ROHHAD-синдром - причины, симптомы, диагностика и лечение
Добавил пользователь Валентин П. Обновлено: 09.01.2026
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Для корректной оценки результатов ваших анализов в динамике предпочтительно делать исследования в одной и той же лаборатории, так как в разных лабораториях для выполнения одноименных анализов могут применяться разные методы исследования и единицы измерения.
Синонимы: компрессионная невропатия, компрессионно-ишемическая невропатия, ловушечная невропатия, капканный синдром, Tunnel syndrome
Туннельные синдромы: причины появления, симптомы, диагностика и способы лечения.
Определение
Туннельные синдромы - это группа заболеваний, причиной которых является локальная компрессия (защемление) и ишемия (нарушение кровоснабжения) периферических нервов, характеризующаяся болью и/или снижением их функций (двигательных и чувствительных).
Туннельные невропатии среди всех заболеваний периферической нервной системы составляют до 40%, причем чаще страдают лица наиболее трудоспособного возраста - 30-50 лет. В литературе описано более 30 форм туннельных невропатий.
Причины появления туннельных синдромов
Головной и спинной мозг получают и отсылают информацию к мышцам, железам и рецепторам. Информация, поступающая к внутренним органам, проходит по нервам. Для большинства периферических нервов характерны три особенности, защищающие их от физической деформации:
- волнообразный ход ненатянутого нерва;
- эластичность;
- расположение нервов относительно суставов предохраняет их от избыточных растяжений во время движений конечностей.
Туннели являются естественными анатомическими структурами, образованными мышцами, связками, фасциями, сухожилиями, костями.
Основным предрасполагающим фактором развития туннельной нейропатии является узость того или иного анатомического туннеля, через который проходят нервы. Сдавлению подвергаются не только нервы, но и сосуды, идущие параллельно нервным стволам, поэтому некоторые туннельные синдромы являются нейроваскулярными.
Широкое распространение туннельных синдромов - это прямое следствие профессиональной деятельности, связанной с длительной статической нагрузкой (монотонной работой за компьютером у офисных работников, профессиональной деятельностью музыкантов, художников, спортсменов). Основным фактором местного патологического воздействия является перенапряжение связочного аппарата и мышц, окружающих нерв. Наибольшая доля туннельных синдромов (80%) связана с поражением верхних конечностей.
Второе место по распространенности занимают посттравматические периферические нейропатии - они развиваются на фоне или после перенесенной инфекции, чаще вирусной этиологии.
Туннельные невропатии часто наблюдаются при эндокринных заболеваниях (сахарном диабете, гипотиреозе, акромегалии), патологиях суставов (подагре, деформирующем остеоартрозе, ревматоидном артрите), объемных образованиях самих нервов (шванноме, невроме), при паранеопластических полиневропатиях (саркоме, липоме), депозитарных невропатиях (амилоидозе), наследственной склонности к параличам от сдавления, а также при некоторых физиологических изменениях гормонального статуса (беременности, лактации, климаксе), приеме пероральных контрацептивов. К нарушению функции периферических нервов могут приводить механическая травматизация, ишемия (которая может быть первичной или возникать вслед за компрессией или одновременно с ней), венозный застой, отек тканей.
Известны семейные формы туннельных невропатий, обусловленные, по-видимому, как избыточным анатомическим сужением тех или иных каналов, так и наследуемыми аномалиями (дополнительными мышцами и сухожилиями, рудиментарными костными шпорами и фиброзными тяжами).
Туннельные синдромы разделяются в зависимости от пораженных нервов на нейропатии черепных нервов, нейропатии плечевого пояса и верхних конечностей, нейропатии тазового пояса и нижних конечностей.
- Карпальный синдром (запястный туннельный синдром) является самой распространенной формой компрессионно-ишемической невропатии, встречающейся в клинической практике. Развивается вследствие сдавливания срединного нерва в том месте, где он проходит через запястный канал под поперечной связкой запястья - удерживателем сухожилий сгибателей пальцев.
- Кубитальный синдром развивается при компрессии локтевого нерва на уровне локтевого сустава.
- Фибулярный синдром - невропатия малоберцового нерва при его сдавлении на уровне коленного сустава, а именно в области головки малоберцовой кости.
- Тарзальный синдром - невропатия нижней конечности, которая возникает в результате сдавления большеберцового нерва, проявляющаяся болью в области голеностопного сустава по внутренней стороне стопы.
- В случае краниальных нейропатий поражаются глазодвигательный, тройничный, лицевой нервы.
По срокам развития туннельные синдромы подразделяют на острые (развиваются от нескольких дней до 4-х недель), подострые (развиваются в течение нескольких недель) и хронические, в т.ч. рецидивирующие (развиваются в течение нескольких месяцев или лет).
Симптомы туннельных синдромов
Тяжесть повреждения нерва, вызванного острой или хронической компрессией, зависит от силы и продолжительности компрессии, а также от размеров нервного волокна, его положения в нервном стволе, количества и размеров пучков нервных волокон.
Полная картина туннельного синдрома включает в себя чувствительные (боль, парестезии, онемение), двигательные (снижение функции, слабость, атрофии) и трофические нарушения.
Возможны различные варианты клинического течения. Чаще всего заболевание дебютирует болью или другими чувствительными расстройствами. Реже - начало характеризуется двигательными нарушениями. Трофические изменения обычно выражены незначительно и только в запущенных случаях.
Сначала боль появляется во время движения (нагрузки), а по мере развития патологии возникает и в покое. Для туннельных синдромов характерны чувство онемения, покалывания по ходу поврежденного нерва, ощущение прохождения электрического тока (электрический прострел), жгучая боль. Двигательные нарушения возникают вследствие поражения двигательных ветвей нерва и проявляются снижением силы, быстрой утомляемостью. В некоторых случаях прогрессирование заболевания приводит к атрофиям, развитию контрактур («когтистая лапа», «обезьянья лапа»). При компрессии артерий и вен возможно развитие сосудистых расстройств, что проявляется побледнением, снижением локальной температуры кожного покрова или появлением синюшности и отечности в области поражения.
