Понтоцеребеллярная гипоплазия и атрофия у ребенка: причины, классификация
Добавил пользователь Дмитрий К. Обновлено: 21.01.2026
Спинальные мышечные атрофии - группа клинически и генетически гетерогенных наследственных заболеваний, вызванных прогрессирующей дегенерацией мотонейронов передних рогов спинного мозга (1). Начало заболевания варьирует от рождения до взрослого возраста. Является орфанным заболеванием (2)
Этиология и патогенез: Генетическое заболевание, при котором возможны все типы наследования (аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный). Ген SMN ответственен за развитие спинальной мышечной атрофии детского возраста с аутосомно-рецессивным типом наследования (1,6)
Название протокола: Спинальные мышечные атрофии у детей
Код(ы) по МКБ-10:
| МКБ-10 | |
| Код | Название |
| G 12 | Спинальная мышечная атрофия и родственные синдромы |
| G 12.0 | Детская спинальная мышечная атрофия, I тип (Верднига-Гоффманна) |
| G 12.1 | Другие наследственные спинальные мышечные атрофии |
| G 12.2 | Болезнь двигательного нейрона |
| G 12.8 | Другие спинальные мышечные атрофии и родственные синдромы |
| G 12.9 | Спинальная мышечная атрофия неуточненная |
Дата разработки/пересмотра протокола: 2018 год.
Сокращения, используемые в протоколе:
| АД | аутосомно-доминантный |
| АР | аутосомно-рецессивный |
| ГЭФР | гастроэзофагеальный рефлюкс |
| ЖКТ | желудочно-кишечный тракт |
| ИВЛ | инвазивная вентиляция легких |
| КТ | компьютерная томография |
| КФК | креатинфосфокиназа |
| МРТ | Магниторезонансная томография |
| НИВЛ | неинвазивная вентиляция легких |
| ПЦР | полимеразная цепная реакция |
| РКИ | рандомизированное клиническое исследование |
| СМА, SMA | спинальная мышечная атрофия |
| УЗИ | ультразвуковое исследование |
| ЭМГ | электромиография |
| MLPA | multiplex dependent probe amplification |
| SMN | сокр. от Survival Motor Neuron (ген «выживаемости мотонейрона» - англ.) |
Пользователи протокола: детские неврологи, неонатологи, педиатры, врачи общей практики, реабилитологи, детские хирурги-ортопеды, анестезиологи-реаниматологи, пульмонологи, клинические генетики.
Категория пациентов: дети.
Шкала уровня доказательности:
| А | Высококачественный мета-анализ, систематический обзор РКИ или крупное РКИ с очень низкой вероятностью (++) систематической ошибки результаты которых могут быть распространены на соответствующую популяцию. |
| В | Высококачественный (++) систематический обзор когортных или исследований случай-контроль или Высококачественное (++) когортных или исследований случай-контроль с очень низким риском систематической ошибки или РКИ с невысоким (+) риском систематической ошибки, результаты которых могут быть распространены на соответствующую популяцию. |
| С | Когортное или исследование случай-контроль или контролируемое исследование без рандомизации с невысоким риском систематической ошибки (+). Результаты, которых могут быть распространены на соответствующую популяцию или РКИ с очень низким или невысоким риском систематической ошибки (++ или +), результаты которых не могут быть непосредственно распространены на соответствующую популяцию. |
| D | Описание серии случаев или неконтролируемое исследование или мнение экспертов. |
| GCP | Наилучшая клиническая практика. |

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
- Спинальная амиотрофия I типа (спинальная амиотрофия раннего детского возраста, или болезнь Верднига-Гоффмана).
- Спинальная амиотрофия II типа (спинальная амитрофия детского возраста, промежуточная форма).
- Спинальная амитрофия III типа (болезнь Кугельберга-Веландера).
- Бульбоспинальная амиотрофия Кеннеди (форма взрослых).
- Дистальная.
Диагностика
МЕТОДЫ, ПОДХОДЫ И ПРОЦЕДУРЫ ДИАГНОСТИКИ (1,3,4,5-14)
Диагностические критерии
Жалобы: практические отсутствие или позднее формирование двигательных навыков.
- СМА 1 типа характеризуется дебютом ранее 6 месяцев, симптомокомплексом «вялого» ребенка, колоколообразной формой грудной клетки, выраженной гипотонией, арефлексией, фасцикуляциями языка и проблемами с дыханием. Пациенты обычно погибают до 2 лет от дыхательной недостаточности, возникающей в результате интеркурентных инфекций.
- СМА II типа характеризуется началом заболевания в возрасте от 6 мес. до 1,5 года и более медленным прогрессированием, тип наследования аутосомно-рецессивный. У больных также отмечается симптомокомплекс «вялого» ребенка, гипотония, арефлексия, фасцикуляции языка и проблемы с дыханием. Такие пациенты максимально способны самостоятельно сидеть, и у них развиваются многочисленные контрактуры крупных суставов
- СМА III типа развивается в возрасте от 1,5 лет, в большинстве случаев прогрессирует медленно, тип наследования аутосомно-рецессивный. эти пациенты могут самостоятельно ходить. Пациенты обычно имеют слабость в подвздошных, четырехглавых и аддукторных мышцах, гипотонию, гипорефлексию и фасцикуляции языка. Некоторые из пациентов этой группы со временем утрачивают способность к самостоятельному передвижению.
- СМА IV типа (бульбоспинальная амиотрофия Кеннеди, форма взрослых) проявляется в среднем на 4 десятилетии жизни со слабости бульбарных мышц (дисфагия, дизартрия) с последующим присоединением слабости проксимальных отделов конечностей, слабость мимической мускулатуры, атрофии и фасцикуляции в языке, генерализованные фасцикулякуции, крампи, постуральный тремор, сенсорная полиневропатия. Часто характерны гинекомастия, снижение половой функции, гипогонадизм, нарушение сперматогенеза, бесплодие, сахарный диабет. Тип наследования - Х-сцепленный рецессивный.
- Изменения нервно-мышечной системы: фибрилляции мышц языка, генерализованная мышечная гипотония, атрофии мышц и фасцикуллярные подергивания в мышцах спины, туловища, проксимальных (реже в дистальных) отделах верхних и нижних конечностей; гипорефлексия вплоть до арефлексии;
- Изменения костно-суставной системы: деформация грудной клетки, деформация позвоночника (кифосколиоз), контрактуры суставов, патология стоп.
