Прионовые болезни у ребенка
Добавил пользователь Владимир З. Обновлено: 01.02.2026
Медленные инфекции ЦНС — поражения центральной нервной системы вирусными вирионами или инфекционными прионами, возникающие после длительного скрытого (инкубационного) периода. Клинически характеризуются парезами, гиперкинезами, расстройством мозжечковых функций, психическими нарушениями, когнитивным снижением до глубокой деменции. Диагностика осуществляется при помощи неврологического обследования, церебральной томографии, анализа спинномозговой жидкости, определения антивирусных антител в крови. Лечение проводится симптоматическими средствами.
Общие сведения
Понятие медленные инфекции ЦНС включает целый ряд неврологических болезней, обусловленных вирионами (вирусными частицами) и прионами (вирусоподобными белками). Первые данные были опубликованы в 1954 году в Исландии учёным, длительно наблюдавшем неописанные ранее заболевания овец, поражающие ЦНС. Автор дал им название медленные инфекции. В 1957 году появилось описание нового заболевания — куру, распространённого среди жителей Новой Гвинеи. Болезнь полностью соответствовала критериям медленных инфекций и открыла список подобных патологий у человека, который продолжает пополняться. Медленные инфекции ЦНС — редко встречающаяся группа нозологий, точные данные о заболеваемости не собраны. Одни формы распространены повсеместно, другие имеют эндемичный характер.
Причины медленных инфекций ЦНС
Изучение свойств возбудителей позволило установить вирусный характер инфекций. Ранее ошибочно предполагалось, что возбудителями выступают специфические вирусные агенты. В последующем удалось определить два этиологических фактора возникновения патологии: вирусы и прионы.
- Вирусы. В настоящее время опровергнута теория специфической этиологии, подтверждена роль обычных вирусов: полиомавируса, флавивируса, цитомегаловируса, вирусов кори, краснухи, простого герпеса. Медленные инфекционные процессы в ЦНС развиваются вследствие персистирования вируса в организме на протяжении многих лет после перенесённого в типичной форме заболевания. Заражение может происходить воздушно-капельным, алиментарным, парентеральным, трансплацентарным путём.
- Прионы. Являются белками, обладающими некоторыми свойствами вирусов, в отличие от последних не имеют ДНК или РНК. Инфекционные прионы вызывают развитие болезни путём трансформации аналогичных нормальных белков нервных клеток в патологические. Инфицирование происходит при употреблении в пищу недостаточно термически обработанного мяса заражённых животных, трансплантации содержащих патогенные прионы тканей, гемотрансфузиях, нейрохирургических вмешательствах.
Доподлинно неизвестно, что становится причиной многолетнего персистирования вирусов, которые остаются в организме переболевших обычной инфекцией пациентов. Возможными причинами считают дефектное строение вирионов, недостаточность иммунной системы, сопровождающуюся пониженной выработкой антител, активацию пролиферативных процессов внутри поражённых вирусами клеток.
Патогенез
Общей патогенетической характеристикой, объединяющей различные медленные инфекции, выступает длительное скрытое развитие патологии, сопровождающееся накоплением возбудителя в церебральных тканях. После перенесённого вирусного заболевания (чаще внутриутробно или в раннем детстве) возбудители остаются в клетках головного мозга в неактивной форме. Причины и механизмы их активации не установлены. Перейдя в активную фазу, возбудители вызывают постепенное развитие воспалительных изменений в ЦНС.
Попавший в клетку прион взаимодействует с находящимся внутри неё геном, что приводит к синтезу аналогичных прионов вместо нормальных клеточных белков. Продолжительный скрытый период обусловлен временем, требующимся для попадания прионов в головной мозг, длительным процессом внутриклеточного накопления синтезируемых патологических белков. Результатом аномального белкового синтеза являются метаболические изменения, приводящие к гибели нейрона.
Морфологическая картина медленных инфекций достаточно вариабельна. Наиболее часто в тканях ЦНС наблюдается формирование очагов глиоза, демиелинизирующихся участков. При истинно вирусной этиологии процесса типично образование периваскулярных лимфоцитарных инфильтратов, фокусов астроцитоза. Морфологические изменения захватывают различные области мозга, часто носят распространённый характер.
Классификация
Медленные инфекции ЦНС имеют различную клиническую картину, однако отмечаются отдельные особенности течения болезней, связанные с их вирусным или прионным генезом. С учетом этого обстоятельства в неврологии заболевания подразделяются по этиологическому принципу на:
- Вирионные — вызванные типичными вирусами. Сопровождаются выработкой специфичных противовирусных антител. Наиболее распространены подострый склерозирующий панэнцефалит, прогрессирующая мультифокальная лейкоэнцефалопатия, краснушный панэнцефалит.
- Прионные — вызванные белками-прионами. Близкое сходство инфекционных прионов с внутриклеточными белками организма обуславливает практически полное отсутствие иммунного ответа при их внедрении. Большинство случаев составляет болезнь Крейтцфельдта-Якоба. К прионным инфекциям относятся также фатальная семейная инсомния, куру, синдром Герстмана.
Симптомы медленных инфекций ЦНС
Общей чертой заболеваний этой группы является медленное незаметное начало без температурной реакции. Характерен продромальный период, в котором отмечается раздражительность, эмоциональная неуравновешенность, рассеянность больного, лёгкие расстройства координации, шаткость во время ходьбы. Период клинической манифестации отличается постепенным нарастанием симптоматики, длящимся 1-3 недели. Типичны экстрапирамидные и пирамидные нарушения, атаксия, психические расстройства, когнитивное снижение.