Диагностика туннельных синдромов
Как правило, диагноз устанавливается на основании характерных клинических проявлений. Удобным для клинициста является использование ряда клинических тестов, которые позволяют дифференцировать различные виды туннельных синдромов.
При наличии у больного признаков компрессионно-ишемического поражения периферических нервов необходимо проведение стандартного комплекса исследований, который включает:
- общий соматический осмотр с определением основных витальных функций;
- неврологический осмотр с использованием специфических тестов; (добавочные ребра, костные мозоли, костные отростки, переломы, вивихи и др.);
- нейровизуализацию (магнитно-резонансную или компьютерную томографию) участка компрессии нервного ствола, позвоночника и др.;
- электрофизиологические методы исследования (электронейромиографию для определения скорости проведения импульса по нерву и уточнения уровня поражения нерва);
- допплерографию сосудов конечностей с выполнением сгибательно-разгибательных проб;
Ультразвуковое сканирование артерий верхних конечностей в комплексной диагностике сердечно-сосудистых заболеваний.
Синдром Титце ( Реберно-хрящевой синдром , Реберный хондрит )
Синдром Титце - заболевание из группы хондропатий, сопровождающееся асептическим воспалением одного или нескольких верхних реберных хрящей в области их сочленения с грудиной. Проявляется локальной болезненностью в месте поражения, усиливающейся при давлении, пальпации и глубоком дыхании. Как правило, возникает без видимых причин, в ряде случаев может отмечаться связь с физическими нагрузками, операциями в области грудной клетки и т. д. Диагноз выставляется на основании жалоб и данных осмотра после исключения более серьезных патологий с помощью рентгенографии, УЗИ, КТ и других исследований. Лечение обычно консервативное: НПВС, блокады, физиотерапия.
МКБ-10

Общие сведения
Синдром Титце (реберно-хрящевой синдром, реберный хондрит) - асептическое воспаление одного или нескольких реберных хрящей в области их соединения с грудиной. Обычно страдают II-III, реже - I и IV ребра. Как правило, процесс захватывает 1-2, реже - 3-4 ребра. В 80% случаев отмечается одностороннее поражение. Заболевание сопровождается припухлостью и болью, порой - иррадиирующей в руку или грудную клетку. Причины развития до конца не изучены. Лечение консервативное, исход благоприятный.
Болезнь обычно развивается в возрасте 20-40 лет, хотя зафиксировано и более раннее начало - в возрасте 12-14 лет. По данным большинства авторов мужчины и женщины страдают одинаково часто, однако некоторые исследователи отмечают, что во взрослом возрасте синдром Титце чаще выявляется у женщин.

Причины
Хотя причины возникновения синдрома Титце в настоящий момент до конца не выяснены, существует несколько теорий, объясняющих механизм развития этого заболевания. Наиболее популярной является травматическая теория. Многие пациенты, страдающие синдромом Титце, являются спортсменами, занимаются тяжелым физическим трудом, страдают острыми или хроническими заболеваниями, сопровождающимися тяжелым надсадным кашлем, или имеют травму ребер в анамнезе.
Сторонники этой теории считают, что из-за прямой травмы, постоянных микротравм или перегрузки плечевого пояса повреждаются хрящи, на границе костной и хрящевой части возникают микропереломы. Это становится причиной раздражения надхрящницы, из малодифференцированных клеток которой образуется новая хрящевая ткань, несколько отличающаяся от нормальной. Избыточная хрящевая ткань сдавливает нервные волокна и становится причиной возникновения болевого синдрома. В настоящее время травматическая теория наиболее признана в научном мире и имеет больше всего подтверждений.
Инфекционно-аллергическая теория. Последователи данной теории находят связь между развитием синдрома Титце и перенесенными незадолго до этого острыми респираторными заболеваниями, спровоцировавшими снижение иммунитета. В пользу этой теории также может свидетельствовать более частое развитие заболевания у лиц, страдающих наркотической зависимостью, а также у пациентов, в недавнем прошлом перенесших операции на грудной клетке.
Алиментарно-дистрофическая теория. Предполагается, что дегенеративные нарушения хряща возникают вследствие нарушения обмена кальция, витаминов группы С и В. Эту гипотезу высказывал сам Титце, впервые описавший данный синдром в 1921 году, однако в настоящее время теория относится к категории сомнительных, поскольку не подтверждается объективными данными.
Симптомы
Пациенты предъявляют жалобы на острые или постепенно нарастающие боли, которые локализуются в верхних отделах грудной клетки, рядом с грудиной. Боли обычно бывают односторонними, усиливаются при глубоком дыхании, кашле, чихании и движениях, могут отдавать в плечо, руку или грудную клетку на стороне поражения. Иногда болевой синдром кратковременный, однако, чаще бывает постоянным, длительным и беспокоит пациента годами. При этом отмечается чередование обострений и ремиссий. Общее состояние в период обострения не страдает. При осмотре определяется выраженная локальная болезненность при пальпации и надавливании. Выявляется плотная, четкая припухлость веретенообразной формы размером 3-4 см.
Диагностика
Диагноз синдрома Титце выставляется специалистом в сфере травматологии и ортопедии на основании клинических данных, после исключения других заболеваний, которые могли стать причиной появления болей в грудной клетке. И одним из основных симптомов, подтверждающих диагноз, становится наличие характерной четкой и плотной припухлости, не выявляемой больше ни при одном заболевании.
В ходе дифференциальной диагностики исключают острую травму, заболевания сердечно-сосудистой системы и внутренних органов, которые могли вызвать подобную симптоматику, в том числе - различные инфекционные заболевания и уже упомянутые злокачественные новообразования. При необходимости пациента направляют на анализы крови, МРТ, КТ, УЗИ и другие исследования.