- Нарушение дыхательной функции: как результат нарушения откашливания и глотания, гиповентиляции во время ночного сна, недоразвития\деформации грудной клетки, частных инфекционных заболеваний вследствие нарушения эвакуации секреторного отделяемого из дыхательных путей.
- Дисфункция ЖКТ: нарушения глотания (в т.ч. из-за бульбарного синдрома) нарушения моторики ЖКТ, которые включают запоры, задержку эвакуации содержимого желудка и потенциально опасный для жизни гастроэзофагальный рефлюкс (ГЭФР).
- Болевой синдром: как последствие патологии опорно-двигательного аппарата, остеопении и переломов.
- Нарушение роста и гипо-/гипертрофия.
- Нарушения чувствительности: не характерно.
- Нарушения психоречевого и когнитивного развития: не характерно
- Дети, которые не могут сидеть без посторонней помощи («лежачие пациенты»);
- Дети, которые могут самостоятельно сидеть, но не могут ходить без посторонней помощи («сидячие пациенты»);
- Дети, которые могут самостоятельно ходить («ходячие пациенты»).
- Общий анализ крови и мочи: специфических изменений нет.
- Биохимический анализ крови: уровень КФК может быть нормальным или слегка повышенным. Интерпретация для оценки задержки двигательного аппарата:
- в некоторых случаях целесообразна ликворограмма - повышение количества белка на 25 % и более.
- Электромиография (ЭМГ): ритмичные потенциалы фасцикуляций с амплитудой до 300 мкВ и частотой 5-35 Гц (ритм частокола). Скорости проведения импульса по периферическим двигательным волокнам могут быть как нормальными, так и немного сниженными за счет вторичных денервационных изменений. Для детей 1 и 2 типов имеет меньшее диагностическое значение, чем для иных типов.
- УЗИ и МРТ мышц: признаки жирового замещения мышечной ткани.
- Генетические тесты - предназначены для установления диагноза СМА, прогнозирования и выбора терапевтических подходов. Отсутствие полных копий SMN1 подтверждает диагноз, информация о копиях SMN2 является важной для прогноза и выбора терапии.
- Количественная ПЦР: идентифицирует гомозиготную делецию SMN1, но не позволяет подсчитать количество копий SMN1 и SMN2.
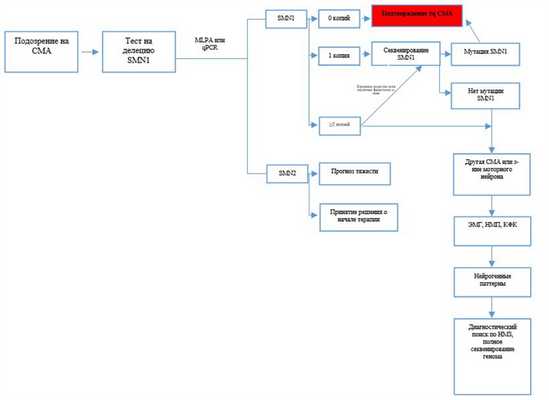
Диагностический алгоритм:
Примечания: ЭМГ - электромиография, НМП- нервно-мышечная проводимость; КФК - креатинфосфокиназа; НМЗ- нервно-мышечные заболевания
Зависимость клинической картины СМА от количества копий генов SMN1 и SMN2.
| Тип СМА | Количество копий генов | Особенности клинической картины |
| СМА | Полное отсутствие обоих генов SMN1 и SMN2 | Летальная ситуация |
| СMA тип 0 | Нет SMN1 гена и 1 копия SMN2 гена. | Тяжелая мышечная слабость, смерть наступает до 1 месяца |
| СMA тип I | Преимущественно делеции SMN1 или несколько миссенс мутаций в SMN1; SMN2 обычно 2 копии. | Симптомокомплекс «вялого» ребенка, смерть наступает до 2 лет. |
| СMA тип II | Мутации превращают ген SMN1 в SMN2; Копий гена SMN2 > 3 копий; Могут встречаться миссенс точечные мутации. | Могут самостоятельно сидеть. |
| СMA тип III | Копий гена SMN2 > 3 копий; Могут встречаться миссенс точечные мутации. | Могут самостоятельно ходить. |
| Специалист | Цель |
| Детский невролог | Клиническая диагностика заболевания, направление на генетическую и параклиническую диагностику. Разработка краткосрочного и долгосрочного плана ведения и реабилитации. Координатор мультидисициплинарной команды специалистов, мониторинг и оценка эффективности комплексного плана лечения и реабилитации. Принятие решения о назначении специфической терапии. |
| Генетик | Генетическая верификация диагноза. Медико-генетическое консультирование семьи, информирование о методах пренатальной и предимплатанционной диагностики. |
| Педиатр | Диагностика и коррекция нарушений со стороны внтуренних органов. Мониторинг физического, соматического и нутритивного статуса. |
| Пульмонолог/специалист по респираторной поддержке | Диагностика нарушений дыхательной системы, разработка и реализация плана лечения и долгосрочной курации в случае их наличия |
| Анестезиолог-реаниматолог (детский) | Диагностика нарушений дыхательной системы пациентов, нуждающихся в проведении неинвазивной вентиляции легких (НИВЛ), коррекция водно-электролитного обмена и белкового статуса на фоне дефицита массы тела тяжелой степени. |
| Гастроэнтеролог | Диагностика и коррекция нарушений пищеварительной системы, разработка и реализация плана лечения и долгосрочной курации в случае их наличия. |
| Диетолог | Решение вопросов подбора и реализации диеты |
| Ортопед | Диагностика нарушений костно-суставной системы, консервативная коррекция патологии позвоночника, суставов, стоп; хирургическая коррекция. Подбор ортезов/туторов и иных необходимых приспособлений. |
| Реабилитолог (в т.ч. специалист ЛФК) | Разработка и реализация комплексной реабилитации (в т.ч. двигательной). Обучение семьи пациента. |
| Психолог | Квалификация психологических нарушений, разработка и реализация плана лечения и долгосрочной курации в случае их наличия. Семейное психологическое консультирование |
Дифференциальный диагноз
Дифференциальный диагноз и обоснование дополнительных исследований
Дифференциальная диагностика СМА и других поражений спинного мозга
| Диагноз Признак | Боковой амиотрофический склероз | Вертеброгенная шейная миелопатия | Сирингомиелия |
| Клиника | Периферические парезы, высокие сухожильные периостальные рефлексы, фибрилляции, фасцикулляции мышц, поражение черепно-мозговых нервов (V-XII), атрофии | Периферические парезы и расстройства чувствительности в зонах иннервации шейных сегментов, доминируют симптомы ишемического поражения двигательных структур шейного утолщения | Дистальные атрофии, мышечный тонус и рефлексы снижены, болевой синдром, расстройства чувствительности диссоциированного типа, вазомоторные и трофические нарушения, дизрафический статус |
| Течение | Прогредиентное | Медленно прогрессирующее | Медленно прогрессирующее |
| Рентгенография | Без особенностей | Шейного отдела - выраженные явления остеохондроза и сужение позвоночного канала | Кифосколиоз, добавочные ребра, не заращение дужек шейных и поясничных позвонков, ассимиляция атланта с затылочной костью, базилярная импрессия |
Другие редкие формы спинальных мышечных атрофий представлены в таблице. Некоторые из них имеют единичные описания в мировой литературе, многие из которых специфичны для конкретных изолятов.