Экстрапирамидные симптомы включают гиперкинезы (атетоз, тремор, дистонические синдромы), иногда — брадикинезию, паркинсоническую скованность. Пирамидные двигательные расстройства протекают в виде прогрессирующих геми- и тетрапарезов. Возможно поражение черепных нервов, проявляющееся парезом лицевой мускулатуры, тугоухостью, нарушением зрения, затруднением глотания и пр. Психические отклонения характеризуются эпизодами эйфории, фобий, бреда, спутанного сознания, отрывочных галлюцинаций. Все медленные инфекции сопровождаются постепенным распадом интеллектуальных функций (памяти, мышления, внимания) с исходом в глубокую деменцию. Нарушения речи обусловлены одновременно сенсомоторной афазией и когнитивным дефицитом. В терминальной стадии наблюдается мутизм — речь отсутствует полностью.
Симптоматика каждой отдельной инфекции имеет свои особенности. Для болезни Крейтцфельдта-Якоба, краснушного панэнцефалита характерна мозжечковая атаксия. Отличительным клиническим проявлением фатальной бессонницы выступает инсомния, доводящая пациентов до психического и физического истощения. Базовым симптомом болезни куру является тремор, типична насильственная улыбка. Синдром Герстмана-Штраусслера-Шейнкера протекает с мышечной гипотонией и угнетением сухожильных рефлексов.
Диагностика
Поскольку медленные инфекции — редкие заболевания, диагностировать их непросто. Неспецифическая клиническая симптоматика, трудности выделения вируса-возбудителя, инфекционного приона осложняют диагностику. Диагностический поиск осуществляется в рамках следующих исследований:
- Сбор анамнеза. Большое значение имеет расспрос относительно перенесённых в прошлом (возможно внутриутробно) инфекций, операций с пересадкой тканей. Опрос включает выявление продромальных симптомов, особенностей дебюта патологических проявлений.
- Оценка неврологического статуса.Неврологи исследует двигательную, чувствительную, рефлекторную, когнитивную сферы, координацию. На основании полученных данных формируется картина многоочагового поражения, свидетельствующая о диффузном характере патологических изменений церебральных тканей.
- Нейровизуализация. Проводится при помощи МРТ, КТ, МСКТ головного мозга. Томография определяет мультифокальное поражение мозга в виде демиелинизации, дегенерации, атрофии. Наблюдается расширение желудочков, говорящее о наличии гидроцефалии.
- Исследование цереброспинальной жидкости. Материал получают при люмбальной пункции. Отсутствие воспалительных изменений спинномозговой жидкости позволяет исключить типичные нейроинфекции. Проводятся ПЦР-исследования, направленные на выявление ДНК вероятных возбудителей, анализ на наличие противовирусных антител. В случае вирионного генеза инфекции эти методики дают возможность верифицировать возбудителя у 70-90% больных.
- Анализ крови на антитела. Информативен в случае вирусной этиологии. Осуществляется с определением противокоревых, противокраснушных антител. Диагностически значимы повторные исследования, демонстрирующие нарастание титра в период активации вируса.
- Биопсия мозга. Выполняется при крайней необходимости. Исследование биоптатов позволяет выявить интранейрональные скопления прионов. Однако в ходе биопсии существует вероятность забора участка неизменённых тканей.
При наличии показаний проводятся консультации смежных специалистов: офтальмолога, психиатра, инфекциониста, генетика. Необходима дифференциальная диагностика с энцефалитами, имеющими подострое начало, сосудистой деменцией, хроническими энцефалопатиями, болезнью Альцгеймера.
Лечение медленных инфекций ЦНС
Эффективная терапия не разработана. Предложенные схемы лечения противовирусными фармпрепаратами оказались безрезультатными. Назначается симптоматическая терапия, которая позволяет облегчить состояние пациентов, но не может замедлить прогрессирование патологического процесса. Она включает противоотёчные, нейропротекторные, витаминные, психотропные, противосудорожные средства. Поиск эффективных способов лечения продолжается.
Прогноз и профилактика
Медленные инфекции ЦНС остаются смертельными заболеваниями. Гибель пациентов вследствие тотального поражения мозга происходит в среднем в течение 1-2 лет от момента развития клинических симптомов. Наибольшая продолжительность жизни наблюдается у больных синдромом Герстмана — 3-5 лет. Профилактические мероприятия сводятся к предупреждению распространения вирусных инфекций, поддержанию должного уровня иммунитета. В отношении кори и краснухи возможна специфическая профилактика, которая проводится путём обязательной вакцинации детей соответствующими вакцинами. Способы предупреждения прионных заболеваний не найдены, поскольку отсутствуют методики определения прионов в трансплантируемых тканях, препаратах крови.
О прионах впервые заговорили не так давно: в 1960 году. Через полтора десятка лет за изучение этой группы заболеваний была вручена первая Нобелевская премия. Потом еще несколько, но прионы все еще остаются во многом загадкой.
Вместе с MedAboutMe попробуем заглянуть в лаборатории ученых, чтобы понять, что такое прионы и что с ними делать.
Немного информации
Первыми заговорили о прионах Джон С. Гриффит, математик, и Тиквах Альпер, радиобиолог. Они изучали некую инфекционную частицу, которая оказалась слишком мала для вируса, и слишком устойчива к ионизирующему излучению.
Оказалось, что эти частицы представляют собой некий белок, обладающий особенными свойствами.
Белки, как известно, представляют собой цепочку аминокислот, свернутую в трехмерную структуру. Прионный белок свернут неправильно, его структура отличается от нормальной белковой молекулы. Но самое неприятное — прионы умеют «перекручивать» нормальные белки в такие же неправильные структуры, вызывая настоящую цепную реакцию. «Перекрученные» прионами белки собираются в амилоидные комплексы, в которых они укладываются плотными «пачками», образующими белковые волокна — фибриллы. Эти белковые нити растут в обе стороны, а если их разорвать — каждый кусочек начинает расти с обеих сторон.