При рентгенологическом исследовании в динамике удается обнаружить нерезкие изменения структуры хряща. На начальных этапах патология не определяется. Через некоторое время становится заметным утолщение и преждевременное обызвествление хряща, появление костных и известковых глыбок по его краям. Еще через несколько недель на передних концах костной части пораженных ребер появляются небольшие периостальные отложения, отчего ребро немного утолщается, а межреберное пространство - суживается. На поздних стадиях обнаруживается слияние хрящевых и костных отрезков ребер, деформирующий остеоартроз реберно-грудинных сочленений и костные разрастания.
Рентгенография при синдроме Титце не имеет самостоятельного значения в момент постановки диагноза, поскольку первые изменения на рентгенограммах становятся заметны лишь спустя 2-3 месяца с начала заболевания. Однако это исследование играет большую роль при исключении всевозможных злокачественных опухолей, как первичных, так и метастатических.
В сомнительных случаях показана компьютерная томография, которая позволяет выявлять изменения, характерные для синдрома Титце на более ранних стадиях. Также в ходе дифференциальной диагностики со злокачественными новообразованиями может выполняться сканирование Tc и Ga и пункционная биопсия, при которой определяются дегенеративные изменения хряща и отсутствие элементов опухоли.
Особую настороженность из-за своей широкой распространенности у взрослых больных вызывают возможные сердечно-сосудистые заболевания и в первую очередь - ишемическая болезнь сердца. Для ИБС характерны кратковременные боли (в среднем приступ стенокардии длится 10-15 минут), в то время как при синдроме Титце боли могут сохраняться на протяжении часов, дней и даже недель. В отличие от синдрома Титце, при ишемической болезни болевой синдром купируется препаратами из группы нитроглицерина. Для окончательного исключения сердечно-сосудистой патологии выполняется ряд анализов и инструментальных исследований (ЭКГ и проч.).
Синдром Титце также приходится дифференцировать от ревматических заболеваний (фиброзита, спондилоартрита, ревматоидного артрита) и местных поражений хрящей и грудины (костохондрита и ксифоидалгии). Для исключения ревматических болезней выполняется ряд специальных анализов. О костохондрите свидетельствует отсутствие гипертрофии реберного хряща, о ксифоидалгии - усиливающиеся при надавливании боли в области мечевидного отростка грудины.
В ряде случаев синдром Титце по своей клинической картине может напоминать межреберную невралгию (и для того, и для другого заболевания характерны длительные боли, усиливающиеся при движениях, чихании, кашле и глубоком дыхании). В пользу синдрома Титце свидетельствует менее выраженный болевой синдром, наличие плотной припухлости в области реберных хрящей и отсутствие зоны онемения по ходу межреберного промежутка. Изменения в биохимическом составе крови, общих анализах крови и мочи при синдроме Титце отсутствуют. Иммунные реакции в норме.
Лечение синдрома Титце
Лечение осуществляется ортопедами-травматологами. Больные находятся на амбулаторном наблюдении, госпитализация, как правило, не требуется. Пациентам назначают местное лечение с использованием мазей и гелей, содержащих нестероидные противовоспалительные препараты. Применяются также компрессы с димексидом. При выраженном болевом синдроме прописывают НПВП и обезболивающие препараты для приема внутрь.
При стойких болях в сочетании с признаками воспаления, которые не удается купировать приемом анальгетиков и нестероидных противовоспалительных препаратов, хороший эффект обеспечивает введение новокаина с гидрокортизоном и гиалуронидазы в пораженную область (блокада грудной мышцы). Кроме того, применяется физиотерапевтическое лечение, рефлексотерапия и мануальное воздействие.
Крайне редко, при упорном течении заболевания и неэффективности консервативной терапии требуется оперативное лечение, которое заключается в поднадкостничной резекции ребра. Хирургическое вмешательство проводится под общим или под местным обезболиванием в условиях стационара.
2. Синдром Титце - междисциплинарный клинический случай / Шестерня П.А., Васильева А.О., Шкиль Л.М., Онищенко С.Б., Михайлова К.О., Никитина М.А. // Сибирское медицинское обозрение - 2017 - №2
ROHHAD-синдром ( РОХХАД-синдром )
ROHHAD-синдром - это крайне редкое заболевание, которое проявляется ожирением, гипоталамической дисфункцией, гиповентиляцией по центральному типу. Болезнь возникает вследствие разнообразных генетических мутаций, иммунологических нарушений. Патология манифестирует в дошкольном возрасте внезапным повышением аппетита и набором веса, позже присоединяются дыхательные и эндокринные расстройства. Диагностика ROHHAD-синдрома требует проведения полисомнографии, изучения уровня газов и гормонов крови, визуализации головного мозга с помощью КТ, МРТ. Пациентам назначается комплексная симптоматическая терапия, в тяжелых случаях требуется проведение ИВЛ.



РОХХАД-синдром был впервые описан в клинической эндокринологии в 1965 г. Современное название он получил благодаря американскому детскому эндокринологу, который в 2007 г. опубликовал работу с описанием 15 клинических случаев заболевания. Аббревиатура ROHHAD образована от английской фразы, которая в переводе на русский означает «быстро прогрессирующее ожирение с гипоталамической дисрегуляцией, гиповентиляцией и вегетативной дисфункцией». По данным на 2021 г., в мире известно около 100 пациентов с подтвержденным ROHHAD-синдромом.

Этиологическая структура точно не установлена, что связано с редкостью встречаемости синдрома и малым сроком клинических наблюдений. Многие ученые подчеркивают роль нейроэндокринных опухолей в этиологии заболевания, вследствие чего аббревиатуру иногда дополняют термином NET (Neural Endocrinological Tumor). Наследственный характер патологии пока не изучен. Существует 3 основных теории развития РОХХАД-синдрома:
- Генетическая. Нарушения связывают с мутациями гена PHOX2B в локусе 4p12 - важного регулятора транскрипции. Обсуждается роль еще 11 генов, которые контролируют формирование нервной системы и нейроэндокринные процессы, однако точные данные по их значению в этиопатогенезе отсутствуют.