Дифференциальная диагностика проксимальных аутосомно-рецессивных СМА и других форм СМА
Понтоцеребеллярная гипоплазия и атрофия у ребенка: причины, классификация
Понтоцеребеллярные гипоплазии (ПЦГ) - группа редких моногенных нейродегенеративных заболеваний, характеризующихся гипоплазией мозжечка и варолиева моста головного мозга, формирующихся внутриутробно.
Клинические проявления большинства генетических вариантов ПЦГ возникают с рождения и характеризуются прогрессирующей микроцефалией, расстройствами дыхания, экстрапирамидными и пирамидными симптомами, изменениями мышечного тонуса, судорогами и задержкой психомоторного развития.
В научно-консультативном отделе ФГБНУ «Медико — генетический научный центр» было проведено обследование 8 пациентов (6 девочек и 2 мальчика) в возрасте от 2 мес до 5 лет из неродственных семей.
Целью исследования стало создание первого описания клинико-генетических характеристик ПЦГ 2А и 4 типа, обусловленных мутациями в гене TSEN54, и разработать рекомендации по совершенствованию алгоритма диагностики заболеваний из группы понтоцеребеллярных гипоплазий.
К настоящему времени выделено 20 аутосомно-рецессивных генетических вариантов ПЦГ, обусловленных мутациями в 18 генах. Три варианта ПЦГ - типы 2А, 4 и 5 - являются аллельными, обусловленными различными мутациями в гене TSEN54. Выделение этих 3 генетических вариантов основано на различии в тяжести клинических проявлений и типах мутаций в гене TSEN54. Анализ мутаций в гене TSEN54 у российских больных с клиническими проявлениями ПЦГ не проводился.
На основании анамнеза, неврологического осмотра, признаков поражения мозжечка и ствола мозга, результатов проведения секвенирования экзома у всех пациентов выявлено ПЦГ, обусловленная мутациями в гене ТSEN54 в гомозиготном и компаунд-гетерозиготном состоянии.
Особенности клинических проявлений и характер тяжести течения заболевания позволили у 5 больных диагностировать ПЦГ 2А типа, у 3 больных - ПЦГ 4 типа. Различие между этими генетическими вариантами обусловлено тяжестью клинических проявлений. Все дети с ПЦГ рождались в срок с нормальными росто-весовыми показателями и достаточно высокими оценками по шкале Апгар.
Первые признаки заболевания возникали с рождения или в первые дни жизни и представляли собой дыхательные расстройства в виде эпизодов апноэ, вялого сосания и / или судорог. У 3 детей с ПЦГ 4 типа клинические симптомы были наиболее выражены и требовали искусственной вентиляции легких и зондового кормления. Возникшие у них судорожные пароксизмы были фармакорезистентными и сопровождались появлением неврологической симптоматики в виде спастического тетрапареза и тремора. У 5 больных микроцефалию выявили с рождения, у 3 больных диагностировали в первые месяцы жизни. У всех пациентов отмечали полиморфные судороги и грубую задержку темпов психомоторного развития. Спастический тетрапарез наблюдался у 7 больных, у 1 ребенка — умеренно-выраженная диффузная мышечная гипотония в сочетании с дистоническими атаками.
Было установлено и то, что 85 % случаев тсенопатий обусловлено мутациями в гене TSEN54, которые наиболее часто диагностируются у больных с клиническими проявлениями ПЦГ 2А типа, при этом у подавляющего большинства обнаруживается гомозиготная мутация с. 919G>T, приводящая к замене аланина на серин в 307‑м положении белковой молекулы. В результате проведения функционального анализа показано, что аминокислотная замена ослабляет функцию белка, но не прекращает ее, что приводит к возникновению умеренно выраженных симптомов заболевания. Наличие этой мутации в компаунд-гетерозиготном состоянии с другими типами мутаций приводит к появлению выраженных клинических признаков заболевания, возникающих с рождения или внутриутробно и приводящих к гибели больных в первые месяцы или годы жизни.
У большинства наблюдаемых пациентов с клиническими и МРТ-признаками ПЦГ выявлена мутация с.919G>T (p.Ala307Ser). У 5 больных с ПЦГ 2А типа она обнаружена в гомозиготном состоянии, а у 3 больных с более тяжелыми клиническими проявлениями, соответствующими ПЦГ 4 типа, - в компаунд-гетерозиготном состоянии, в 1 случае - с мутацией с.670_671delAA (p.Lys224fs) и в 2 - с мутацией c.1264C>T (p.Gln422fs).
Учитывая, что мутации в гене TSEN54 обеспечивают возникновение самых распространенных вариантов аутосомно-рецессивных ПЦГ, можно сделать заключение, что у больных с типичными клиническими и МРТ- признаками заболевания диагностический поиск следует начинать с анализа этой мутации. Это позволит снизить временные и экономические затраты при проведении подтверждающей молекулярно-генетической диагностики. При отсутствии данной мутации необходимо осуществлять дифференциальную диагностику с другими генетическими вариантами ПЦГ, моногенными заболеваниями, которые сопровождаются прогрессирующей атрофией мозжечка, такими как спиноцеребеллярные атаксии 2 и 7 типов, болезнь нарушения гликозилирования дистрогликанов 1А типа, редкими наследственными синдромами, при которых обнаруживается сочетание гипоплазии мозжечка с другими пороками развития мозга.