Никакой собственной информации эти частицы в организм не привносят: просто изменяют структуру присутствующих, естественных, «родных» белков, в результате чего те также становятся инфекционными частицами.
На сегодняшний день известно несколько заболеваний, вызываемых прионами. Это почесуха овец (скрейпи), губчатая энцефалопатия коров (коровье бешенство), хроническую изнуряющую болезнь (CWD) лосей и оленей у животных.
У людей к прионным заболеваниям относят новогвинейскую куру, болезнь Герстмана-Штраусслера-Шейнкера, очень редкую болезнь — фатальную семейную бессонницу, и наиболее распространенную, но при этом тоже очень редкую болезнь Крейтцфельдта-Якоба. Недавно появилась информация о том, что прионные молекулы белка обнаружены также в растительном мире — у растения Arabidopsis thaliana.
Нобелевская премия 1976 года была вручена именно за работу по изучению болезни куру и доказательство ее прионной природы. Это заболевание было распространено среди коренных обитателей Новой Гвинеи. Оказалось, что заражение происходило при ритуальных актах каннибализма: у этого народа было принято поедать мозг умерших. Прекращение практики антропофагии привело к исчезновению болезни.
Все формы заболевания имеют длительный инкубационный период, во время которого не проявляется никаких симптомов. А когда признаки появляются — помочь больному уже ничем нельзя. Прионы постепенно разрушают мозг, превращая его в некое подобие старой губки со множеством дырочек. Человек или животное постепенно утрачивают контроль за телом, оказываются не в состоянии двигаться, питаться. Болезнь заканчивается полным распадом личности и гибелью.
Все известные ныне проявления прионных болезней у млекопитающих вызваны одним и тем же белком PrP. Хотя структура этого белка похожа у разных видов животных и у человека, некоторые различия все-таки существуют, что объясняет наличие так называемого «видового барьера». Это означает, что обычно не происходит передачи прионов от жиотвных одного вида другим. Обычно — не означает никогда. Так, например, губчатая энцефалопатия коров и болезнь Крейтцфельдта-Якоба у человека вызывается одним и тем же прионом. Передача инфекции возможна при употреблении в пищу мяса больного животного.
Откуда берутся прионы?

Эти загадочные «неправильные белки» могут передаваться от родителей детям, и долгое время вести себя тихо и никак не проявляться. На долю наследственной передачи приходится около 10% зафиксированных случаев.
Прионы могут передаваться различными путями от зараженных животных и людей, доказана алиментарная форма передачи инфекции. Так происходит примерно в 5 случаях из 100. Нормальный белок может «перекрутиться» в прион спонтанно, после чего начнется патологический процесс, в который будут вовлекаться все новые и новые молекулы белка PrPC, превращаясь в искаженную форму PrPSc. Именно так возникает 85% случаев заболевания.
Две эти формы абсолютно идентичны по последовательности аминокислот, но имеют разные пространственные формы. При этом патологическая форма чрезвычайно устойчива к денатурации химическими веществами и физическому воздействию, включая радиацию.
Накопление прионов в клетке нарушает ее работу, а затем приводит к гибели.
Причины спонтанного изменения нормального белка все еще остаются загадкой, хотя некоторые факторы, вызывающие превращение, уже известны. В частности, запустить прионную болезнь может перенесенное заболевание коронавирусной инфекцией, о чем сообщили в ноябре 2021 года врачи из Санкт-Петербурга. О том же сообщается в статьях ученых из разных стран: постковидные осложнения вызывают запуск прионной болезни. Высказываются опасения, что пандемия коронавируса может стать началом другой эпидемии — прионной болезни, и это уже очень серьезно.
До пандемии частота случаев болезни Крейтцфельдта-Якоба не превышала 1: 1000000000. Как будет после — пока не может сказать никто.
Чем опасны прионы?
Эти инфекционные частицы очень малы даже для вирусов. Они не несут в себе генетической информации, не вызывают образования антител. Их очень сложно обнаружить до того, как начнут появляться симптомы, а тогда уже мало что можно сделать.
Прионы легко передаются.
Летом 2021 года во Франции сразу пять лабораторий остановили программы изучения прионов после того, как произошло несколько случаев заражения сотрудников. Такие случаи были и прежде: например, в 2010 году в одной из лабораторий нечаянно поранилась Эмили Жоме, работавшая с прионами. Первые симптомы болезни Крейтцфельдта-Якоба появились через 7 лет, а в 2019 году женщина скончалась.
В описанном случае болезнь не проявлялась в течение нескольких лет, все это время в организме происходили необратимые изменения. И все это время опасным заболеванием могли заразиться окружающие люди.
Как правило, заболевание представляет собой прогрессирующую деменцию, сопровождающуюся миоклонусом и атаксией. В начале заболевания могут отмечаться также различные зрительные расстройства, эпилептиформные приступы, изменения поведения (агрессия, бессонница, тревожность, галлюцинации и т. д.)
Определенные подвижки есть. Американским исследователям удалось «сбить с толку» прионы, подсунув им так называемые «антисмысловые олигонуклеотиды», мешающие прионам воспроизводить себе подобные молекулы. В результате инъекций у лабораторных мышей развитие болезни замедлилось на 98%, что можно считать очень хорошим результатом.
Тем не менее, замедлить — не означает победить. Изучение прионов необходимо продолжать, с соблюдением всех мер безопасности. Потому что если существует вероятность распространения опасной инфекции, человечество должно быть готовым противостоять ей.
Прионы — белки с эффектом царя Мидаса

Прионы — одна из самых больших загадок в биологии и медицине. Возможно, когда-то именно эти неживые вещества, ведущие себя подобно микроорганизмам, привели к исчезновению неандертальцев. Сегодня же они являются объектом огромного интереса со стороны врачей и ученых. MedAboutMe рассказывает о нюансах прионных инфекций и медленных смертельных болезнях, которые они вызывают.
