первые годы жизни.
Множественная сульфатазная недостаточность — заболевание, которое легко спутать с МПС 2-го типа, если не выполнить объективную лабораторную диагностику. Клиническая симптоматика заболевания почти идентична симптоматике МПС-2, но результаты лабораторных исследований показывают принципиальные отличия. Так, при множественной сульфатазной недостаточности в крови снижен уровень не только идуронат-2-сульфатазы, но и других ферментов, в частности арилсульфатаз А, В, С. ДНК-диагностика показывает мутации в гене SUMF1.
Шаг 3. Исключение иных наследственных заболеваний или синдромальных состояний, схожих с болезнями накопления, в том числе МПС
Микроделеция Xq28, включающая ген IDS. Заболевание обусловлено делецией в хромосомной области, которая затрагивает ген IDS. Мутации этого гена лежат в основе МПС 2-го типа, поэтому неудивительно, что клинические признаки микроделеции Xq28 практически совпадают с симптомами МПС-2, включая высокий уровень ГАГ в моче. Примечательно, что ферментозаместительная терапия, назначаемая обычно при МПС-2, в этом случае также может давать положительную динамику. Но формально это все-таки не МПС-2, поскольку заболевание вызвано делецией, затрагивающей ген IDS, а не мутацией в нем.
Синдром Беквита — Видемана. Достаточно распространенный синдром, имеющий схожую с МПС симптоматику: интеллектуальный дефицит, органомегалию, грыжи. В то же время молекулярно-генетическое исследование показывает совершенно отличную от МПС картину: это синдромальное состояние, обусловленное изменениями в регионе 11р15, включающем гены факторов роста и гены-супрессоры опухолевого роста.
Х-сцепленная демиелинизирующая лейкодистрофия со спондилоэпифизарной дисплазией. Для этого заболевания характерны клинические признаки, схожие с симптоматикой болезней накопления: гаргоилизм, изменения скелета по типу множественного дизостоза (сколиоз, воронкообразная деформация грудной клетки), интеллектуальный дефицит, вальгусная деформация нижних конечностей, рецидивирующие отиты, тугоухость и другие. При этом уровень ГАГ в моче в пределах нормы, отсутствует дефицит лизосомных ферментов. Молекулярно-генетическое исследование указывает на специфическую мутацию в гене AIFM1, что и позволяет установить верный диагноз.
Шаг 4. Дифдиагностика с заболеваниями, имеющими схожую с МПС клиническую симптоматику
И, наконец, необходимо исключить заболевания, под которые маскируются МПС: ювенильный ревматоидный артрит; детский церебральный паралич; гипертензионно-гидроцефальный синдром; периферическая нейропатия; гепатит неясного генеза; гепатолиенальный синдром; врожденный порок сердца; хронический отит, аденоидит, тонзиллит; хронический обструктивный бронхит; муковисцидоз; бронхиальная астма; гипотиреоз; иммунодефицитное состояние; помутнение роговицы.
Надо отметить, что часть этих диагнозов в контексте МПС лишь симптомы, например, помутнение роговицы, порок сердца, аденоидит или отит. Гепатит неясного генеза — симптом органомегалии, увеличения печени; муковисцидоз, хронический обструктивный бронхит — проявления поражения нижних дыхательных путей у пациентов с МПС.
Поэтому за постановкой любого диагноза должен стоять тщательный сбор анамнеза, включая неонатальный и семейный, а также объективная лабораторная диагностика. В случае с МПС этот принцип исключительно важен. Вот лишь пару примеров. Раньше детям с МПС часто выставляли детский церебральный паралич как окончательный диагноз. И это на фоне отсутствия отягощенного неонатального анамнеза, хронической гипоксии, повышения мышечного тонуса, позотонических и патологических рефлексов, характерных для ДЦП.
К диагнозам, которые прежде часто «подменяли» истинный, относится и гипотиреоз. Отчасти заболевание действительно напоминает МПС (крупный язык, задержка психоречевого развития, увеличенная щитовидная железа по данным УЗИ могут навести на мысль о болезни накопления). Однако у пациентов с МПС увеличение щитовидной железы обусловлено отложением ГАГ в тканях органа, которое при этом не дает никакого гормонального дисбаланса.
Болезнь I-клеток (муколипидоз типа II)
Болезнь I-клеток (муколипидоз типа II) - заболевание, которое связано с нарушением расщепления мукополисахаридов и сфингогликолипидов. В связи с этим клинически оно проявляется симптомами, свойственными мукополисахаридозам и болезни Ниманна-Пика (спланхномегалией, грубыми чертами лица, задержкой умственного развития и неврологическими расстройствами).