К настоящему времени идентифицировано несколько десятков таких синдромов, основными из которых являются умственная отсталость с гипоплазией мозжечка и необычным лицом (OMIM: 300486), умственная отсталость с гипоплазией мозжечка и варолиева моста с микроцефалией (OMIM:300749), лисэнцефалия 7 типа с гипоплазией мозжечка (OMIM: 616342).
Авторы исследования предполагают следующий диагностический алгоритм:
при наличии у больного специфических клинических проявлений и МРТ-признаков ПЦГ диагностика должна начинаться с анализа мутации с.919G>T (p.Ala307Ser). При ее обнаружении в гомозиготном состоянии диагноз считается уточненным. При обнаружении мутации в гетерозиготном состоянии поиск этиологического фактора необходимо продолжить с помощью секвенирования экзома нового поколения.
В результате проведенного исследования показано, что, так же как в европейской популяции, у российских больных с наиболее распространенными вариантами ПЦГ 2А и 4 типа есть мажорная мутация с.919G>T (p.Ala307Ser) в гене TSEN54, которая регистрируется в гомозиготном или компаунд-гетерозиготном состоянии. Полученные результаты позволяют сделать заключение о необходимости в первоочередном анализе этой мутации у больных с клиническими и МРТ-признаками ПЦГ.
Источник: Клинико-генетические характеристики понтоцеребеллярной гипоплазии, обусловленной мутациями в гене TSEN54 (OMIM: 277470) Дадали Е.Л., Акимова И.А., Семенова Н.А., Гусева Д.М., Щагина О.А., Чухрова А.Л., Канивец И.В., Коростелев С.А.
Понтоцеребеллярная гипоплазия
Понтоцеребеллярная гипоплазия представляет собой группу довольно редких наследственных прогрессирующих нейродегенеративных заболеваний. На сегодняшний день описано восемь типов понтоцеребеллярной гипоплазии. Частота заболеваний неизвестна. Все формы заболевания имеют ряд общих клинических признаков, включающих в себя аномальное развитие головного мозга, проблемы с двигательной активностью, задержку развития, умственную неполноценность, прогрессирующую микроцефалию и церебральные проявления различной степени. Заболевание проявляется с рождения, в ряде случаев первые признаки отмечаются уже внутриутробно. Больные, как правило, погибают в раннем детском возрасте.
Понтоцеребеллярная гипоплазия, тип 1 характеризуется понтоцеребеллярной гипоплазией в сочетании с потерей двигательных нейронов передних рогов спинного мозга, что клинически делает заболевание схожим со спинальной мышечной атрофией, в частности, со спинальной мышечной атрофией, тип I. МРТ исследования выявляют мозжечковую гипоплазию различной степени выраженности. Так же могут быть вовлечены мост и другие отделы головного мозга. У больных понтоцеребеллярной гипоплазией, тип 1 наблюдаются тяжелая гипотония, парезы, дисфагия, дыхательная недостаточность, задержка психомоторного развития. У многих пациентов представлены признаки внутриутробного начала заболевания, такие как врожденные контрактуры и многоводие. Микроцефалия, как правило, при рождении отсутствует, однако развивается позже. Больные понтоцеребеллярной гипоплазией, тип 1 погибают в первый год жизни.
Понтоцеребеллярная гипоплазия, тип 2 является наиболее изученной формой заболевания. К основным признакам понтоцеребеллярной гипоплазии, тип 2 относят экстрапирамидальную дискинезию и дистонию, в ряде случаев - спастичность. Другие клинические признаки представлены нарушением дыхания с рождения, формированием генерализованного клонуса в неонатальном периоде, спазмами мускулатуры. При МРТ-исследовании выявляется бабочкоподобный паттерн мозжечка с гладкими, резко уменьшенными полусферами и относительно малым червем. В 40% случаев наблюдается средняя или тяжелая атрофия коры головного мозга. Двигательные нейроны передних рогов спинного мозга в патологический процесс не вовлекаются. Продолжительность жизни варьирует от неонатального до взрослого периода, однако, большинство пациентов не доживает до пубертата.
Понтоцеребеллярная гипоплазия, тип 3 известна как церебеллярная атрофия с прогрессирующей микроцефалией. Больные имеют невысокий рост. Заболевание проявляется спазмами мускулатуры, гипотонией, симптомами, обусловленными атрофией зрительного нерва.
Понтоцеребеллярная гипоплазия, тип 4 или оливопонтоцеребеллярная гипоплазия клинически схожа с понтоцеребеллярной гипоплазией, тип 2, но имеет более тяжелое течение. Многоводие, продолжительный генерализованный клонус, врожденные контрактуры, первичная гиповентиляция легких с последующей необходимостью искусственной поддержки дыхания - признаки поражения уже во внутриутробном периоде у таких пациентов. На МРТ присутствуют яркие признаки патологии структур головного мозга (С-образная форма ядер олив продолговатого мозга, скопление ликвора в перицеребральном пространстве, признаки задержки развития неокортекса и др.), так же свидетельствующие о раннем, неонатальном, развитии заболевания.
Понтоцеребеллярная гипоплазия, тип 5 имеет внутриутробное начало развития, характеризующееся спазмо-подобной активностью в сочетании с тяжелой оливопонтоцеребеллярной гипоплазией и тяжелым повреждением червя мозжечка.
У больных понтоцеребеллярной гипоплазией, тип 6 наблюдается гипоплазия мозжечка, энцефалопатия, дисфагия, спазмы мускулатуры, прогрессирующая микроцефалия и генерализованная гипотония, сопровождающаяся спастичностью. В ходе биохимических исследований было выявлено снижение активности митохондриальных комплексов I, III, IV в мышцах.
Понтоцеребеллярная гипоплазия, тип 7. Признаками заболевания являются задержка психомоторного развития, гипотония, нарушения дыхания, аномалия развития половых органов. МРТ демонстрирует наличие гипоплазии мозжечка и атрофии головного мозга.
Понтоцеребеллярная гипоплазия, тип 8 характеризуется тяжелым нарушением психомоторного развития, гипотонией, спастичностью, различными зрительными нарушениями. При МРТ-исследовании выявляются гипоплазия мозжечка, утоньшение мозолистого тела, увеличение количества белого вещества.