XX век — столетие прионов

XX век можно по праву назвать «столетием прионов»: за это время ученые нашли причину загадочной болезни и столкнулись с массовыми ее проявлениями, которые показали всю серьезность проблемы.
Хотя сами прионы были открыты сравнительно недавно, свою историю в медицине прионные инфекции ведут с XVIII века. Первые упоминания об овечьей почесухе (скрепи) в Англии датируются 1732 годом. Явление было столь распространено, что его даже обсуждали в Британском парламенте. В 1899 году ученые доказали инфекционную природу заболевания: то есть тот факт, что необычное поведение овец, которые в результате умирали в судорогах, заразно и передается через повреждения на коже и пищу. Среди характерных признаков скрепи были очень длительный инкубационный период и специфические поражения тканей и органов.
В 1954 году Б. Сигурдссон представил научному сообществу результаты своих изысканий в области скрепи у исландских овец. А через 3 года ученые описали аналог этой болезни — у аборигенов Новой Гвинеи. Инфекция распространялась в ходе ритуального каннибализма: в знак уважения к умершему соплеменнику аборигены съедали его мозг и, вместе с опытом и мудростью, получали прионы (если таковые у него имелись). Сами жители Новой Гвинеи называли эту болезнь «куру» и считали ее результатом действия злых духов.
На протяжении следующих 30 лет ученые высказывали самые разные догадки на тему прионов. Сама идея «размножения» белковых тел без помощи ДНК или РНК звучала абсурдно и противоречила базовым основам молекулярной биологии. До сих пор существуют приверженцы вирусной теории происхождения прионов. Но в 1982 году Стенли Прузинер объявил об обнаружении инфекционного агента, состоящего практически полностью из белка. Через два года прион был изолирован, а в 1997 году Прузинер получил Нобелевскую премию по физиологии и медицине.
В 1986 году в Великобритании разразилась эпидемия трансмиссивной губчатой энцефалопатии рогатого скота. Источником инфекции стала мясо-костная мука, которой выкармливали телят. Изначально при ее производстве сырье (кости, остатки туш животных) обрабатывалось при температуре до 130°С, но в конце 1970-х годов производители изменили технологию и снизили температуру обработки до 110°С. Это и привело к массовому заражению коровьим бешенством. Попутно оказалось, что многие другие виды млекопитающих, которым перепадала мясо-костная мука или части зараженных коров, также болеют губчатой энцефалопатией.
В марте 1996 года англичане сообщили о первых 10 жертвах прионов среди людей. Через месяц стало известно о пострадавших во Франции, а на данный момент число жертв губчатой энцефалопатии среди людей превысило 200 человек только на территории Европы. Число стран, где были зафиксированы случаи коровьего бешенства у животных, к 2006 году достигло 40.
Прионы — форма белковой псевдожизни

Итак, прионы — это группа белков, имеющих определенную трехмерную конфигурацию, которая считается аномальной. При встрече с аналогичным белком нормальной конфигурации прион запускает процесс его превращения в такую же аномальную структуру. Как и легендарный царь Мидас, превращавший в мертвое золото все, к чему он прикасался, прионы трансформируют нормальный белок в себе подобный, способный разносить инфекцию дальше. То есть, стоит подхватить прионную инфекцию, как в организме запускается цепная реакция, медленно, но верно разрушающая ткани мозга (прионы проходят сквозь гематоэнцефалический барьер).
Но, в отличие от царя Мидаса, прионы «переводят на сторону зла» не все белки подряд, а только PrP — прионовый белок (prion protein) или белок, устойчивый к протеазе (protease-resistant protein). Он кодируется геном PRNP (PRioN Protein) и в основном производится в нервной системе, но его можно найти и в других тканях организма. Он имеет две основных изоформы:
За производство прионной формы белка отвечает тот же ген, но имеющий одну или несколько из почти 40 мутаций, известных на сегодняшний день. Постепенно трансформируя подходящие белки, прионы образуют амилоиды — агрегаты белковой природы, сходные с амилоидными бляшками, формирующимися при болезни Альцгеймера. И это приводит к аналогичным последствиям — разрушению нервных клеток. Поэтому прионные инфекции относят к нейродегенеративным заболеваниям.
В ходе уничтожения нейронов на их месте образуются дыры («вакуоли»), а структура ткани мозга начинает в этом месте напоминать губку. Отсюда официальное название этой группы заболеваний — губчатые или спонгиоформные энцефалопатии.
Среди ученых до сих пор не утихают споры на тему, являются ли прионы формой жизни. С одной стороны, они размножаются: был один прион, нашел подходящий белок — стало два приона. С другой стороны, у них нет ДНК (или РНК) — переносчика наследственной информации, как у вирусов, бактерий, грибков и других инфекций.
Для кого опасны прионы?

Прионы опасны для всех форм жизни, у которых есть белок PrP. Прионные болезни уже обнаружены, кроме человека, у овец, коз, коров, норок, оленей, лосей, мулов, кошек (и домашних, и диких), антилоп, страусов и даже у дрожжей. К счастью для всего живого на Земле, прионные инфекции в большинстве своем видоспецифичны. Это значит, что PrP белки разных видов незначительно отличаются друг от друга, поэтому в большинстве случаев у человека нет шансов умереть, скажем, от прионов страуса или дрожжевого грибка.
Но из каждого правила есть исключения. Прионы, губительные для коров, вызывают у человека один из вариантов болезни Крейтцфельдта-Якоба. А подхватить коровий прион можно, съев зараженное мясо. Правда, не каждый человек, употребивший бифштекс коровы, которая скончалась от губчатой энцефалопатии рогатого скота, заболеет. Для заражения необходима комбинация факторов, в число которых входит генетическая предрасположенность к таким инфекциям.