- Эпигенетическая. Наблюдения за монозиготными близнецами показывают, что РОХХАД-синдром может возникать только у одного ребенка. Исследователи связывают это с влиянием эпигенетики - изменения активности определенных генов без нарушений их последовательности, происходящие под влиянием условий жизни.
- Иммунологическая. У части пациентов в спинномозговой жидкости присутствуют антигипофизарные и антигипоталамические антитела, которые потенциально могут повреждать структуры головного мозга, быть пусковым фактором нарушений. Обсуждается связь заболевания с целиакией и прочими аутоиммунными патологиями.
Патогенез
Структурные аномалии гена PHOX2B нарушают миграцию нервного гребня и развитие вегетативной нервной системы. Расстройства автономного контроля витальных функций проявляются неадекватной вентиляцией в ответ на гиперкапнию и гипоксемию. Ситуация усугубляется нарушением дыхательной функций: замедленная передача нервных импульсов мешает двигательной активности диафрагмы и других составляющих дыхательной мускулатуры.
Большинство клинических проявлений РОХХАД-синдрома связывают с поражением гипоталамуса. Он играет ключевую роль в поддержании гомеостаза: регулирует процессы голода и насыщения, теплопродукции и теплоотдачи, жажды и диуреза. При гипоталамической дисфункции развиваются серьезные метаболические нарушения, которые приводят к ожирению, расстройствам водно-электролитного баланса, эндокринной дисрегуляции.
Клинические проявления ROHHAD-синдрома возникают в возрасте от 0 до 9 лет, типичное время манифестации - 2-4 года. До этого периода физическое и психическое развитие детей происходит согласно возрасту, признаки врожденной патологии отсутствуют. РОХХАД-синдром манифестирует возрастанием аппетита вплоть до булимии, быстрым набором массы тела. Дети страдают ожирением, имеют характерную внешность: лунообразное лицо с большими щеками, гипертелоризм, короткая толстая шея.
Вегетативная дисрегуляция при РОХХАД-синдроме проявляется расстройствами ЖКТ с хроническими запорами или диареей, косоглазием, измененным восприятием боли. Спустя несколько месяцев присоединяются водно-электролитные нарушения, уменьшается объем суточной мочи, возможен энурез. Дети с ROHHAD-синдромом более подвержены инфекционным заболеваниям органов дыхания.
Ключевой особенностью болезни являются дыхательные нарушения. Они манифестируют в среднем на шестом году жизни ребенка в виде синдрома обструктивного апноэ во сне, постоянного храпа, альвеолярной гиповентиляции. В типичных случаях симптоматика развивается только ночью, тяжелые варианты заболевания проявляются постоянными респираторными нарушениями. Родители замечают цианотичный оттенок кожи ребенка, приступы слабости и апатичности в течения дня.
Осложнения
Жизнеугрожающим последствием ROHHAD-синдрома являются кардиореспираторные кризы, которые без экстренной медицинской помощи завершаются смертью пациента от остановки сердца. Характерны психические расстройства в виде аутизма, умственной отсталости, поведенческих нарушений. Неврологическая патология представлена судорожными приступами. У 40-56% детей диагностируется ганглионеврома.
Ожирение нередко сопровождается другими эндокринными заболеваниями. Описана связь РОХХАД-синдрома с нарушениями толерантности к глюкозе, ранним началом сахарного диабета, гипертриглицеридемией. На фоне этих изменений в детском возрасте формируется жировая болезнь печени. У большинства больных развивается вторичный гипотиреоз, карликовость, гипогонадотропный гипогонадизм.
Вследствие редкости и многогранности клинических проявлений ROHHAD-синдрома своевременная постановка диагноза затруднена. Пациенты проходят обследование у педиатра, детского эндокринолога, невролога и других специалистов. Значимую роль имеет определение патогномоничных признаков болезни, таких как внезапное ожирение и гиповентиляция. Диагностический поиск включает следующие методы исследования:
- Полисомнография. Диагностика проводится, чтобы определить наличие, длительность и количество приступов апноэ во время сна. Методика также регистрирует ЭКГ, ЭЭГ, электроокулограмму и другие параметры, которые нужны врачу для определения тяжести состояния ребенка.
- Нейровизуализация. Для исключения объемных новообразований и врожденных пороков ЦНС, которые имеют сходные проявления с РОХХАД-синдромом, выполняется КТ или МРТ головного мозга. При судорожных пароксизмах назначается электроэнцефалография.
- Анализ газов крови. Признаком суточной гиповентиляции считается снижение SpO2 менее 95% при одновременном повышении содержания углекислого газа более 50 мм рт. ст. в состоянии бодрствования. Для быстрой оценки дыхательной функции используется пульсоксиметрия.
- Гормональный профиль. Пациентам с ROHHAD-синдромом назначается расширенное исследование, которое включает анализы на гормоны гипофиза и гипоталамуса, щитовидной железы, надпочечников. По показаниям проводится оценка уровня инсулина и гликемического профиля.
Дифференциальная диагностика
При постановке диагноза следует исключить:
- синдром Прадера-Вилли;
- экзогенно-конституциональное ожирение;
- адипозогенитальную дистрофию.
При наличии неврологической симптоматики проводится дифференциальная диагностика с ожирением, спровоцированным опухолями головного мозга. РОХХАД-синдром дифференцируют с другими вариантами центральной гиповентиляции.