Все восемь форм понтоцеребеллярной гипоплазии наследуются по аутосомно-рецессивному типу. В таблице представлены известные на сегодняшний день гены, ответственные за развитие понтоцеребеллярной гипоплазии 1-8 типов.
В Центре Молекулярной Генетики методом прямого автоматического секвенирования проводится поиск мутаций в гене VRK1.
Спинальные амиотрофии ( Спинальные мышечные атрофии )
Спинальные амиотрофии — это генетические заболевания, проявляющиеся мышечной атрофией и обусловленные дегенеративными изменениями спинальных мотонейронов и моторных ядер ствола головного мозга. Общим симптомокомплексом выступают симметричные вялые параличи с атрофиями мышц и фасцикуляциями на фоне интактной чувствительной сферы. Диагностируются спинальные амиотрофии по данным семейного анамнеза, неврологического статуса, ЭФИ нервно-мышечного аппарата, МРТ позвоночника, ДНК-анализа и морфологического исследования мышечного биоптата. Лечение малоэффективно. Прогноз зависит от формы спинальной мышечной атрофии и возраста ее дебюта.
Общие сведения
Спинальные амиотрофии (спинальные мышечные атрофии, СМА) — наследственно обусловленные заболевания, в основе которых лежит дегенерация мотонейронов спинного мозга и ствола головного мозга. Описаны в конце XIX века. Их частота составляет 1 случай на 6-10 тыс. новорожденных. Около 85% спинальных мышечных атрофий составляют проксимальные формы с более выраженной слабостью и атрофиями проксимальных мышечных групп конечностей. На долю дистальных форм приходится лишь 10% СМА. На сегодняшний день спинальные амиотрофии представляют практический интерес для целого ряда дисциплин: детской и взрослой неврологии, педиатрии, генетики.
Причины
Благодаря современной генетике установлено, что возникающие дегенеративные процессы двигательных нейронов обусловлены мутациями в генах SMN, NAIP, H4F5, ВTF2p44, расположенных на 5-ой хромосоме в локусе 5q13. Несмотря на то, что спинальные амиотрофии детерминируются аберрациями одного хромосомного локуса, они представляют собой группу разнородных нозологий, одни из которых проявляются в младенческом возрасте, а другие манифестируют у взрослых. В большинстве случаев амиотрофии наследуются аутосомно-рецессивно.
Патогенез
Генетические мутации приводят к развитию дегенеративных изменений в передних рогах спинного мозга. Нарушается иннервация и нейротрофика поперечно-полосатой мускулатуры. В результате постепенно возникает атрофия мышечной ткани. Преимущественное поражение отдельных групп мышц (проксимальных или дистальных частей верхних или нижних конечностей) у различных форм спинальных амиотрофий отличается. Характерно отсутствие нарушений чувствительности.
Общепринятым считается разделение спинальных мышечных атрофий на детские и взрослые. Детские СМА классифицируются на ранние (дебютирующие в первые месяцы жизни), более поздние и ювенильные. Детские спинальные амиотрофии представлены:
- амиотрофией Верднига-Гоффманна;
- ювенильной формой Кугельберга-Веландера;
- хронической инфантильной СМА;
- синдромом Виалетто-ван Лэре (бульбоспинальная форма с глухотой);
- синдромом Фацио-Лонде.
Взрослые формы СМА манифестируют в возрасте от 16 до 60 лет и отличаются более доброкачественным клиническим течением. К СМА взрослого возраста относятся:
- бульбоспинальная амиотрофия Кеннеди;
- скапулоперонеальная;
- лицелопаточноплечевая и окулофарингеальная формы;
- дистальная СМА;
- мономелическая СМА.
Выделяют также изолированные и сочетанные спинальные амиотрофии. Изолированные СМА характеризуются преобладанием поражения спинальных мотонейронов, которое во многих случаях является единственным проявлением заболевания. Сочетанные спинальные амиотрофии представляют собой редкие клинические формы, при которых симптомокомплекс амиотрофии комбинируется с другой неврологической или соматической патологией. Описаны сочетания СМА с врожденными пороками сердца, глухотой, олигофренией, понтоцеребеллярной гипоплазией, врожденными переломами.
Симптомы спинальных амиотрофий
Общим для спинальных мышечных атрофий является симптомокомплекс симметричного вялого периферического паралича: слабость, атрофия и гипотония мышечных групп одноименных конечностей (чаще вначале обеих ног, а затем и рук) и туловища. Пирамидные нарушения не типичны, но могут развиваться на поздних стадиях. Расстройства чувствительности отсутствуют, функция тазовых органов сохранена. Обращает внимание более выраженное поражение проксимальных (при проксимальных СМА) или дистальных (при дистальных СМА) мышечных групп. Типично наличие фасцикулярных подергиваний и фибрилляций.
Болезнь Верднига-Гоффмана
Встречается в 3-х клинических вариантах. Врожденный вариант дебютирует в первые 6 мес. жизни и является наиболее злокачественным. Его симптомы могут проявляться еще во внутриутробном периоде слабым шевелением плода. Дети с рождения имеют мышечную гипотонию, не способны переворачиваться и держать голову, при более позднем дебюте — не могут сидеть. Патогномонична поза лягушки — ребенок лежит с разведенными в стороны и согнутыми в коленях и локтях конечностями.
Амиотрофии имеют восходящий характер — вначале возникают в ногах, затем вовлекаются руки, позже — дыхательная мускулатура, мышцы глотки и гортани. Сопровождается задержкой психического развития. К 1,5 годам наступает смертельный исход.
Ранняя спинальная амиотрофия манифестирует до 1,5 лет зачастую после инфекционного заболевания. Ребенок утрачивает двигательные способности, не может стоять и даже сидеть. Периферические парезы сочетаются с контрактурами. После вовлечения дыхательных мышц развивается дыхательная недостаточность и застойная пневмония. Летальный исход обычно происходит в возрасте до 5-ти лет. Поздний вариант дебютирует после 1,5 лет, отличается сохранением двигательной способности до 10-летнего возраста. Летальный исход наступает к 15-18 годам.
Ювенильная спинальная амиотрофия Кугельберга-Веландера
Характеризуется дебютом в период от 2 до 15 лет. Начинается с поражения проксимальных мышц ног и тазового пояса, затем захватывает плечевой пояс. Около четверти пациентов имеют псевдогипертрофии, что делает клинику сходной с проявлениями мышечной дистрофии Беккера. В плане дифдиагностики большое значение имеет наличие мышечных фасцикуляций и данные ЭМГ. Течение амиотрофии Кугельберга-Веландера доброкачественное без костных деформаций, в течение ряда лет пациенты остаются способными к самообслуживанию.