Сегодня выделяют следующие разновидности прионных болезней человека:
Упоминавшаяся выше болезнь гвинейских аборигенов, сегодня практически полностью искорененная в силу отказа племен от каннибализма.
Крайне редкое, наследственное заболевание с инкубационным периодом от 5 до 30 лет.
Тоже очень редкое и передающееся по наследству заболевание, в результате которого человек погибает от необратимых повреждений мозга на фоне бессонницы.
Она же — коровье бешенство или трансмиссивная спонгиоформная энцефалопатия и др. Эта болезнь существует в нескольких модификациях. Выделяют спорадическую (sCJD), наследственную (fCJD) и ятрогенную (iCJD) формы. А с 1955 года, после вспышки коровьего бешества в Великобритании — новый вариант nvCJD.
Статистика, собранная в отношении болезни Крейтцфельдта-Якоба, показывает, что только в 1-5% случаев человек может получить прионную инфекцию путем заражения извне, например, при пересадке органов и тканей. В 10-15% случаев заражение является наследственным заболеванием. И, наконец, в 85% случаев причина внезапного развития болезни остается неизвестной. Чаще всего, она поражает людей в возрасте от 40 до 69 лет.
Уничтожить прионы: миссия невыполнима

Иммунная система ничего не имеет против прионной версии белка, принимая PrPSc за «своего» и не подозревая о его вреде для здоровья . Поэтому никакого сопротивления со стороны организма аномальный белок не встречает. Инкубационный период прионных болезней может длиться десятками лет, за что они вошли в список «медленных инфекций». Но с момента появления первых симптомов времени у жертвы практически не остается, и она довольно быстро погибает.
Ученые давно бьются над проблемой ранней диагностики прионных заболеваний, но пока их поиски не увенчались особым успехом. В биологических жидкостях, то есть в крови и моче, прионы присутствуют в столь малых концентрациях, что определить их довольно сложно. Самый точный метод их выявления — анализ образцов тканей мозга, но его можно провести только посмертно. О ранней и массовой диагностике, таким образом, пока речи не идет.
Лекарств против прионов не существует. Они настолько просты, что близки к совершенству в плане противостояния любых угроз. Это всего лишь хорошо упакованный белок. Его невозможно уничтожить кислотой, УФ-излучением, ферментами, разрушающими другие белки, и даже ионизирующая радиация бессильна против прионных частиц — настолько они малы.
Можно ли защитить здоровье человека?
Организм человека сам производит белок PrP, неправильная конфигурация которого приводит к смертельному заболеванию. Почему бы не избавиться от этого белка? Если найти способ «выключить» ген, который его кодирует — человечество и иные живые существа, подверженные прионным заболеваниям, были бы надежно защищены от них. Но нет. Сделать это невозможно по той простой причине, что данный ген и данный белок жизненно необходимы. При их отсутствии (или «отключении») вокруг нервных волокон перестает формироваться миелиновый слой — своего рода, изолирующая защитная оболочка для отростков нервных клеток. А это приводит к болезням периферической нервной системы и, как следствие, к параличам.
Меры профилактики: запрет на мясо и лекарства

Особое внимание уделяется использованию различных органов и тканей крупного рогатого скота для производства лекарств, косметических средств и медицинских изделий. Безопасных частей зараженного животного не существует. Это, кстати, привело к тому, что в Великобритании по итогам эпидемии было запрещено производство и использование большого числа лекарств, производимых из тканей и органов коров. И в России под запрет попали некоторые препараты (кортикотропин, лактин и питуитрин для инъекций и др.). Некоторые лекарственные средства (актовегин, солкосерил) запрещены в одних странах (США) и разрешены в других (Россия).
Справится ли человечество с прионами — покажет время. А пока банальный совет: не покупайте мясо, чистота которого не подтверждена санитарными сертификатами. Прионные инфекции встречаются крайне редко, и жить с ними можно очень долго, но исход у них один.
Как проявляются прионные болезни

В последние годы многие интернет-ресурсы заполонили статьи о вреде и пользе прионов. Эти противоречивые молекулы представляют собой особый класс вид инфекционных агентов (белки с измененной структурой, которые не содержат нуклеиновых кислот). Их свойством является способность увеличивать численность при помощи живых клеток подобно вирусам. Прионы способны также вызывать болезни, поражающие преимущественно нервную систему.
Что такое прионные болезни?
Прионные болезни (прионопатии) — это группа заболеваний с нейродегенеративным механизмом, которые невозможно вылечить. Под действием неизвестных факторов (как врожденных, так и приобретенных) нормальный клеточный прионный белок изменяет форму и превращается в патологический. Считается, что такие заболевания могут возникать в результате генетических мутаций: реже заражение происходит при контакте с биологическим материалом больного человека.
Новообразованные прионы накапливаются в организме, провоцируя появление основных симптомов. На это могут понадобиться многие годы, поэтому средний возраст заболевших всегда не младше 30 лет. Обратное возвращение белка в нормальную форму невозможно: поэтому все недуги, вызываемые прионами, являются смертельными. При исследовании мозгового вещества пациента с подобным заболеванием обнаруживаются морфологические изменения: мозг имеет губчатую структуру с множеством микроскопических точечных отверстий.
Куру: механизм заражения и симптомы
Куру — одна из самых редких прионопатий. До середины прошлого столетия куру часто встречалась среди коренного населения Папуа-Новой Гвинеи. У членов племени была традиция поедания частей тела умершего человека: считалось, что таким образом родственникам усопшего передается его сила. Чаще всего заболеванию были подвержены женщины и дети, так как именно им доставался наиболее зараженный прионами орган — мозг.
Куру опасна тем, что первые признаки недуга могут проявиться даже через 20-50 лет после употребления зараженного биологического материала. Основные симптомы недуга:
- спонтанные неконтролируемые движения тела и конечностей в виде подергивания;
- непроизвольный смех и сардоническая улыбка;
- резкие смены настроения;
- быстро прогрессирующее слабоумие и нарушение ориентации в пространстве.