Лечение ROHHAD-синдрома
Пациентам с РОХХАД-синдромом требуется помощь мультидисциплинарной команды врачей при участии детских психологов, коррекционных педагогов и членов семьи. Для коррекции нутритивного статуса назначается диета с ограничением по калорийности, повышенным содержанием легкоусвояемых и витаминизированных продуктов. Также рекомендована лечебная гимнастика для предупреждения набора веса. Основные направления симптоматического лечения:
- Респираторная поддержка. С учетом тяжести состояния и возраста пациента может использоваться лицевая маска, назальные канюли, неинвазивная вентиляция с положительным давлением. При кардиореспираторных кризах больного переводят на классическую ИВЛ.
- Гормонотерапия. Заместительное лечение тиреоидными и стероидными гормонами применяется при сопутствующих эндокринных заболеваниях. Для контроля водно-солевого обмена возможно ограничение приема жидкости, добавление мочегонных препаратов.
- Дополнительная фармакотерапия. Для коррекции нарушений ЖКТ показаны ферменты, слабительные средства, противодиарейные препараты. Контроль нервно-психической симптоматики достигается благодаря антиконвульсантам, антипсихотикам, нейрометаболическим средствам.
Прогноз и профилактика
Летальность достигает 50-60%, основной причиной смерти является сердечно-легочная недостаточность. Однако прогноз улучшается при раннем обращении к врачу, подборе комплексной терапии и хорошем уровне комплаенса. Дальнейшее изучение этиопатогенеза РОХХАД-синдрома дает надежду на появление более эффективных методов терапии. Семьям, в которых есть больной ребенок, при планировании следующей беременности обязательно генетическое консультирование.
2. Синдром ROHHAD (обзор литературы и клинический случай)/ Л.А. Балыкова, Н.В. Ивянская, Е.С. Самошкина, С.А. Ивянский// Практическая медицина. - 2018. - №8.
3. Синдром ROHHADNET/ М.А. Караева, И.Б. Журтова, Е.М. Орлова, Л.И. Ширяева// Ожирение и метаболизм. - 2011. - №3.
4. ROHHAD (Rapid-onset Obesity with Hypoventilation, Hypothalamic Dysfunction, Autonomic Dysregulation) Syndrome-What Every Pediatrician Should Know About the Etiopathogenesis, Diagnosis and Treatment: A Review/ Lazea C, Sur L, Florea M.// Int J Gen Med. - 2021
ROHHAD-синдром - причины, симптомы, диагностика и лечение
Синдром Толоса — Ханта (СТХ) — редкое заболевание, которое проявляется сочетанием дисфункции одного или нескольких краниальных нервов, болью в области глазницы. У детей СТХ диагностируют чрезвычайно редко из-за отсутствия настороженности специалистов. Правильно поставить диагноз можно только после тщательного обследования с использованием МРТ или мультиспиральной компьютерной томографии. В статье представлено описание клинического наблюдения СТХ у ребенка первого десятилетия жизни, у которого похожие эпизоды птоза верхнего века, боли в области правого глаза наблюдались и ранее, однако причину их появления установить не удавалось. Подробно описаны катамнез заболевания, клинические проявления, неврологический статус и данные дополнительных методов обследования. Особенности клинического течения с обострениями и ремиссиями, быстрым регрессом симптоматики на фоне применения пульс-терапии глюкокортикостероидами, исключение специфического гранулематозного воспаления наружной стенки кавернозного синуса по данным МРТ позволили диагностировать идиопатический СТХ. Особо отмечена важность всестороннего неврологического и соматического обследования пациентов с клиническими проявлениями СТХ в связи с многообразием причин возникновения данного синдрома.
Ключевые слова: педиатрия, синдром Толоса — Ханта, офтальмоплегия, верхняя глазничная щель, кавернозный синус, глюкокортикостероиды.
E.V. Shishkina 1 , T.N. Bazilevskaya 2 , R.F. Izokhvatova 2 , I.V. Novikova 2 , Yu.V. Panfilova 2 , M.Yu. Galaktionova 1 , D.A. Maiseenko 1
1 Prof. V.F. Voino-Yasenetsky Krasnoyarsk State Medical University, Krasnoyarsk,
Russian Federation
2 Krasnoyarsk Interregional Children’s Clinical Hospital, Krasnoyarsk, Russian Federation
Tolosa-Hunt syndrome (THS) is a rare disease manifested with one or more cranial nerve disorders and orbital pain. In children, THS is diagnosed very rarely due to the lack of vigilance among physicians. Careful examination including MRI or helical CT help diagnose this disease. This paper discusses case history of THS in a child in the first ten years of life who has previously experienced similar episodes of ptosis and right-sided orbital pain; however, the cause of these symptoms has remained elusive. Disease anamnesis, clinical signs, neurological status, and additional diagnostic data are described in detail. Clinical course of THS with its relapses and remissions, rapid symptom regress after corticosteroid pulse therapy, and no specific granulomatous inflammation of the lateral wall of the cavernous sinus as demonstrated by MRI suggest idiopathic THS. The importance of in-depth neurological and general examination of patients with clinical signs of THS due to a variety o f the causes of this syndrome is highlighted.
Keywords: pediatrics, Tolosa-Hunt syndrome, ophthalmoplegia, superior orbital fissure, cavernous sinus, corticosteroids.
Введение
Синдром Толоса — Ханта (СТХ) — редкое заболевание, характеризующееся сочетанием дисфункции одного или нескольких краниальных нервов (глазодвигательных, первой или второй ветви тройничного нерва, зрительного нерва) с болевым синдромом различной степени выраженности в области глазницы или периорбитальной области.
Впервые данный синдром был описан испанским неврологом E.S. Tolosa в 1954 г. и дополнен в 1961 г. группой американских нейрохирургов во главе с W.E. Hunt, так исторически закрепилось современное название данного клинического синдрома — СТХ [1, 2].
В клинической практике СТХ одинаково часто встречается у мужчин и у женщин, в основном в пожилом и старческом возрасте. Наиболее редко СТХ возникает в течение первых двух десятилетий жизни, описание таких клинических случаев носит спорадический характер [3]. Встречаемость СТХ составляет 1-2 случая на 1 000 000 взрослого населения, в детской практике статистических данных нет [4].