Что провоцирует / Причины Болезни I-клеток (муколипидоза типа II):
Заболевание обусловлено нарушением функций лизосомных гидролаз. Тип наследования - аутосомно-рецессивный.
Патогенез (что происходит?) во время Болезни I-клеток (муколипидоза типа II):
В фибробластах больных с болезнью I-клеток отмечается дефицит b-гексозаминидазы, арилсульфатазы А и b-глюкоронидазы, в жидких средах организма (сыворотке крови, моче, цереброспинальной жидкости) активность лизосомных гидролаз повышена. Расщепление мукополисахаридов, гликолипидов и гликопротеидов снижено. Предполагают, что нарушается поглощение ферментов лизосомой вследствие изменений посттрансляционной модификации. В молекулах этих ферментов отсутствует маннозо-6-фосфатная маркерная группировка, узнаваемая специфическим рецептором, который обеспечивает внутриклеточный транспорт ферментов в лизосомы. Дефект маркерного центра лизосомных ферментов обусловлен недостаточностью фосфотрансферазы, локализованной в пластинчатом комплексе аппарата Гольджи. При электронной микроскопии фибробластов определяются необычные цитоплазматические включения. Такие же изменения отмечаются в аксонах периферических нервов, печени, почечных клубочках и эпителии канальцев почек.
При патологоанатомическом исследовании обнаруживают утолщение клапанов сердца, инфильтрацию миокарда вакуолизированными фибробластами и пролиферацию соединительной ткани стенки аорты.
Симптомы Болезни I-клеток (муколипидоза типа II):
Уже с рождения отмечаются гепатомегалия, гротескные черты лица, гипертрофия десен, кифосколиоз, расширение межреберных промежутков. Характерны врожденный вывих бедра, грыжи, двусторонняя косолапость; выражены контрактуры суставов. Часто наблюдаются респираторные вирусные инфекции. Кожа грубая, толстая, вокруг глаз кожа отечна, имеется незначительное помутнение роговиц. Отмечается прогрессирующая задержка физического и психомоторного развития. Дети, как правило, погибают в возрасте до 5 лет.
Диагностика Болезни I-клеток (муколипидоза типа II):
Диагноз ставят на основании клинической картины, различий в активности лизосомальных гидролаз в жидких средах организма и фибробластах. Дифференциальный диагноз проводят с синдромом Гурлер (мукополисахаридоз тип I), при котором экскреция мукополисахаридов с мочой в пределах нормы.
Лечение Болезни I-клеток (муколипидоза типа II):
Лечение не разработано. Для предотвращения рождения больного ребенка осуществляют пренатальную диагностику болезни I-клеток, которая возможна лишь в специализированных клиниках.
К каким докторам следует обращаться если у Вас Болезнь I-клеток (муколипидоз типа II):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Болезни I-клеток (муколипидоза типа II), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Поражение нервной системы при герпетических инфекциях

Галина Авдей, доцент кафедры неврологии и нейрохирургии ГрГМУ, кандидат мед. наук. 02.07.2022 Фото носит иллюстративный характер. Из открытых источников.
Беспокойство врачей все больше вызывает вирус, симптомы которого изучал еще Гиппократ почти два с лишним тысячелетия тому назад. Сегодня 99 % населения Земли заражены вирусом герпеса, а смертность из-за него, по данным ВОЗ, на втором месте после гриппа.
Этиология и основные типы
Первичный контакт с вирусом герпеса обычно происходит в раннем детстве. К 5-6 годам ребенок уже успевает подхватить герпес. Эпидемиологические исследования показали, что к 15-летнему возрасту инфицировано 80 % детей, а к 30 годам 90 % населения имеют антитела к вирусам герпеса того или иного типа.
Вирус размножается в носоглотке, через лимфу попадает в кровь, разносится по организму и находит недосягаемое убежище: в узлах периферической нервной системы спинного мозга. Основными путями передачи герпеса являются контактный (физический контакт с зараженным), воздушно-капельный (через частицы слюны при чихании и кашле), половой (генитальный герпес, во время незащищенного полового контакта), бытовой (через предметы совместного пользования), вертикальный (от беременной женщины к ребенку во время беременности или родов).
У большинства людей вирус в дремлющем состоянии, никак себя не проявляет. Но при неблагоприятных условиях (при переохлаждении, злоупотреблении алкоголем, табакокурении, внезапном стрессе, травме кожи в периоральной области, переутомлении или недосыпании) вирус активизируется и вызывает поражение кожи, слизистых оболочек, верхних дыхательных путей, глаз, урогенитального тракта, нервной системы, внутренних органов.