Бульбоспинальная амиотрофия Кеннеди
Наследуется рецессивно сцеплено с Х-хромосомой, манифестирует только у мужчин после 30-летнего возраста. Типично медленное, относительно доброкачественное течение. Дебютирует с амиотрофии проксимальных мышц ног. Бульбарные расстройства появляются через 10-20 лет и благодаря медленному прогрессированию не вызывают нарушения витальных функций. Может наблюдаться тремор головы и рук. Патогномоничным симптомом выступают фасцикулярные подергивания в периоральных мышцах. Зачастую отмечается эндокринная патология: атрофия яичек, снижение либидо, гинекомастия, сахарный диабет.
Дистальная СМА Дюшенна-Арана
Может иметь как рецессивный, так и доминантный тип наследования. Дебют приходится чаще на 20-летний возраст, но может произойти в любой период до 50 лет. Амиотрофии начинаются в кистях рук и приводят к формированию «когтистой кисти», затем охватывают предплечье и плечо, в связи с чем рука приобретает вид «руки скелета». Парезы мышц голеней, бедер и туловища присоединяются гораздо позже. Описаны случаи манифестации заболевания монопарезом (поражением одной руки). Прогноз благоприятный, за исключением случаев сочетания данного вида СМА с торсионной дистонией и паркинсонизмом.
Скапуло-перонеальная СМА Вюльпиана
Манифестирует в период от 20 до 40 лет амиотрофиями плечевого пояса. Типичны «крыловидные лопатки». Затем присоединяется поражение перонеальной группы мышц (разгибатели стопы и голени). В ряде случаев вначале поражаются перонеальные мышцы, а затем плечевой пояс. Спинальная амиотрофия Вюльпиана отличается медленным течением с сохранностью способности передвигаться спустя 30-40 лет от ее дебюта.
Осложнения
К наиболее частым неблагоприятным последствиям можно отнести постоянные падения и ассоциированные с ними патологические переломы, что связано с постепенно нарастающей мышечной атрофией мышц нижних конечностей. При бульбоспинальной амиотрофии Кеннеди нередко наблюдаются осложнения со стороны эндокринной и репродуктивной систем - эректильная дисфункция, первичное бесплодие, сахарный диабет.
Более тяжелые состояния встречаются реже, в основном при болезни Верднига-Гоффмана или на поздних стадиях других амиотрофий. Вследствие выраженной атрофии мышц глотки и дыхательной мускулатуры пища попадает в дыхательные пути (аспирация), развивается дыхательная недостаточность. Возможны грубые костно-суставные деформации, утрата способности к ходьбе и самообслуживанию. В единичных случаях обнаруживается рак грудной железы.
Больных со спинальными амиотрофиями курируют врачи-неврологи, а при манифестации в детском возрасте - педиатры или неонатологи. При необходимости может потребоваться привлечение для консультации других специалистов - эндокринологов и генетиков. Для диагностики некоторых разновидностей амиотрофий большое значение имеют анамнестические данные, а именно возраст появления симптоматики (например, болезнь Верднига-Гоффмана всегда дебютирует у детей до 6 месяцев, а амиотрофия Кугельберга-Веландера - после 2 лет).
Во время осмотра пациента обращается внимание на снижение общего мышечного тонуса, ослабление или утрату сухожильных рефлексов, скелетно-мышечные деформации. У части больных отмечается псевдогипертрофия икроножных мышц. Чтобы подтвердить или исключить диагноз назначается следующее дополнительное обследование:
- Лабораторные исследования. В лабораторных анализах практически все показатели находятся в пределах нормальных значений. Исключение составляет концентрация креатинфосфокиназы, которая у небольшой части пациентов может быть высокой.
- ЭМГ. При проведении игольчатой электромиографии обнаруживаются признаки дегенерации двигательных спинномозговых нейронов - снижение скорости и амплитуды вызванных потенциалов действия, регистрация спонтанной биоэлектрической активности в покое (фасцикуляций, фибрилляций), «ритм частокола».
- Биопсия мышц. При гистологическом исследовании мышечной ткани отмечаются типичные для амиотрофии изменения: некроз миофибрилл, разрастание жировой и соединительной ткани, чередование атрофии и гипертрофии в сочетании с интактными участками мышечной ткани.
- Спирометрия. При поражении дыхательной мускулатуры во время выполнения функции внешнего дыхания выявляются рестриктивные нарушения в виде снижения жизненной емкости легких.
- Генетический анализ. Основной метод диагностики, позволяющий достоверно установить диагноз спинальной амиотрофии. С помощью полимеразной цепной реакции обнаруживаются генетические мутации.
Дифференциальная диагностика
Дифференциальный диагноз спинальных амиотрофий необходимо проводить с другими наследственными нейродегенеративными заболеваниями, поражающими мышечную ткань. К таким патологиям можно отнести:
- псевдогипертрофическую мышечную дистрофию Дюшенна;
- ювенильный боковой амиотрофический склероз;
- детский церебральный паралич.
Лечение спинальных амиотрофий
Немедикаментозная терапия
Все без исключения больные должны быть госпитализированы в стационар. В тяжелых ситуациях (например, при дыхательной недостаточности вследствие слабости дыхательных мышц) пациентов переводят в отделение реанимации и интенсивной терапии и подключают к аппарату ИВЛ. Все мероприятия направлены на облегчение состояния пациента. Несмотря на это, комплексный подход и строгое соблюдение рекомендаций врачей позволяют улучшить качество жизни больного. Виды консервативной терапии, применяющиеся для терапии спинальных амиотрофий:
- Обеспечение питания. При значительном нарушении акта глотания особое внимание уделяется вопросу кормления. Консистенция пищи должна быть полутвердой, положение пациента вертикальным. Может понадобиться установка назогастрального зонда.
- ЛФК. С целью повышения тонуса мышц и замедления их атрофии рекомендуются регулярные физические нагрузки. Наиболее эффективно совмещение активных (выполняются специалистом) и пассивных упражнений (выполняются самим пациентом).
- Физиолечение. Для активации метаболизма в мышечных тканях назначаются сеансы электростимуляции модулированным током, грязевые аппликации, электрофорез.