Клинические проявления развиваются остро или подостро после перенесенной вирусной инфекции, переохлаждения, стресса. Как правило, первым неврологическим симптомом становится боль различной интенсивности, локализующаяся ретробульбарно, в лобной, надбровной или височной областях. Спустя несколько дней (реже одновременно) присоединяются диплопия, косоглазие и ограничение подвижности глазного яблока на стороне боли. Развитие тотальной офтальмоплегии сопряжено с поражением всех нервов, проходящих через верхнюю глазничную щель, и встречается в 25% случаев СТХ [5].
Основой для постановки диагноза служат катамнестические данные о периодическом возникновении односторонних симптомов поражения верхней глазной щели и/или кавернозного синуса. Рутинные общеклинические и биохимические анализы не информативны. При анализе цереброспинальной жидкости в 50% наблюдается норма или незначительное повышение уровня белка до 1 г/л с редким минимальным изменением цитоза, основным методом подтверждения СТХ являются МРТ или мультиспиральная компьютерная томография [6].
Практика показывает, что сходные клинические признаки наблюдаются при широком круге неврологических и соматических заболеваний: бактериальном, вирусном и грибковом воспалении наружной стенки кавернозного синуса или мозговых оболочек; первичных либо вторичных опухолях мозга и орбиты (аденома гипофиза, менингиома крыла основной кости, краниофарингиома, невринома, метастазы в головной мозг и/или орбиту); сосудистых мальформациях (артериовенозные аневризмы внутренней сонной артерии, каротидно-кавернозные соустья и др.) и диссекциях ветвей внутренней сонной артерии; тромбозе; лимфоме; эпидермоидных кистах кавернозного синуса; орбитальном миозите; саркоидозе; некоторых заболеваниях крови; офтальмической мигрени; системных аутоиммунных заболеваниях, таких как гранулематоз Вегенера, системная красная волчанка; при болезни Крона. По этой причине пациенты с характерной клинической картиной требуют тщательного обследования для исключения другой органической патологии [7, 8].
Чаще всего в основе СТХ лежат аутоиммунные механизмы формирования гранулем (гранулемы) в области наружной стенки кавернозного синуса и/или в проекции верхней глазной щели, выявляемых при нейровизуализации с помощью МРТ головного мозга или при проведении трансназальной биопсии. Особого внимания заслуживают случаи так называемого идиопатического СТХ, когда воспалительные или аутоиммунные причины отсутствуют, данные МРТ в норме либо имеющиеся изменения неспецифичны — за такими пациентами требуется катамнестическое наблюдение с целью исключения иного генеза заболевания [9].
Приводим собственный клинический опыт постановки диагноза СТХ у ребенка, полагая, что описание заболевания в детском возрасте представляет несомненный интерес для практикующих неврологов и педиатров.
Клиническое наблюдение
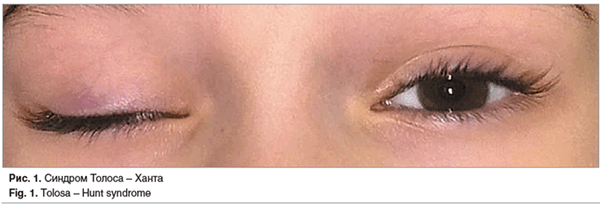
Пациентка С., 6 лет и 3 мес. На момент госпитализации предъявляла жалобы на боли в области правого глаза, слезотечение, опущение верхнего века, головокружение, повышение температуры тела до 37,2-37,4 °C. Данные клинические проявления сохранялись в течение трех дней.
Из анамнеза заболевания, со слов матери, известно, что похожее состояние у ребенка возникло в третий раз. В возрасте 1 год и 2 мес. девочка была госпитализирована в стационар с такими же очаговыми симптомами. Однако, учитывая, что на момент госпитализации у девочки отмечались симптомы ОРВИ, подъем температуры тела до субфебрильных цифр и незначительное повышение уровня белка до 0,6 г/л в спинномозговой жидкости без увеличения цитоза, был выставлен диагноз: энцефалит вирусной этиологии, с преимущественной локализацией в задней черепно-мозговой ямке, с офтальмоплегическим синдромом. При нейровизуализации головного мозга с помощью МРТ отмечены только признаки незавершенной миелинизации. Осмотр окулиста патологии не выявил. На фоне лечения с использованием глюкокортикостероидов (ГКС) в течение 2 нед. очаговая симптоматика регрессировала.
Повторно схожие симптомы наблюдались в возрасте 3 года и 2 мес. На фоне полного здоровья у ребенка возникла боль в области орбиты правого глаза с птозом верхнего века. Девочка была госпитализирована с подозрением на острое нарушение мозгового кровообращения. Проведено МРТ-обследование: органических признаков поражения головного мозга не выявлено. Патология органа зрения исключена. Был выставлен диагноз: острая нейропатия глазодвигательного нерва неуточненной этиологии. На фоне двухнедельного лечения, которое, как и в предыдущий раз, включало ГКС, клинические проявления полностью регрессировали.
После выписки из стационара находилась под наблюдением невролога по месту жительства, получала курсы ноотропной и дегидратирующей терапии.
Из анамнеза жизни известно, что ребенок родился от второй доношенной беременности, за время беременности у женщины дважды были эпизоды острой респираторной инфекции без повышения температуры, с легкими катаральными явлениями, получала симптоматическое лечение. Роды физиологические самостоятельные. В первый год жизни девочка росла и развивалась в соответствии с возрастом. На диспансерном учете у узких специалистов не состояла.
В возрасте четырех лет пациентке поставлен диагноз бронхиальной астмы, по поводу которой получает специфическую терапию, включая курсы гормональной терапии (ГКС). На момент госпитализации бронхиальная астма в стадии ремиссии. Вакцинирована согласно прививочному календарю РФ.