В настоящее время известно 8 основных типов герпесвирусов (см. таблицу).
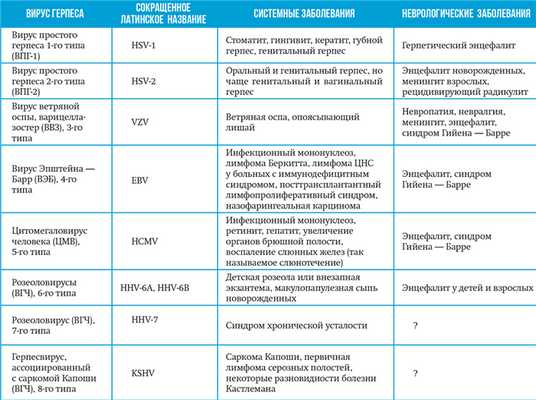
Таблица. Заболевания, вызываемые вирусами герпеса. Самый распространенный, а потому и опасный — герпес 1-го и 2-го типа (ВПГ-1 и ВПГ-2). Инфицированность ВПГ-1 достигает в европейских странах 50-80 %, ВПГ-2 — 10-25 %. Антитела к ВПГ-1 либо к ВПГ-2 обнаружены у 95 % жителей США. Вирусом Эпштейна — Барр инфицировано до 50 % молодых людей (до 18 лет) в развитых странах. В США антитела к ВЭБ в крови обнаруживают у 95 % лиц, достигших 35-летнего возраста. Частота выделения ВГЧ-7 у детей в возрасте старше 36 месяцев — 100 %.
Клинические проявления герпетической инфекции отличаются значительным разнообразием. Они зависят от локализации поражения, распространенности, состояния иммунной системы, типа вируса, а также от механизмов заражения.
Важное место в структуре заболеваний вирусной этиологии принадлежит поражению нервной системы. Приблизительно 20 % случаев обусловлены герпесвирусной инфекцией. При этом у 14-30 % пациентов длительно сохраняются остаточные явления вплоть до инвалидизации, а летальность составляет от 5 % до 70 % в зависимости от клинической формы поражения нервной системы.
По мнению академика, доктора мед. наук Н. Д. Львова, вирус простого герпеса (ВПГ-1) присутствует примерно у каждого третьего человека.
Герпетические энцефалиты
ВПГ-1 может вызывать различные заболевания нервной системы, но наиболее тяжелое из них — герпетический энцефалит. Частота — 2-2,5 случая на 1 млн населения в год. Наиболее часто проявляется в двух возрастных группах: между 5 и 30 годами и у лиц старше 50 лет. ВПГ-1 поражает передние отделы головного мозга (лобные и височные доли).
У 2/3 больных наблюдается острое начало с высокой температурой тела в течение 3-4 дней, нередко с респираторными симптомами, с последующим присоединением неврологической симптоматики. Частым симптомом является нарушение сознания вплоть до комы в течение нескольких дней вследствие быстро нарастающего отека мозга.
Характерная черта герпетического энцефалита — поражение височной доли с одной стороны, что проявляется изменениями личности со снижением интеллектуально-мнестических функций (иногда до уровня деменции) и психическими расстройствами (от неадекватного поведения до делириозного состояния).
Возможны нарушение ориентации в пространстве и времени, проявления агрессивности, слуховые и зрительные галлюцинации, сумеречное состояние сознания. Частым признаком заболевания является судорожный синдром —эпилептические припадки генерализованного или очагового характера. Их частота достигает 40 %. Наблюдаются джексоновские, оперкулярные, вегетативные и психомоторные пароксизмы. Очаговые поражения нервной системы проявляются моно- и гемипарезами, нарушением корковых функций (афазия, гемианопсия, апраксия, аграфия, акалькулия), поражением черепных нервов.
При исследовании цереброспинальной жидкости выявляют лимфоцитарный плеоцитоз (от десятков до сотен клеток в 1 мкл) и повышенный уровень белка при нормальном содержании сахара.
При электроэнцефалографическом исследовании на фоне дизритмии имеет место характерная диффузная медленноволновая высокоамплитудная активность и очаговое поражение височно-лобных отделов мозга. При МРТ-исследовании головного мозга обнаруживаются очаги поражения с преобладанием изменений в передних отделах и преимущественным вовлечением коры.
Летальность при герпетическом энцефалите достигает 40-80 %, а у большинства выживших сохраняются грубые органические дефекты.