- Массаж. Для улучшения кровообращения и лимфооттока в мышцах выполняются различные виды массажа - ручной (стимулирующий, расслабляющий) и аппаратный (вибромассаж).
- Ортопедическое лечение. Для предупреждения и коррекции костных деформаций и суставных контрактур рекомендуется использование ортопедических приспособлений: корсетов, ортезов, ортопедической обуви.
- Респираторная поддержка. Нередко возникает необходимость в устранении кислородной недостаточности. В зависимости от тяжести состояния больного назначаются ингаляции кислорода через лицевую маску/назальную канюлю или неинвазивная вентиляция легких через портативные аппараты ИВЛ.
Для достижения максимального эффекта лечение должно проводиться непрерывно и подбираться индивидуально для конкретного пациента.
Медикаментозная терапия
Этиотропная терапия с доказанной эффективностью на сегодняшний день отсутствует. Лекарственные препараты, применяющие для лечения спинальных амиотрофий, следующие:
- Метаболические средства. Для улучшения обменных процессов в мотонейронах и мышечных тканях применяются препараты, стимулирующие функцию митохондрий (коэнзим Q10, янтарная кислота), ноотропы (пирацетам, гамма-аминомасляная кислота), L-карнитин.
- Вальпроаты и Кленбутерол. Исследования показали, что противоэпилептические препараты из группы производных вальпроевой кислоты и агонист бета-адренергических рецепторов Кленбутерол способны увеличивать образование белка выживаемости мотонейронов (SMN) и, соответственно, улучшать клиническое течение заболевания.
- ИПП и прокинетики. Пациентам с гастроэзофагальным рефлюксом, который нередко развивается при нарушении глотания, назначаются ингибиторы протонной помпы (эзомепразол) и средства, стимулирующие моторику желудочно-кишечного тракта (итоприд).
- Муколитики и отхаркивающие средства. У больных со слабостью дыхательной мускулатуры для устранения таких проблем как слабое отхаркивание и скопление в дыхательных путях густой мокроты применяются препараты, разжижающие мокроту (ацетилцисетин) и стимулирующие ее отхаркивание (терпингидрат).
- Гормональные препараты. Людям с возникшими эндокринологическими осложнениями показаны инсулин, сахароснижающие препараты (метформин), тестостерон и антиандрогены (дутастерид).
Хирургическое лечение
При развитии грубых деформаций грудной клетки и позвоночника или крайне выраженных контрактур суставов показаны ортопедические операции. Лежачим больным, страдающим постоянно рецидивирующими пневмониями, выполняется трахеостомия. При гастроэзофагеальном рефлюксе, резистентном к медикаментозному лечению, прибегают к лапароскопической фундопликации Ниссена.
Экспериментальное лечение
Прогноз
Прогноз всецело зависит от клинического варианта СМА и возраста ее манифестации. Наиболее неблагоприятный прогноз имеют детские спинальные амиотрофии, при начале в младенческом возрасте они зачастую приводят к летальному исходу в течение первых 2-х лет жизни ребенка. Спинальные амиотрофии взрослого возраста отличаются способностью больных самостоятельно обслуживать себя в течение многих лет, а при медленном прогрессировании имеют благоприятный прогноз не только для жизни, но и для трудоспособности пациентов (при создании для них оптимальных условий труда).
Профилактика
Специфических методов первичной профилактики не существует. Единственный способ предотвратить возникновение болезни - пренатальная диагностика (обнаружение мутаций в ворсинах хориона или амниотической жидкости) с прерыванием беременности. Предупредить развитие осложнений и максимально сохранить работоспособность позволяет своевременное начало комплексной терапии.
Гипоплазия эмали
Гипоплазия эмали - недостаточное развитие поверхностного слоя (эмали) молочных или постоянных зубов. Крайне выраженной формой гипоплазии эмали является аплазия - ее полное отсутствие. Проявляется изменением формы и внешнего вида зубов, наличием белесых или депигментированных участков, бороздок, углублений, при аплазии - болевыми ощущениями на различные раздражители. Наиболее часто гипоплазия эмали ведет к развитию глубокого кариеса, пульпита, формированию неправильного прикуса.
Гипоплазия эмали - это врожденный порок недоразвития зуба или его тканей связанный с нарушения обмена веществ у плода. Аплазия эмали является крайним выражением гипоплазии и проявляется полным отсутствием эмалевого покрытия или же отсутствием зуба.
Причины развития
Возникают гипоплазии эмали из-за серьезных нарушений обмена веществ в организме плода. И основной причиной является либо патология закладок клеток зародыша, либо неблагоприятные факторы, негативно влияющие на плод.
Не вполне корректно утверждение о том, что гипоплазия эмали возникает только из-за нарушения минерального обмена и участки гипоплазии - это ни что иное, как зона деминерализации. Если бы это было основной причиной гипоплазий эмали, то патология не была столь широко распространена. На сегодняшний день заболеваемость постоянно растет, так как вредные факторы начинают действовать на зачатки зубов еще задолго до формирования и рождения плода. Выводы о том, что токсикозы и инфекционные заболевания у беременной приводят к аномалиям развития плода, вполне обоснованны. Исследования подтверждают, что аплазии эмали и другие аномалии зубов чаще встречаются у детей, чьи матери перенесли во время беременности ОРВИ, краснуху, токсоплазмоз. Или же если часть беременности протекала с серьезными токсикозами. Недоразвитие эмали наблюдается у недоношенных детей и у детей, получивших травмы во время родов. Предрасполагающими факторами являются энцефалопатии, атопические дерматиты, рахит и другие нарушения обмена кальция.
Гипоплазия эмали в той или иной степени обнаруживается практически у половины детей дошкольного и младшего школьного возраста. При этом она носит системный характер и чаще наблюдаются поражение нескольких зубов. Гипоплазия эмали с глубокими изменениями диагностируют у 40% клинически здоровых детей.
Гипоплазия эмали диагностируется на молочных и на постоянных зубах, при этом чаще она встречается именно на постоянных зубах, что ведет к увеличению риска заболеваний зубов в старшем возрасте. Нарушения эмалевого покрытия молочных зубов связаны с патологиями, протекающими в организме женщины во время беременности. Тогда как гипоплазия эмали постоянных зубов обусловлена нарушениями процессов обмена веществ в организме ребенка, которые развиваются, начиная с 5-6 месяца жизни. А поскольку заболевания в период первого года жизни встречаются гораздо чаще, чем серьезные патологии во время беременности, то соответственно гипоплазия эмали постоянных зубов превалирует, что является серьезной проблемой.
Локализация и групповая принадлежность зубов при гипоплазии эмали зависит от того, в каком возрастном периоде ребенок перенес заболевание, которое и стало главным патогенетическим звеном. Так, заболевания, перенесенные в первые месяцы жизни, приводят к гипоплазии эмали режущего края центральных резцов и бугров шестых зубов. Это связано с формированием данных зубов на 5-6 месяце жизни ребенка.
Если же заболевания вызывают глубокие изменения в обмене веществ ребенка или продолжается в течение длительного времени, то участки гипоплазии эмали наблюдаются по всей длине коронки и на поверхности зуба. Неровная структура эмали указывает на длительность и волнообразность заболеваний, перенесенных в период формирования зубов. Тяжесть перенесенных заболеваний сказывается и на глубине изменений эмали. Так, незначительные патологии могут проявляться лишь меловидными пятнами, а тяжелые заболевания могут вызвать недоразвитие эмали вплоть до ее аплазии.
При местной гипоплазии эмали поражаются только часть зубов. Обычно ее возникновение связано с локализованными нарушениями обмена веществ вблизи зачатков постоянных зубов. Воспалительные процессы в области верхушки корней молочных зубов способствуют возникновению местной гипоплазии эмали. Такой вид гипоплазии чаще встречается на малых коренных зубах, потом как их зачатки расположены между корнями молочных моляров.
Клинические проявления
Системная гипоплазия
В зависимости от тяжести, системная гипоплазия эмали может проявляться изменением цвета эмали, ее недоразвитием или же полным отсутствием.
Изменение цвета эмали проявляется в виде симметричных белых пятен различных форм, которые расположены на одноименных зубах. Меловые пятна обнаруживаются на вестибулярной поверхности и не сопровождаются неприятными или болезненными ощущениями. Диагностическим признаком является то, что наружный слой эмали на пораженном участке гладкий, блестящий и не изменяет цвет при взаимодействии с красителями. На протяжении жизни пятно не изменят ни цвет, ни форму.
Более тяжелые проявления гипоплазии чаще не заметны при обычном осмотре. Волнистая, точечная и бороздчатая эмаль проявляется после высушивания поверхности. При тщательном осмотре стоматологу становятся заметны чередования небольших валиков и углублений с неизмененной эмалью. Чаще эта форма гипоплазии проявляется в виде точечных углублений в эмали, которые располагаются на разном уровне. В первое время эти участки имеют нормальный цвет, но по мере дальнейшего роста зуба эти участки постепенно пигментируются. В некоторых случаях гипоплазия эмали проявляется в виде одиночной гиперпигментированной полосы на коронке зуба. Иногда эта бороздка довольно глубока и происходит заметное уменьшение размера коронки зуба в виде перехвата, такую форму гипоплазии называют бороздчатой. Очень редко наблюдается лестничная гипоплазия, когда на коронке зубов формируется несколько бороздок. Но характерным является то, что даже при тяжелых формах таких гипоплазий эмали, ее целостность не нарушена.
Реже других изменений встречается аплазия эмали на определенном участке. При этом болевой синдром формируется при контакте с раздражителем и проходит после его устранения. Клинически данная патология проявляется отсутствием эмали на части коронки зуба, но чаще на дне чашеобразного углубления, либо в бороздке, охватывающей коронку зуба. Часто при аплазии эмали имеется и недоразвитие дентина. Это проявляется изменениями формы зубов, характерной для данной группы.
Изменения формы зубов при гипоплазиях эмали
- Зубы Гетчинсона. При данной патологии верхние центральные резцы имеют отверткообразную или бочкообразную форму. Их размер у шейки несколько больше, чем у режущей поверхности и имеется полулунная выемка у режущего края. Ранее полагалось, что данный симптом наблюдается только при врожденном сифилисе, однако позже установили, что аномалия возникает и при иных причинах.
- Зубы Пфлюгера. При данном симптоме поражены первые моляры, размер коронки у щеки больше, чем у жевательной поверхности. Бугры недоразвиты, что придает зубам схожесть с конусом.
- Зубы Фурнье. Клинически выглядят так же, как и при симптоме Гетчисона, но без полулунной выемки.
Местная гипоплазия эмали.
Такие гипоплазии возникают на постоянных зубах из-за вовлечения в воспалительный процесс зачатков зубов или вследствие механической травмы развивающего зачатка. Клинически этот тип гипоплазий проявляется в виде белых или желтовато-коричневых пятен, а чаще в виде точечных углублений по всей поверхности. В некоторых случаях эмаль коронки зуба полностью или частично отсутствует.
Гипоплазии эмали зубов приводят к тому, что микробы более агрессивно воздействуют на дентин, беспрепятственно проникая в него и вызывая глубокий кариес. Вторым серьезным осложнением является поражение других тканей зуба - цемента, дентина и пульпы, так как гипоплазии эмали редко протекают изолированно. У многих детей впоследствии формируется неправильный прикус.
Лечение
Тактика лечения зависит от степени выраженности гипоплазии, так при одиночных пятнах и неглубоких поражениях эмали этиотропное лечение не проводится. Проводят профилактику кариеса и уделяют больше внимания уходу за полостью рта. Иногда, если пятна расположены на вестибулярной поверхности зубов, то они становятся косметическим дефектом, так как хорошо видны во время разговора. Для их устранения проводят пломбирование композиционными материалами. Если присутствуют изменения эмали в виде точечных углублений и перехватов, то тоже устраняют с помощью пломбирования.
Ярко-выраженные дефекты эмали и дентина являются показанием для ортопедического лечения с установкой металлокерамических коронок. Профилактикой гипоплазии эмали является гармоничное развитие ребенка, для того чтобы не допустить серьезных заболеваний в период формирования иммунитета.
Читайте также:
- Причины и возбудитель дизентерии. Механизмы развития дизентерийной инфекции
- Ранения поджелудочной железы
- Некроз кости после облучения. Гистохимия кости после радиационного поражения
- Тактика при повреждениях тазового кольца. Диагностика, лечение
- Приобретенная гемангиома конъюнктивы на широком основании: признаки, гистология, лечение, прогноз