Данные общего и биохимического анализов крови, общего анализа мочи без особенностей. Консультирована офтальмологом: грубых нарушений глазодвигательной функции не выявлено. На глазном дне изменений нет.
МРТ-исследование головного мозга с ангиографией сосудов головного мозга. Данных за наличие изменений очагового характера в веществе мозга не получено. МР-ангиограмма: асимметрия диаметров интракраниальных сегментов позвоночных артерий (D>S); непрямолинейный ход основной артерии; вариант строения виллизиева круга в виде неполной задней трифуркации правой внутренней сонной артерии и снижения кровотока по правой задней соединительной артерии.
Электроэнцефалографический видеомониторинг: без патологии. Осмотрена эндокринологом: исключена эндокринная патология. Исключена миастения, локальная (глазная) форма. Рентгенография грудной клетки: без очаговых и инфильтративных изменений в легких.
С учетом жалоб при поступлении, анамнеза заболевания (третий эпизод офтальмоплегии), клинических проявлений и данных дополнительного обследования выставлен окончательный диагноз: приобретенная идиопатическая правосторонняя офтальмоплегия, СТХ.
За время нахождения в отделении проводилось комплексное лечение: пульс-терапия ГКС (дексаметазона натрия фосфат 4 мг/сут) в течение 12 дней, холиномиметики с антихолинэстеразным действием (неостигмина метилсульфат 0,6 мл/сут) и витамины с микроэлементами. На фоне лечения клинические проявления полностью регрессировали.
Представленное наблюдение СТХ является яркой демонстрацией отсутствия настороженности врачей, работающих с детьми, в отношении данного синдрома. Первичные офтальмопарез или офтальмоплегия характеризуются многообразием вариантов течения и синдромов, часто встречающихся в нейроофтальмологической практике, одним из которых является СТХ.
В работах как зарубежных, так и отечественных авторов СТХ освещается как редкий синдром во взрослой практике, а его возникновение в педиатрии считается практически невозможным. Встречаются единичные описательные статьи с клиникой данного синдрома у детей в различных возрастных группах [10]. Учитывая, что варианты болевой офтальмоплегии многочисленны и этиология их различна, СТХ ставят как диагноз исключения.
Особенности катамнеза клинического течения данного заболевания с обострениями и ремиссиями, быстрым регрессом симптоматики на фоне применения пульс-терапии ГКС, исключение специфического гранулематозного воспаления наружной стенки кавернозного синуса в результате проведения нескольких МРТ позволили нам диагностировать идиопатический СТХ.
Заключение
Синдром Толоса — Ханта, при всем многообразии нейроофтальмологических синдромов, является редко диагностируемой патологией не только в педиатрической, но и во взрослой практике. В представленном клиническом наблюдении манифестация заболевания в очень раннем возрасте, сопровождавшаяся признаками ОРВИ, фактически исключила даже саму возможность заподозрить данную патологию. Только благодаря тщательному обследованию пациентки с использованием лабораторных и нейровизуализационных методик, сопоставлению клинической картины и результатов исследований, анализу рецидивирующего характера течения заболевания стало возможным поставить правильный диагноз.
Сведения об авторах:
Базилевская Тамара Николаевна — врач-невролог КГБУЗ «КМДКБ № 1»; 660015, Россия, г. Красноярск, ул. Ленина, д. 149; ORCID iD 0000-0002-5874-5342.
Изохватова Рамзия Фаридовна — врач-невролог КГБУЗ «КМДКБ № 1»; 660015, Россия, г. Красноярск, ул. Ленина, д. 149; ORCID iD 0000-0001-5819-6112.
Новикова Ирина Валерьевна — врач-невролог КГБУЗ «КМДКБ № 1»; 660015, Россия, г. Красноярск, ул. Ленина, д. 149; ORCID iD 0000-0003-3511-5764.
Панфилова Юлия Валерьевна — врач-невролог КГБУЗ «КМДКБ № 1»; 660015, Россия, г. Красноярск, ул. Ленина, д. 149; ORCID iD 0000-0002-6354-2776.
Галактионова Марина Юрьевна — д.м.н., доцент, заведующая кафедрой поликлинической педиатрии и пропедевтики детских болезней с курсом последипломного образования ФГБОУ ВО КрасГМУ им. проф. В.Ф. Войно-Ясенецкого Минздрава России; 660022, Россия, г. Красноярск, ул. Партизана Железняка, д. 1; ORCID iD 0000-0001-7437-0512.
Маисеенко Дмитрий Александрович — к.м.н., доцент кафедры акушерства и гинекологии Института последипломного образования, ФГБОУ ВО КрасГМУ им. проф. В.Ф. Войно-Ясенецкого Минздрава России; 660022, Россия, г. Красноярск, ул. Партизана Железняка, д. 1; ORCID iD 0000-0003-1569-5098..
About the authors:
Tamara N. Basilevskaya — neurologist, Krasnoyarsk Interregional Children’s Clinical Hospital No. 1, 149, Lenin Str., Krasnoyarsk, 660015, Russian Federation; ORCID iD 0000-0002-5874-5342.
Ramziya F. Izokhvatova — neurologist, Krasnoyarsk Interregional Children’s Clinical Hospital No. 1, 149, Lenin Str., Krasnoyarsk, 660015, Russian Federation; ORCID iD 0000-0001-5819-6112.
Irina V. Novikova — neurologist, Krasnoyarsk Interregional Children’s Clinical Hospital No. 1, 149, Lenin Str., Krasnoyarsk, 660015, Russian Federation; ORCID iD 0000-0003-3511-5764.
Yuliya V. Panfilova — neurologist, Krasnoyarsk Interregional Children’s Clinical Hospital No. 1, 149, Lenin Str., Krasnoyarsk, 660015, Russian Federation; ORCID iD 0000-0002-6354-2776.