ВПГ-2 обычно вызывает энцефалиты у новорожденных и детей первого полугода жизни. Заражение происходит при рождении от матерей, пораженных генитальным герпесом, хотя не исключено и трансплацентарное инфицирование. Симптоматика энцефалита, вызванного ВПГ-2, в значительной степени идентична проявлениям энцефалита, обусловленного ВПГ-1.
Если энцефалит составляет компонент генерализованного инфекционного процесса, то обычно выявляются симптомы недостаточности надпочечников, признаки поражения печени, легких, перикарда. Нередко на коже и слизистых оболочках появляются герпетические высыпания. Изменения на ЭЭГ носят обычно диффузный характер в связи с незрелостью детского мозга и особенностями его реактивности.
Энцефалиты, вызванные вирусом ветряной оспы (ВВЗ), характеризуются более благоприятным течением, редким развитием коматозных состояний, относительно невысокой летальностью. Наряду с поражением коры в патологический процесс вовлекаются структуры ствола мозга, мозжечка, подкорковых узлов, и поэтому в отличие от энцефалитов, вызванных вирусом простого герпеса, довольно часто обнаруживаются стволовые, церебеллярные, подкорковые симптомы и синдромы. Для них характерно присутствие симптомов ганглионевритов, а также поражение кожи и слизистых оболочек.
Применение противогерпетических препаратов в первые 3 суток высыпаний способствует уменьшению выраженности постгерпетической невралгии у 42 % больных.
Герпетические менингиты
Наблюдаются у взрослых на фоне первичной генитальной инфекции. Протекают как серозные, асептические, с кожными высыпаниями и без них. В отличие от менингитов другой этиологии не имеют четкой сезонности и обычно не сопровождаются выраженными признаками респираторного заболевания. Температура тела редко превышает 38 °С, головные боли чаще средней интенсивности. У всех больных довольно долго (до 2 недель) сохраняется ригидность затылочных мышц, а у многих определяется диссоциация между частотой и выраженностью ригидности затылочных мышц и симптомом Кернига. В неврологическом статусе довольно часто обнаруживается очаговая микросимптоматика.
Менингиты, обусловленные ВВЗ, также протекают как асептические, но общемозговые и общеинфекционные симптомы, признаки интоксикации более выражены, чем при менингитах, вызванных ВПГ-1. Сроки санации спинномозговой жидкости во многих случаях превышают месяц.
Все менингиты заканчиваются благоприятно.
Ганглионевриты
Наиболее частой формой герпетических поражений периферической нервной системы являются ганглионевриты, особенно вызванные ВВЗ. Чаще развиваются у пожилых в связи со снижением эффективности иммунного ответа. Обычно развернутой картине ганглионеврита предшествуют общеинфекционные симптомы — общее недомогание, вялость и разбитость, повышение температуры тела. На фоне общеинфекционных проявлений внезапно возникают интенсивные боли в зоне одного или нескольких сегментов. Боли подчас настолько мучительны, что нередко их расценивают как симптом острого хирургического заболевания. Характерно сочетание боли с зудом, чувством жжения, увеличение регионарных лимфоузлов. Спустя несколько часов или дней после возникновения болей, парестезий, гиперестезий на коже и слизистых оболочках на фоне эритемы в виде пояса на туловище или продольной полосы на конечностях появляются папулы, а затем везикулы, заполненные прозрачной серозной, позже — мутной, гнойной, иногда геморрагической жидкостью. Высыпания продолжаются в течение нескольких дней и располагаются по ходу одного или нескольких кожных сегментов. Постепенно пузырьки подсыхают, образуют корочки, которые отходят в течение нескольких недель, редко — месяцев. На коже остаются темно-бурая пигментация и рубцы.
Чаще поражаются ганглии грудной локализации, на втором месте по частоте — краниальные (ганглии тройничного нерва). В патологический процесс вовлекается первая ветвь тройничного нерва. Появление герпетических высыпаний на роговице сопряжено с возможностью развития стойких и резких нарушений зрения, вплоть до слепоты. Через 4-7 недель после острого периода офтальмического герпеса может возникнуть контралатеральный гемипарез с плеоцитозом и повышением уровня белка в спинномозговой жидкости. Причиной служит инфаркт мозга. Патогенез этого синдрома связывают с непосредственным вирусным поражением сосудистой стенки с развитием ангиита.
Осложнения
Редким осложнением офтальмического герпеса является синдром Гийена — Барре. Возможно развитие и ретробульбарного неврита.
Поражение ганглия коленца, наряду с герпетическими высыпаниями в наружном слуховом проходе и на барабанной перепонке, нередко сопровождается вовлечением в патологический процесс лицевого и кохлеовестибулярного нервов (синдром Рамсея — Ханта).
После острого периода у пожилых людей часто (30-40 %) формируется стойкая, мучительная резистентная к лечению постгерпетическая невралгия. В основном она обусловлена гибелью мякотных волокон или их демиелинизацией, задержкой нормальных процессов ремиелинизации вследствие органических ресурсов вегетативного (сосудисто-трофического) обеспечения в организме пожилых людей.
Противогерпетические препараты эффективны при лечении невропатии лицевого нерва, так как ВПГ-1 вызывает не только клеточное, но и аксональное повреждение нерва.
Герпетические радикулоганглионевриты отличаются от дискогенной радикулопатии:
- возникновением неврологической симптоматики в непосредственной связи с высыпаниями на коже и общеинфекционными проявлениями;
- отчетливым распространением боли за пределы корешковых зон;
- сочетанием болей с зудом и чувством жжения;
- сочетанием нескольких видов нарушений чувствительности и частым появлением гиперпатии;
- чувствительными расстройствами вне зоны герпетических высыпаний;
- нерезкой выраженностью симптомов тонического напряжения;
- умеренным ограничением объема движений в позвоночнике;
- длительным преобладанием в клинической картине симптомов раздражения;
- полным регрессом радикулярных симптомов вне рецидивов герпеса;
- отчетливым клиническим эффектом противовирусной терапии.
Распространение патологического процесса за пределы периферической нервной системы сопряжено с поражением мозговых оболочек и вещества мозга. Поэтому радикуло(поли)ганглионеврит, полирадикулоневрит, мультирадикулоганглионеврит могут протекать с менингеальным, энцефалитическим и (или) миелитическим синдромом. А также возможны многочисленные варианты одновременного поражения нервной системы и других органов и систем.
Достоверное участие ВГЧ-7 и ВГЧ-8 в патологии нервной системы в настоящее время не доказано.
Диагностика и лечение
При МРТ выявляются очаги воспаления и отека в лобных и височных долях. На ЭЭГ — периодическая островолновая активность в височной доле на фоне локального или диффузного замедления ритма.
Для лабораторной диагностики герпетической инфекции ценным являются цитологический метод, иммуноферментный анализ и хемилюминесценция, выявление вирус-специфических антител к герпес-вирусам (HSV, EBV, CMV, HHV6, VZV); авидность IgG, иммуноблоттинг (референс-метод), ПЦР, оценка иммунного, интерферонового и цитокинового статусов и госпитального комплекса (HDsAg, anti-HCV total, RW, anti-HIV-1,2).
Золотым стандартом в диагностике асимптомной или атипичной герпетической инфекции является выделение вируса на культуре ткани (чувствительность 80-100 %, специфичность 100 %).
В повседневной практике осуществление этого метода значительно затруднено вследствие сложности проведения, дороговизны и длительного промежутка времени, в течение которого проводится исследование. Поэтому наиболее приемлем, чувствителен и специфичен метод ПЦР. В течение 7 часов возможно с высокой степенью достоверности (до 98 %) диагностировать герпетическую инфекцию центральной нервной системы. Материалом для исследования на герпес являются кровь, мазки из глотки, содержимое пузырьков, язв, моча.
В лечении применяют противовирусные препараты (ациклические нуклеозиды), проводят иммунотерапию (иммуномодуляторы, интерфероногены, вакцинация), заместительную терапию (иммуноглобулины, интерфероны) и симптоматическую терапию (НПВП, адаптогены, антиоксиданты).
Излечиться полностью от герпеса невозможно — вирус остается в организме человека на всю жизнь после первичного инфицирования. Все средства лишь убирают симптомы и позволяют восстановить и поддержать иммунитет.
ВОЗ принимает меры для повышения осведомленности о герпесвирусной инфекции и ее симптомах, расширения доступа к противовирусным препаратам, содействует проведению исследований, направленных на разработку новых средств профилактики и борьбы с этой инфекцией.
Читайте также:
- Прогноз аортальной недостаточности. Лечение недостаточности клапана аорты
- Методика удаления поднижнечелюстной слюнной железы
- Потенцированный наркоз под управляемым дыханием. Обеспечение операции на средостении
- Оперативный доступ при сочетанной травме. Временный гемостаз и эвакуация крови при сочетанной травме.
- Прямое измерение артериального давления. Инвазивное измерение артериального давления