Средняя продолжительность жизни больных с куру не превышает одного года. Это связано с прогрессирующим поражением нервной системы.
Заболевание Крейтцфельда-Якоба

Эта патология в 90% случаев обусловлена употреблением в пищу зараженной говядины, реже причиной развития болезни Крейтцефельда-Якоба является наследование мутантного гена (от 5 д 15 %). Недуг чаще встречается у мужчин старше 45-60 лет. Заболевание развивается постепенно: сначала пациент обращает внимание на ухудшение памяти, снижение способности к концентрации. Постепенно присоединяются спутанность сознания и спонтанные движения различных мышечных групп, нарушается походка. Диагностика недуга осуществляется с помощью КТ, МРТ, электроэнцефалографии. В среднем от момента возникновения первых проявлений до момента гибели проходит от 5 месяцев до 2,5 лет.
Смертельная бессонница: почему страдает мозг?
Другой, не менее редкой формой прионопатий является смертельная бессонница. Каждый человек хотя бы раз в своей жизни сталкивался с обычным нарушением сна, которое возникает при сбое привычного режима. Однако смертельная бессонница — это совершенное другое состояние, которое приводит к медленному разрушению не только нервной системы, но и психики человека. Основные проявления болезни:
- нарушения сна;
- панические атаки и склонность к истерии;
- галлюцинации и бред;
- невозможность самообслуживания;
- неадекватное, агрессивное поведение;
- увеличение артериального давления.
Смерть наступает через 9-70 месяцев после того, как обнаруживаются первые симптомы.
Прионопатия с изменением чувствительности к воздействию протеаз
Эта прионопатия является одним из недавно обнаруженных заболеваний, связанных с образованием в организме аномального белка. Согласно данным статистики, она поражает около 2-4 людей на 100 миллионов человек. Средний возраст заболевших — 70 лет. В основе этой прионопатии лежит изменение чувствительности прионов к действию ферментов организма — протеаз. Это приводит к усиленному расщеплению аномальных белков и прогрессированию болезни. Такая прионопатия начинается с изменений в психике больного: он становится агрессивными, вспышки злости и гнева чередуются с приступами эйфории, нарушается речь, страдает координация. Пациенты постепенно теряют способность к коммуникации и самообслуживанию. Продолжительность жизни людей с подобным недугом составляет не более 24 месяцев.
Заболевание Герстманна—Штреусслера—Шейнкера

Прионопатия Герстманна—Штреусслера—Шейнкера передается по наследству. Особенностью этой болезни является более раннее начало (в возрасте до 40 лет), а также относительно медленное течение. Пациенты предъявляют жалобы на неуклюжесть, нарушения походки, снижение слуха. Также в первую очередь страдает речь: больной не может внятно сформулировать предложения, постоянно теряет мысль в своих рассуждениях. Постепенно поражается мышечная система, может развиваться полная неподвижность верхних конечностей. Смерть обычно наступает в течение 5 лет после постановки диагноза от нарушения работы дыхательных мышц или сопутствующих осложнений (пневмония, гангрена).
Существует ли профилактика прионных заболеваний?
Защитить мозг человека от наследственных прионопатий не представляется возможным. Однако в случае с приобретенной формой заболевания рекомендуется соблюдать определенные правила безопасности (в особенности людям, которые так или иначе контактируют с биологическим материалом), позволяющие уменьшить риск возникновения недуга. Профилактика прионопатий включает в себя:
- использование средств индивидуальной защиты (масок, перчаток, защитных костюмов);
- обработка предметов многоразового использования в автоклаве при высокой температуре на протяжении часа;
- химическая обработка инструментов (с помощью гидроксида натрия), контактировавших с биологическими материалами.
Читайте далее

Мозг ребенка и жиры пищи: зачем нужны омега-3 кислоты?
Питание многих детей к школьному возрасту существенно отличается от здорового, сбалансированного рациона.
Прионы: исследования таинственных молекул продолжаются

Новость
Путь прионов
Автор
Редакторы
Статья на конкурс «био/мол/текст»: Прионные заболевания — феномен, открытый в двадцатом веке, и в нем же начавший играть большую роль: увеличение продолжительности жизни в развитых странах привело к тому, что все больше людей стало доживать до «своего Альцгеймера» или «своего Паркинсона». Природа нейродегенеративных заболеваний продолжает оставаться туманной, и ученые пока исследуют только отдельные их аспекты — например, причину развития именно в старческом возрасте или способность передаваться от одних видов живых существ другим.
«Био/мол/текст»-2012
Спонсор конкурса — дальновидная компания Thermo Fisher Scientific.
Всё началось с того, что в 20 веке учёные заинтересовались природой необычных заболеваний человека и животных: куру, Крейтцфельда-Якоба, скрэпи. Заметное сходство патологии этих болезней дало основание для гипотезы об их инфекционности, что впоследствии было экспериментально подтверждено. Тогда возник вопрос о возбудителе данных заболеваний. Прежде чем был найден ответ, были выявлены необычайные свойства возбудителей: они не размножаются на искусственных питательных средах, устойчивы к высокой температуре, формальдегиду, различным видам излучений, действию нуклеаз. Очистка инфекционного материала и его изучение позволило провозгласить о том, что «во всём виноват» белок, который 30 лет назад получил название прион (от англ. pr[otenacious infect]ion — белковая инфекция).
Так, известные американские учёные — вирусолог и врач Д.К. Гайдушек, раскрывший инфекционную природу прионных болезней, в 1976 г. и биохимик С.Б. Прузинер, который определил прионы и разработал прионную теорию, в 1997 г., — были удостоены Нобелевских премий. Их работы стали импульсом для последующих исследований, благодаря которым были изучены новые виды прионных инфекций. Но, даже несмотря на неугасающий интерес к «прионной теме», образование прионов до сегодняшнего дня остаётся загадкой.
Биологическая сущность прионов
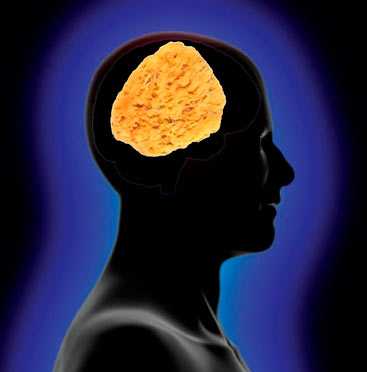
Рисунок 1. Метафора нейродегенеративного поражения мозга — это губка, в которую превращается нервная ткань в результате массовой гибели нейронов.
Молекула приона не является чем-то экзотическим: в «нормальной» форме она имеется на поверхности нервных у каждого из нас. При этом мы отлично себя чувствуем, и наши нервные клетки живы и здоровы. Однако это всё до тех пор, пока наш нормальный белок не «переродится» в аномальную форму. А если это случится, то приведёт к ужасающим последствиям: инфекционная форма прионов имеет свойство «склеиваться» с другими молекулами и, мало того, «конвертировать» их в эту же самую форму, вызывая «молекулярную эпидемию». В результате этой полимеризации на нервной клетке появляются токсичные белковые бляшки , и она погибает [1]. На месте погибшей клетки образуется пустота — вакуоль, заполненная жидкостью. С течением времени будет исчезать один нейрон за другим, а в мозге — образовываться всё больше «дыр», пока, наконец, мозг не превратится в губку (рис. 1), за чем неминуемо последует смерть.
Существует упрощенное представление, что полимеризованные прионные фибриллы «протыкают» нейрон, что вызывает его гибель. На самом деле это не совсем так: предшествующие фибриллярной стадии сферические агрегаты прионов также обладают токсичностью (по крайней мере, для болезни Альцгеймера): «Альцгеймеровский нейротоксин: ядовиты не только фибриллы». — Ред.
Но как может нормальный природный белок (обозначается PrP C ) вдруг стать патологическим (PrP Sc ; Sc — от слова «scrapie»)? Что должно произойти? Как и в случае «обычной» инфекции, для такой трансформации необходима встреча с молекулой инфекционного приона. Существуют два пути передачи этой молекулы: наследственный (за счёт мутаций в гене, кодирующего белок) и инфекционный. То есть внедрение приона может произойти неожиданно — например, при употреблении в пищу недостаточно хорошо прожаренного или сваренного мяса (в котором должна присутствовать форма PrP Sc ), при переливании крови, при трансплантации органов и тканей, при введении гормонов гипофиза животного происхождения.
И тогда происходит удивительное событие: нормальные молекулы белка, контактируя с патологическими, сами превращаются в них, изменяя свою пространственную структуру (механизм трансформации остаётся загадкой и по сей день) [1]. Таким образом прион, как самый настоящий инфекционный агент, заражает нормальные молекулы, запуская цепную реакцию, разрушительную для клетки.
Некоторые сведения о прионах
- прионный белок включает 254 аминокислотных остатка и «весит» 33-35 килодальтон [2];
- ген, кодирующий белок PrP, найден у человека, млекопитающих и птиц [1];
- для полного уничтожения прионного белка нужна температура не менее 1000 градусов [1]!
- возможно, прионы принимают участие в межклеточном узнавании и клеточной активации [3];
- не исключено, что функцией прионов является подавление возрастных процессов [3];
- при развитии клинических проявлений прионных заболеваний нет ни признаков воспаления, ни изменений в крови;
- предполагается, что прионы участвуют в развитии шизофрении и миопатии;
- механизм действия прионов и их превращения из нормальной формы в патологическую остаётся неясным.
Условия возникновения заболеваний
Условия возникновения прионовых болезней уникальны. Они могут формироваться по трём сценариям: как инфекционные, спорадические и наследственные поражения. В последнем варианте главную роль играет генетическая предрасположенность [2].
Знаменитый исследователь прионов Стэнли Прузинер (Stanley Prusiner) выделяет две поразительные особенности, присущие таким нейродегенеративным заболеваниям, как болезнь Крейтцфельда-Якоба, болезнь Альцгеймера и болезнь Паркинсона. Первая заключается в том, что более 80% случаев заболевания — спорадические (то есть, случайные, возникающие «сами собой»). Вторая: несмотря на то, что большое количество мутантных белков, специфичных к определённой болезни, экспрессируется в процессе зародышевого развития, формы наследования этих нейродегенеративных заболеваний проявляются позже. Это предполагает, что некоторые процессы происходят во время старения, которое «дает волю» болезнетворным белкам [5]. Более 20 лет назад автор утверждал, что данный процесс включает случайный рефолдинг (пересворачивание) белка в неправильно свёрнутый, что соответствует переходу в инфекционное состояние — прион.
Интересные факты насчет болезни Альцгеймера: ее вероятность может повышаться вследствие хронического недосыпания («Новый шаг к пониманию болезни Альцгеймера: возможно, недосыпание является одним из факторов риска»), а сам альцгеймеровский нейропептид (β-амилоид Aβ) может быть частью системы врожденного иммунитета («Возможно, β-амилоид болезни Альцгеймера — часть врождённого иммунитета»). — Ред.
В последнее десятилетие интерес к этой теме возобновился в связи с возможностью развития диагностики и эффективной терапии [5]. Появилось множество различных объяснений для возрастных нейродегенеративных болезней, — например, окислительная модификация ДНК, липидов и/или белков; соматические мутации; измененный врождённый иммунитет; экзогенные токсины; несоответствия ДНК—РНК; нарушение работы шаперонов; отсутствие одного из аллелей гена [5]. Альтернативным комплексным разъяснением служит то, что различные группы белков могут формировать прионы. Несмотря на то, что небольшое количество прионов может быть удалено посредством путей белковой деградации, их чрезмерное накопление с течением времени позволяет прионам самостоятельно распространяться в организме (рис. 2), что приводит к нарушению деятельности центральной нервной системы [5].
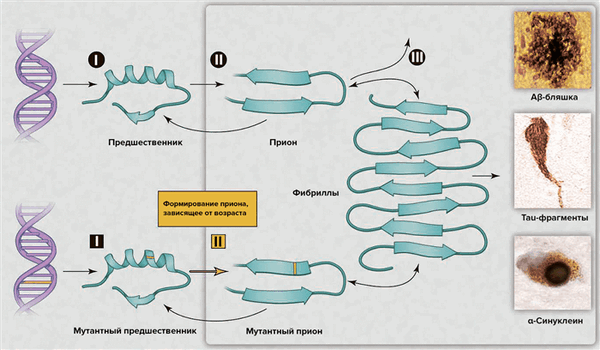
Рисунок 2. Процессы нейродегенерации, вызванной прионами. Сверху: накопление «нормального» прионного белка повышает его вероятность перехода в токсичную конформацию, которая описывается бóльшим содержанием β-структуры. Прионы наиболее патогенны в форме олигомеров; после образования фибрилл токсичность снижается. В зависимости от того, о каком конкретно прионном белке идет речь, в патологическом состоянии он может образовывать бляшки, клубки или тельца включения. Возможные пути лекарственного вмешательства: (I) снижение концентрации «нормального» белка-предшественника; (II) ингибирование образования прионной формы; (III) уничтожение токсичных агрегатов. Снизу: Наследственная старческая нейродегенерация объясняется двумя событиями: наличием мутантной формы предшественника и образованием из него приона, готового к олиго- и полимеризации с образованием токсичных форм.
Группы риска прионных заболеваний
Вот кого прионные заболевания могут настичь с наибольшей вероятностью:
- работники пищевой промышленности;
- ветеринары;
- патологоанатомы;
- хирурги;
- пациенты трансплантолога;
- каннибалы;
- лица, в семье которых были замечены синдромы Герстманна—Штрейслера—Шейнклера или фатальной инсомнии.
Лабораторная диагностика и лечение
Диагностика базируется на внутримозговом заражении мышат или хомяков, у которых медленно (до 150 дней) развивается соответствующее заболевание, если пациент был болен [2]. Часто проводится гистологическое исследование головного мозга погибших животных [2].
К сожалению, до настоящего времени еще не разработаны эффективные методы лечения прионовых болезней, хотя попытки предотвратить конформационный переход нормального белка в аномальный производятся. Поэтому самым надёжным способом предупреждения развития инфекционных форм является профилактика [2].
Особенно актуальным становится решение «прионного вопроса» в связи с нарастающей угрозой возникновения эпидемии через инвазивные медицинские операции и даже при приёме лекарственных средств.
Перспективы
По-видимому, интерес к прионам не угаснет до тех пор, пока предположения на их счёт полностью не подтвердятся и не будут найдены эффективные способы лечения прионных заболеваний. В статье [6] говорится о необходимости современного исследования, которое требует тщательного рассмотрения чужеродных прионов в экстраневрональных тканях.
В качестве модельных объектов авторы использовали мышей: две линии, которые трансгенно экспрессировали овечий прионный белок, и одну линию, которая экспрессировала человеческий прионный белок (рис. 3). Задачей было сравнить эффективность межвидовой передачи инфекции посредством тканей мозга и селезёнки. Внутримозговое заражение чужеродным прионным белком выражалось в отсутствии или небольшом количестве инфекционного агента в мозгах этих мышей. Однако инфекционные чужеродные прионы обнаруживались в селезёнке на более ранних этапах заражения в сравнении с моментом, когда были использованы нейротропные прионы, тем самым определяя, что лимфатическая ткань может быть более пермиссивной к распространению чужеродных прионов по сравнению с мозгом.
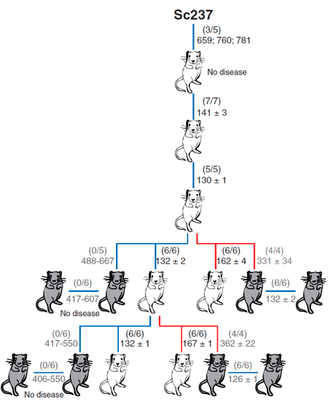
Рисунок 3. Способность приона хомяков Sc237 заражать и передаваться при введении в мозг или селезенку трансгенным мышам, имеющим прионный белок PrP овцы (tg338; белые мыши) или человека (tg7; серые мыши). Число заболевших/инъецированных мышей показано в скобках; ниже приведено среднее время жизни (в днях).
Чем вызвана эта предпочтительная репликация прионов в лимфатических тканях, пока неизвестно. Однако полученные данные показывают, что человек может быть более чувствительным к чужеродным прионам, чем предполагалось ранее на основании присутствия прионов в мозгу, и по этой причине бессимптомный переносчик прионной болезни может быть не распознан. Это ещё раз подтверждает, что такая могущественная биомолекула как прион таит в себе немало загадок, раскрытие которых, возможно, поможет в понимании ряда неразрешимых проблем человечества.
Читайте также:
- Причины кровотечения из прямой кишки и обследование при нем
- Нарушение эластичности стенки и перистальтики. Нарушение положения органа
- Послеоперационная олигоурия в гинекологии. Метаболическая реабилитация после операции
- Глазные инфекции. Причины, диагностика, лечение офтальмологических инфекций
- Синдром Гленара (Glenard)