Marina Yu. Galaktionova — Doct. of Sci. (Med.), Associate Professor, Head of the Department of Polyclinic Pediatrics and Propaedeutics of Children’s Diseases with the Course of Postgraduate Education, Prof. V.F. Voino-Yasenetsky Krasnoyarsk State Medical University, 1, Partizan Zheleznyak Str., Krasnoyarsk, 660022, Russian Federation; ORCID iD 0000-0001-7437-0512.
Синдром Иценко-Кушинга (Синдром Кушинга, Сushing's syndrome)
Синдром Кушинга: причины появления, симптомы, диагностика и способы лечения.
Синдром Иценко-Кушинга - это сочетание клинических симптомов, вызванных хроническим повышением уровня кортизола или родственных ему кортикостероидов в крови. Болезнь Иценко-Кушинга - это синдром Кушинга, причиной которого служит избыточная гипофизарная продукция адренокортикотропного гормона (АКТГ), как правило, обусловленная небольшой доброкачественной опухолью гипофиза - аденомой. Иногда АКТГ производится опухолью, которая не связана с гипофизом, она может находиться где угодно, чаще - в легких и грудной клетке. Порой злокачественные опухоли хорошо маскируются под железы и начинают вырабатывать гормоны альдостерон и кортизол, что, в свою очередь, приводит к повышению их концентраций в человеческом организме. При этом собственные железы понемногу начинают атрофироваться - таким образом организм старается бороться с избытком гормонов.
Синдром Иценко-Кушинга также возникает из-за перепроизводства кортизола надпочечниками или употребления больших доз глюкокортикоидных препаратов таких как преднизолон или дексаметазон при лечении ряда болезней (астмы, ревматоидного артрита и некоторых других аутоиммунных патологических состояний). Заболевание может возникнуть в любом возрасте, но чаще всего в 20-40 лет, оно может быть врожденным или приобретенным. Женщины поражаются в 10 раз чаще, чем мужчины.
У пациентов, страдающих алкоголизмом или тяжелыми депрессивными расстройствами, а также во время беременности, иногда наблюдается небольшое повышение уровня гормонов надпочечников и развивается псевдо-синдром Иценко-Кушинга.
Классификация заболевания. Кодирование по МКБ-10
Синдром Иценко-Кушинга (E24):
E24.0. Болезнь Иценко-Кушинга гипофизарного происхождения (гиперсекреция АКТГ гипофизом, гиперадренокортицизм гипофизарного происхождения);
E24.1. Синдром Нельсона;
E24.3. Эктопический АКТГ-синдром;
E24.4. Кушингоидный синдром, вызванный алкоголем;
E24.8. Другие состояния, характеризующиеся кушингоидным синдромом;
E24.9. Синдром Иценко-Кушинга неуточненный.
Симптомы синдрома Иценко-Кушинга
У большинства больных с различными формами гиперкортицизма: АКТГ-зависимыми (болезнь Иценко-Кушинга, аденома гипофиза, АКТГ эктопический синдром) и АКТГ-независимыми формами (аденома коры надпочечника и/или двусторонняя микро-, макроузелковая гиперплазия) клинические проявления заболевания постоянны и зависят от скорости секреции кортизола надпочечниками.
К классическим признакам синдрома Иценко-Кушинга у взрослых относятся «лунообразное» лицо багрово-красного цвета, часто возникают многочисленные угревидные высыпания, центральное ожирение с одновременной потерей жировой ткани на бедрах, ягодицах и руках, истончение кожи и ломкость капилляров, приводящие к легкому и часто спонтанному образованию синяков. За счет неправильного и неравномерного жироотложения происходит необратимая деформация позвоночника, больные сутулятся, происходит нарушение осанки (кифоз, сколиоз). На бедрах, предплечьях, животе можно увидеть растяжки ярко-красного или даже фиолетового цвета, надключичные жировые подушечки и периферические отеки. Часто происходит разрушение костной ткани, отмечается склонность к переломам. У женщин по причине избытка половых гормонов возникают признаки излишнего оволосения по мужскому типу, появляются существенные перебои менструального цикла. У детей самым ранним признаком служит избыточная масса тела при задержке роста.
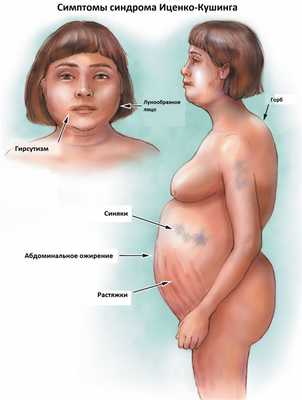
За счет повышенного уровня кортизола могут возникать гипертония, аритмия, поражение сердца и сосудов, высокий уровень глюкозы в крови, снижение зрения, приступы агрессивности, депрессия, нарушения терморегуляции (именно такие больные очень часто потеют, а также могут мерзнуть в жаркую погоду).
Диагностика синдрома Иценко-Кушинга
Ярко выраженный синдром Иценко-Кушинга не представляет особых диагностических трудностей. Достаточно лишь оценить внешний облик человека и провести с ним беседу. Но заболевание с умеренными проявлениями может вызвать у врача ряд проблем. Всегда следует исключать предварительный прием глюкокортикостероидов пациентом (экзогенный синдром Кушинга). Диагноз ставится клинически, а подтверждается данными лабораторных и визуализирующих методов исследований для установления стадии болезни и выяснения первопричины патологии.
Подтверждение избытка кортизола выполняется строго по показаниям врача одним из четырех методов:
- оценка количества кортизола - определение свободного кортизола мочи в двукратных суточных пробах;
Синонимы: Анализ мочи на кортизол; Анализ суточной мочи на свободный кортизол; Кортизол мочи. Hydrocortisone; Urine cortisol; Free Cortisol Urine Test; Urine Cortisaol Test. Краткое описание теста .
Читайте также: