Синдром Альперса (Alpers) - синонимы, авторы, клиника
Добавил пользователь Alex Обновлено: 08.01.2026
(Alpers, 1931) - детское заболевание с аутосомно-рецессивным типом наследования. Симптомокомплекс расстройства образуют следующие основные признаки: 1. локальные или генерализованные судороги; 2. миоклонии; 3. мозжечковый тремор и хореоатетоз; 4. иногда амавроз. Заболевание течёт катастрофическими темпами с быстрым образованием глубокого слабоумия, психического маразма. Смерть наступает при явлениях децеребрационной ригидности или в состоянии эпилептического статуса. Специфического метода лечения и эффективных методов профилактики заболевания ныне не существует, носительство анормального рецессивного гена фенотипически никак себя не обнаруживает. Синонимы: Прогрессирующая диффузная дегенерация серого вещества головного мозга, Семейная дегенерация серого вещества головного мозга в детском возрасте.
(описан американским нейрохирургом В. J. Alpers, 1900-1981; синонимы - прогрессирующая полидистрофия мозга, инфантильная полидистрофия мозга) - заболевание, связанное с дегенеративными изменениями в сером веществе мозга. Начинается, как правило, в младенческом возрасте с мышечной гипотонии. Затем появляются судороги, спастика, миоклонусы. Отмечается задержка физического развития, микроцефалия, атрофия мышц, контрактуры суставов. Со временем развиваются слепота, глухота, слабоумие. Семейная форма заболевания наследуется по аутосомно-рецессивному типу. Прогноз заболевания неблагоприятный. Лечение симптоматическое. B. J. Alpers. Diffuse progressive degeneration of the grey matter of the cerebrum. Archives of Neurology and Psychiatry, Chicago, 1931; 25: 469-505.
(Alpers B.J., 1931), Наблюдается в детском возрасте и характеризуется наличием локальных или генерализованных судорог, миоклонических проявлений, хореоатетоза и тремора мозжечкового генеза. Иногда - амавроз. Течение катастрофическое, быстрое, с образованием глубокого слабоумия. Чем в более раннем возрасте началось заболевание, тем скорее наступает смерть при явлениях децеребрации и эпилептического статуса. Заболевание наследственное (аутосомно-рецессивный тип передачи). Син.: прогрессирующая диффузная дегенерация серого вещества головного мозга, семейная дегенерация серого вещества головного мозга в детском возрасте.
развитие у детей слепоты, судорог, спастического изменения мышечного тонуса, миоклонуса, слабоумия, связанное с дегенеративными изменениями серого вещества мозга; носит семейный характер, наследуется, предположительно, по аутосомно-рецессивному типу.
Альперса синдром (В. J. Alpers, род. в 1900 г., амер. нейрохирург; син. полидистрофия мозга прогрессирующая) — развитие у детей слепоты, судорог, спастического изменения мышечного тонуса, миоклонуса, слабоумия, связанное с дегенеративными изменениями серого вещества мозга; носит семейный характер, наследуется, предположительно, по аутосомно-рецессивному типу.
(В. J. Alpers, род. в 1900 г., амер. нейрохирург; син. полидистрофия мозга прогрессирующая) развитие у детей слепоты, судорог, спастического изменения мышечного тонуса, миоклонуса, слабоумия, связанное с дегенеративными изменениями серого вещества мозга; носит семейный характер, наследуется, предположительно, по аутосомно-рецессивному типу.
Синдром Альперса
Синдром Альперса — наследственная болезнь (аутосомно-рецессивный механизм передачи), характеризующаяся развитием у детей слепоты, локальных или генерализованных судорог, миоклонических проявлений, слабоумия. Течение очень быстрое, с образованием слабоумия. [1] [2] . На сегодняшний день заболевание неизлечимое, и чем младше больной, тем быстрее наступает смерть.
Заболевание может вызываться мутациями ядерного гена POLG1, кодирующего митохондриальную ДНК-полимеразу гамма. [источник не указан 959 дней]
Примечания
- Синдромы по алфавиту
- Неврологические синдромы
Wikimedia Foundation . 2010 .
Полезное
Смотреть что такое "Синдром Альперса" в других словарях:
Синдром Клейне — Левина МКБ 10 G47.847.8 МКБ 9 327.13327.13 OMIM … Википедия
Синдром беспокойных ног — МКБ 10 G25.825.8 МКБ 9 333.94333.94 OMIM … Википедия
Синдром Рея — МКБ 10 G93.793.7 МКБ 9 331.81331.81 DiseasesDB … Википедия
Синдром Клейне — Левина — МКБ 10 G47.8 МКБ 9 327.13 OMIM 148840 DiseasesD … Википедия
Синдром мышечной скованности — МКБ 10 G25.825.8 МКБ 9 333.91333.91 OMIM … Википедия
Синдром Лея — МКБ 10 G31.831.8 МКБ 9 330.8330.8 OMIM … Википедия
Синдром Лейка — МКБ 10 G31.8 МКБ 9 330.8 OMIM 256000 DiseasesDB … Википедия
Альперса синдром — (В. J. Alpers, род. в 1900 г., амер. нейрохирург; син. полидистрофия мозга прогрессирующая) развитие у детей слепоты, судорог, спастического изменения мышечного тонуса, миоклонуса, слабоумия, связанное с дегенеративными изменениями серого… … Большой медицинский словарь
Альперса синдром — (Alpers B.J., 1931), Наблюдается в детском возрасте и характеризуется наличием локальных или генерализованных судорог, миоклонических проявлений, хореоатетоза и тремора мозжечкового генеза. Иногда - амавроз. Течение катастрофическое, быстрое, с… … Толковый словарь психиатрических терминов
Болезнь Альперса ( Синдрома Альперса-Хуттенлохера )
Болезнь Альперса — это редкое митохондриальное заболевание, которое характеризуется прогрессирующей энцефалопатией в сочетании с циррозом печени. Патология возникает вследствие мутации ДНК гамма-полимеразы (POLG1). Синдром проявляется эпилептическими приступами, угнетением психомоторных функций, токсическим поражением печени. Для диагностики используется ЭЭГ, МР-сканирование головного мозга, УЗИ печени, а также лабораторные исследования (биохимический анализ крови, коагулограмма, генетический тест). Терапия поддерживающая: антиконвульсанты, метаболические препараты, парентеральное питание.
МКБ-10
Общие сведения
Заболевание было впервые описано Б. Альперсом в 1931 г. у 4-месячной девочки с резистентными эпилептическими приступами. В 1976 г. П.Хуттенлохер уточнил характер наследования патологии, дополнил описание клинической картины. Частота встречаемости синдрома Альперса-Хуттенлохера составляет 1 случай на 100000 живорожденных детей. Болезнь отличается неблагоприятным течением, быстрым ухудшением состояния пациентов, присоединением соматических заболеваний, поэтому, несмотря на редкость, она не теряет актуальности в современной неврологии.
Причины
Болезнь Альперса обусловлена мутацией гена POLG1 (в локусе 15q25), который отвечает за функционирование гамма-полимеразы в митохондриях. Самые распространенные генные дефекты — A467T и W748S. Заболевание наследуется по аутосомно-рецессивному механизму, поэтому вероятность повторения патологии составляет 25% при следующей беременности. Риск развития синдрома повышается при близкородственных браках, другие предрасполагающие факторы не установлены.
Патогенез
Молекулярную основу заболевания составляет недостаточность гамма-полимеразы — единственной эукариотической ДНК-полимеразы, поддерживающей репликацию ДНК в митохондриях. При синдроме Альперса уровень репликации генетического материала снижается до критического, ухудшается активность митохондриальных ферментов. Дефицит АТФ сказывается на наиболее энергозависимых структурах — органах центральной и периферической нервной системы, печени, ЖКТ.
При болезни Альперса первыми поражаются проводники глубокой чувствительности, расположенные в задних столбах спинного мозга, также нарушаются функции мозжечка. Вовлечение в процесс коры обусловлено патологической электрической активностью в головном мозге. Резистентные к препаратам судорожные приступы обычно провоцируют склероз гиппокампа.
Симптомы болезни Альперса
Типичный возраст манифестации заболевания — от 2 до 4 лет, хотя описаны случаи раннего (3-4 месяца) и позднего начала болезни (до 8 лет). Для синдрома Альперса характерно бимодальное распределение, второй пик приходится на период 17-24 года. До появления первых клинических признаков дети имеют нормальное психомоторное развитие, достаточный уровень интеллекта. После старта неврологической симптоматики происходит резкий регресс ранее приобретенных навыков.
В половине случаев болезнь Альперса манифестирует судорожными приступами. Это могут быть фокальные судороги, миоклонические формы (ритмичные мышечные подергивания без спазмов мускулатуры), генерализованные тонико-клонические судороги, которые называют «большим эпилептическим пароксизмом». У детей первые приступы часто провоцируются фебрильной лихорадкой при инфекционных заболеваниях.
Синдром Альперса характеризуется множественными неврологическими нарушениями. У больных наблюдается шаткость походки, дискоординация движений, расстройства мелкой моторики. Сенсорная полинейропатия проявляется нарушением всех видов чувствительности, парестезиями. Как правило, возникают расстройства зрения (корковая слепота). Беспокоят мигренозные головные боли, в том числе как компонент ауры судорожных приступов.
Важным симптомом болезни является поражение печени. Клинически это проявляется тошнотой, ухудшением аппетита, нарушениями пищеварения. Зачастую больной испытывает слабость, сонливость, у него усугубляется неврологический дефицит. При угнетении синтеза печеночных факторов свертывания открываются спонтанные кровотечения из носа, десен, легких или желудочно-кишечного тракта.
Осложнения
Наиболее опасное последствие синдрома Альперса — фатальный токсический гепатит, который возникает при приеме препаратов из группы вальпроевой кислоты, купирующих судорожные приступы. При этом развивается тяжелая печеночная недостаточность, из-за чего пациенты погибают на протяжении 2-4 месяцев, независимо от проводимого лечения. Поражение гепатобилиарной системы может проявляться изменением архитектоники желчных путей, фиброзом печени.
При прогрессировании заболевания на фоне мышечной гипотонии развивается дисфагия, нарушения перистальтики ЖКТ. Возникает необходимость во введении желудочного зонда или использовании парентерального питания. Изредка поражение пищеварительной системы дополняется панкреатитом. У 10% больных развивается кардиомиопатия, результирующая тяжелой сердечной недостаточностью.
Диагностика
При первичном приеме у детского невролога выявляется атипичная эпилептическая энцефалопатия, которая требует обязательного подтверждения или исключения болезни Альперса. К обследованию больного привлекают генетика, гепатолога, кардиолога. Учитывая полиморфность клинических проявлений, для верификации диагноза назначаются инструментальные и лабораторные методы исследования:
- ЭЭГ. В начале болезни определяется биполярная или гомолатеральная эпилептическая активность в виде спайков и полиспайков. Для периода разгара синдрома Альперса типична диффузная медленная активность преимущественно в областях затылка с региональными акцентами в височно-теменных зонах мозга.
- МРТ головного мозга. На снимках визуализируются зоны умеренного гиперинтенсивного сигнала в затылочных областях церебральной коры. Для резистентных приступов характерно появление участков повышенной интенсивности в таламусе, продолговатом мозге. В базальных ганглиях, стволе мозга и мозжечке визуализируется прогрессирующая атрофия.
- УЗИ брюшной полости. При токсическом гепатите отмечается умеренное увеличение печени, снижение эхогенности ее паренхимы. Для уточнения характера патологии производится биопсия органа, по результатам которой выявляется жировая инфильтрация гепатоцитов, пролиферация желчных протоков, а при длительном существовании болезни — фиброз печени.
- Исследования крови. В биохимическом анализе крови определяется гипогликемия, повышение уровня трансаминаз. На развитие печеночной недостаточности указывает гипербилирубинемия, гипераммониемия, гипоальбуминемия. При оценке коагулограммы снижен протромбиновый индекс.
- Генетический анализ. При подозрении на синдром Альперса проводится полный анализ гена POLG1 методом автоматического секвенирования. Верификация диагноза возможна при обнаружении одной из 60 известных на сегодня мутаций. Изредка тест дополняют измерением числа копий митохондриальной ДНК.
Лечение болезни Альперса
Консервативная терапия
Сложность оказания медицинской помощи страдающим болезнью Альперса связана с отсутствием эффективных этиопатогенетических медикаментов. Лечащим врачом индивидуально для пациента подбирается симптоматическая терапия, которая корректирует неврологические нарушения, стабилизирует параметры гомеостаза. Лечение проводится в неврологических или реанимационных отделениях, применяются следующие группы препаратов:
- Антиоксиданты из группы витаминоподобных веществ. Обладают положительным анаболическим эффектом, улучшают метаболические показатели. Кроме того, являются единственными средствами, позволяющими купировать вальпроат-индуцированную печеночною недостаточность.
- Антиконвульсанты. Для устранения эпилептических пароксизмов рекомендованы современные противосудорожные медикаменты из групп сульфат-замещенных моносахаридов и ламотриджинов, которые не вызывают токсический гепатит. При их неэффективности используются лекарства старшего поколения (барбитураты, бензодиазепины).
- Инфузионные растворы. Для коррекции водно-электролитного обмена назначаются кристаллоидные растворы, по показаниям проводятся вливания препаратов глюкозы, аминокислотных смесей для поддержания адекватного уровня энергетического обмена.
Паллиативная помощь
Поскольку болезнь всегда заканчивается смертью пациента, необходимо обеспечить полноценный уход и медицинскую помощь на терминальном этапе заболевания. Лечение предполагает внутривенную гидратацию и введение питательных смесей, купирование судорог, устранение гипоксии путем гипербарической оксигенации, кислородной поддержки, ИВЛ. В рамках паллиатива проводится социальная и психологическая помощь членам семьи больного.
Прогноз и профилактика
Болезнь Альперса имеет неблагоприятное прогредиентное течение, смерть наступает в течение 3-4 лет от манифестации клинической симптоматики. Комплексная медицинская помощь облегчает состояние пациентов, однако пока не существует способов вылечить болезнь. Основным методом первичной профилактики является медико-генетическое консультирование семейных пар с отягощенной наследственностью.
Решающее значение во вторичной профилактике болезни имеет контроль показателей крови во время лечения больных с эпилепсией с помощью препаратов вальпроевой кислоты. При повышении показателей печеночных проб и снижении уровня тромбоцитов неврологи должны предполагать возможность митохондриальной патологии, своевременно направлять пациента на консультацию в генетический центр.
1. Синдром Альперса-Хуттенлохера во врачебной практике/ М.И. Душар, Г.Р. Акопян, Л.И. Волос, О.Я. Ковалюк// Современная педиатрия. — 2019. — №5.
2. Синдром Альперса-Хуттенлохера/ Т.Т. Батышева, В.М. Трепилец, Л.Я. Ахадова, Г.С. Голосная// Эпилепсия и пароксизмальные состояния. — 2015. — №1.
3. Эпилепсия у детей с митохондриальными заболеваниями: особенности диагностики и лечения/ Н.Н. Заваденко, А.А. Холин// Эпилепсия и пароксизмальные состояния. — 2012. — №2.
Болезнь Альперса
Болезнь Альперса
МКБ-10 коды
Описание
Болезнь Альперса. Это редкое митохондриальное заболевание, характеризующееся прогрессирующей энцефалопатией, связанной с циррозом печени. Патология возникает из-за мутации ДНК-гамма-полимеразы (POLG1). Синдром проявляется эпилептическими припадками, угнетением психомоторных функций, токсическим поражением печени. Для диагностики используются ЭЭГ, МРТ головного мозга, УЗИ печени, а также лабораторные исследования (биохимический анализ крови, коагулограмма, генетический тест). Поддерживающая терапия: противосудорожные препараты, метаболические препараты, парентеральное питание.
Дополнительные факты
Заболевание впервые было описано в 1931 г. Б. Альперсом у 4-месячной девочки с резистентными эпилептическими припадками. В 1976 г. P. Huttenlocher уточнил тип наследования патологии и добавил описание клинической картины. Заболеваемость синдромом Альперса-Хаттенлохера составляет 1 случай на 100 000 живорождений. Заболевание характеризуется неблагоприятным течением, быстрым ухудшением состояния больных и добавлением соматических заболеваний, поэтому, несмотря на свою редкость, не теряет своего значения в современной неврологии.
Болезнь Альперса вызывается мутацией в гене POLG1 (в локусе 15q25), который отвечает за функционирование гамма-полимеразы в митохондриях. Наиболее частыми дефектами генов являются A467T и W748S. Заболевание передается по аутосомно-рецессивному механизму, поэтому вероятность рецидива патологии при следующей беременности составляет 25%. Риск развития синдрома увеличивается при близкородственных браках, другие предрасполагающие факторы не установлены.
Молекулярная основа заболевания - дефицит гамма-полимеразы - единственной эукариотической ДНК-полимеразы, которая поддерживает репликацию ДНК в митохондриях. При синдроме Альперса уровень репликации генетического материала снижается до критического уровня, ухудшается активность митохондриальных ферментов. Дефицит АТФ поражает самые энергозависимые структуры - органы центральной и периферической нервной системы, печень, желудочно-кишечный тракт.
При болезни Альперса в первую очередь страдают проводники с глубоким зондированием, расположенные в задних столбах спинного мозга, а также нарушаются функции мозжечка. Вовлечение коры головного мозга в процесс происходит из-за аномальной электрической активности мозга. Приступы с лекарственной устойчивостью часто вызывают склероз гиппокампа.
Клиническая картина
Типичный возраст начала заболевания составляет от 2 до 4 лет, хотя описаны случаи раннего (3-4 месяца) и позднего начала заболевания (до 8 лет). Синдром Альперса характеризуется бимодальным распределением, второй пик приходится на возраст от 17 до 24 лет. До появления первых клинических симптомов у детей нормальное психомоторное развитие и достаточный интеллект. После появления неврологических симптомов происходит резкое снижение ранее приобретенных навыков.
Болезнь Альперса в половине случаев проявляется судорогами. Это могут быть очаговые припадки, миоклонические формы (ритмические подергивания мышц без мышечных спазмов), генерализованные тонико-клонические припадки, которые именуются «большим эпилептическим пароксизмом». У детей первые приступы часто спровоцированы лихорадкой, связанной с инфекционным заболеванием.
Синдром Альперса характеризуется множественными неврологическими расстройствами. У больных наблюдается нестабильность походки, разлад движений, нарушение мелкой моторики. Сенсорная полинейропатия проявляется нарушением всех видов чувствительности, парестезиями. Как правило, возникают нарушения зрения (корковая слепота). Беспокоят мигрени, в том числе как составная часть судорожной ауры.
Важный симптом заболевания - поражение печени. Клинически это проявляется тошнотой, плохим аппетитом и нарушениями пищеварения. Часто больной ощущает слабость, сонливость, обостряется его неврологический дефицит. При подавлении синтеза факторов свертывания крови в печени открывается спонтанное кровотечение из носа, десен, легких или желудочно-кишечного тракта.
Ассоциированные симптомы: Высокая температура тела. Нарушение походки. Судороги. Судороги в ногах. Тонико-клонические судороги. Фебрильная температура тела. Шаткая походка.
Возможные осложнения
Наиболее опасным последствием синдрома Альперса является токсический гепатит со смертельным исходом, возникающий при приеме препаратов из группы вальпроевой кислоты, снимающих судороги. При этом развивается тяжелая печеночная недостаточность, от которой пациенты умирают в течение 2-4 месяцев независимо от лечения. Повреждение гепатобилиарной системы может проявляться изменением архитектуры желчевыводящих путей, фиброзом печени.
По мере прогрессирования заболевания на фоне мышечной гипотензии развиваются дисфагия и нарушения моторики ЖКТ. Возникает необходимость введения назогастрального зонда или использования парентерального питания. Иногда поражение пищеварительной системы дополняется панкреатитом. Кардиомиопатия развивается у 10% пациентов, приводя к тяжелой сердечной недостаточности.
При первичном обращении к детскому неврологу выявляется атипичная эпилептическая энцефалопатия, требующая обязательного подтверждения или исключения болезни Альперса. К обследованию пациента привлекаются генетик, гепатолог и кардиолог. Учитывая полиморфизм клинических проявлений, для верификации диагноза назначаются инструментальные и лабораторные методы исследования:
• ЭЭГ. В начале болезни определяется биполярная или гомолатеральная эпилептическая активность в виде спайков и полиспайков. Для пикового периода синдрома Альперса характерна диффузная медленная активность, преимущественно в затылочных областях с региональными акцентами в височно-теменных областях мозга.
• МРТ головного мозга. На снимках видны зоны умеренного гиперинтенсивного сигнала в затылочных областях коры головного мозга. Для стойких припадков характерно появление участков повышенной интенсивности в области таламуса, продолговатого мозга. В базальных ганглиях, стволе мозга и мозжечке визуализируется прогрессирующая атрофия.
• УЗИ брюшной полости. При токсическом гепатите наблюдается умеренное увеличение печени, снижение эхогенности ее паренхимы. Для выяснения характера патологии проводится биопсия органа, по результатам которой выявляется жировая инфильтрация гепатоцитов, разрастание желчевыводящих путей и, при длительном существовании заболевания, фиброз печени.
• Кровавые анализы. В биохимическом анализе крови определяется гипогликемия, повышение уровня трансаминаз. На развитие печеночной недостаточности указывает гипербилирубинемия, гипераммонемия, гипоальбуминемия. При оценке коагулограммы протромбиновый индекс снижен.
• Генетический анализ. При подозрении на синдром Альперса проводится полный анализ гена POLG1 с использованием автоматического секвенирования. Верификация диагноза возможна после обнаружения одной из 60 известных на сегодняшний день мутаций. Иногда тест дополняется измерением количества копий митохондриальной ДНК.
Лечение
Сложность лечения людей с болезнью Альперса связана с отсутствием эффективных этиопатогенетических препаратов. Лечащий врач индивидуально подбирает пациенту симптоматическое лечение, которое устраняет неврологические нарушения, стабилизирует параметры гомеостаза. Лечение проводится в неврологических отделениях или отделениях реанимации, используются следующие группы препаратов:
• Антиоксиданты из группы витаминоподобных веществ. Они обладают положительным анаболическим действием, улучшают параметры обмена веществ. Кроме того, это единственный способ остановить печеночную недостаточность, вызванную вальпроатом.
• Противосудорожные препараты. Для устранения судорог рекомендуется использование современных противосудорожных средств из группы моносахаридов и сульфатзамещенных ламотриджинов, не вызывающих токсический гепатит. Если они неэффективны, используются препараты старшего поколения (барбитураты, бензодиазепины).
• Растворы для инфузий. Для коррекции водно-электролитного обмена назначают растворы кристаллоидов, по показаниям вводят препараты глюкозы, смеси аминокислот для поддержания адекватного уровня энергетического обмена.
Поскольку заболевание всегда заканчивается смертью пациента, необходимо оказать комплексную помощь и медицинскую помощь по окончании болезни. Лечение включает внутривенную гидратацию и введение питательных смесей, купирование приступов, устранение гипоксии путем гипербарической оксигенации, подачу кислорода, искусственную вентиляцию легких. В рамках паллиативной помощи членам семьи пациента оказывается социально-психологическая помощь.
Прогноз
Список литературы
1. Синдром Альперса-Хуттенлохера во врачебной практике/ М.И. Душар, Г.Р. Акопян, Л.И. Волос, О.Я. Ковалюк// Современная педиатрия. — 2019. — №5.
2. Синдром Альперса-Хуттенлохера/ Т.Т. Батышева, В.М. Трепилец, Л.Я. Ахадова, Г.С. Голосная// Эпилепсия и пароксизмальные состояния. — 2015. — №1.
3. Эпилепсия у детей с митохондриальными заболеваниями: особенности диагностики и лечения/ Н.Н. Заваденко, А.А. Холин// Эпилепсия и пароксизмальные состояния. — 2012. — №2.
Лечение и прогноз при синдроме Альперса
Одним из наиболее опасных врожденных детских недугов считается синдром Альперса. Коварство патологии заключается в том, что она не проявляется во время внутриутробного развития и диагностируется только после рождения младенца. К сожалению, способов лечения нет, терапия направлена на устранение возникшей симптоматики. Несмотря на то что болезнь диагностируется редко, родителям следует знать основные признаки аномалии, чтобы вовремя помочь ребенку.
Факторы, способствующие возникновению синдрома
Единственной причиной болезни Альперса считается аномальная мутация митохромной ДНК, провоцирующая постепенную дегенерацию серого вещества головного мозга. У больных детей отмечается атрофия мозговой ткани с последующей утратой жизненно важных функций. Способствуют появлению заболевания следующие факторы:
- наличие у одного или обоих родителей генной мутации (патология наследуется по аутосомно-рецессивному типу);
- возникновение случайной генетической аномалии во время внутриутробного развития (отец и мать не имеют скрытой патологии).
Мутация становится причиной того, что митохондрии мозга не могут полноценно функционировать. Клетки теряют способность использовать энергию, получаемую от усвоения питательных веществ, и постепенно погибают.
Редкость выявления синдрома Альперса объясняется тем, что ген, несущий признаки мутации, является рецессивным, и при наличии дефектной хромосомы только у матери или отца обычно не провоцирует возникновение патологического процесса (дети становятся носителями генетической мутации, которая не отражается на здоровье, но передается потомству). Высок риск развития болезни, когда оба родителя являются рецессивными носителями или при осложнениях, возникавших у женщины во время беременности.
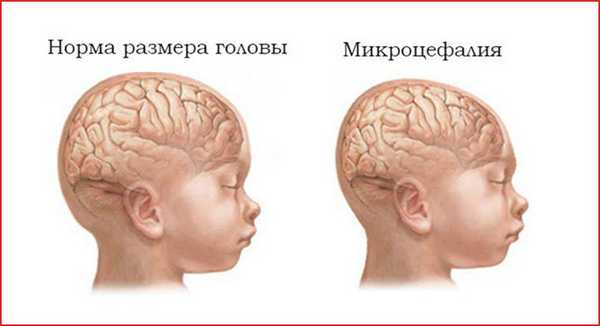
Классификация и проявления недуга
По времени возникновения выделяют 2 вида заболевания:
- Раннее. Патология проявляется у новорожденных и при ней отмечается сильное отставание ребенка в развитии.
- Позднее. У младенцев генетические митохромные нарушения «спят» и активизируются только во время полового созревания, в возрасте 14-16 лет (случается редко).
В подростковом возрасте клиника болезни сопровождается возникновением слабоумия, снижением слуха и зрения или другими мозговыми расстройствами.
У грудничков проявления более тяжелые:
- микроцефалия;
- задержка психического и физического развития;
- мышечная атония;
- частые срыгивания (возможна рвота);
- нарушения координации;
- появление судорог.
Возможны и другие симптомы, связанные с нарушением нормальной работы мозга.
Ребенок с патологией Альперса отстает в развитии от сверстников. У него отмечается снижение общего веса, деформация скелета и ухудшение подвижности суставов.
Диагностические методики
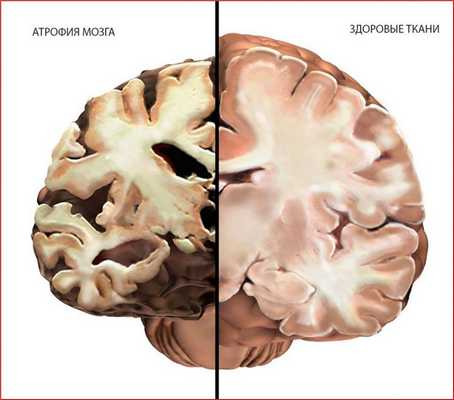
Особенностью синдрома Альперса является стремительное нарастание симптоматики, связанное с деструктивными процессами в мозговой ткани (на фото показан здоровый мозг и мозговая структура малыша с синдромом Альперса).
Но некоторые болезни могут проявляться сходными признаками, поэтому для уточнения диагноза ребенку назначают:
- ЭЭГ. Электроэнцефалограмма позволяет оценить мозговые колебания и выявить возникающие отклонения.
- КТ. Компьютерное сканирование считается самым информативным методом, позволяющим обнаружить очаги деструкции и спрогнозировать возможное ухудшение самочувствия (судороги, снижение слуха, затруднение мышления).
- Биохимия. В крови у детей отмечают повышенное содержание печеночных ферментов. Биохимия мало информативна на ранних стадиях заболевания, когда еще не произошло поражение гепатоцитов.
- УЗИ. Ультразвуковое исследование печени позволяет определить жизнеспособность органа и выявить участки погибших печеночных клеток, замещенных жировой тканью.
- Генный анализ. Выявляют митохондрическую хромосомную мутацию.
Наиболее точным методом диагностики считается генетическое исследование крови и КТ мозговой ткани.
Терапия при заболевании
- Поддержание функций мозговых клеток. Применяют «Актовегин», «Кортексин», «Церебрализин».
- Предотвращение судорожных припадков. Назначают «Депакин», «Конвулекс».
Применяемое лечение негативно влияет на функции гепатоцитов, провоцируя усиление развивающейся печеночной недостаточности. Для повышения работоспособности органа и его защиты используют гепатопротекторы («Гептрал», «Карсил»).
Прогноз болезни
Раннее выявление патологии помогает улучшить качество жизни маленького пациента и снизить тяжесть течения недуга, но прогноз неблагоприятный. Клетки мозга продолжают погибать из-за генетического дефекта митохондрий и нарушения энергообмена. Большинство пациентов с синдромом Альперса умирают по двум причинам:
- судорожный припадок;
- атрофия клеток мозга, отвечающих за жизнеобеспечение организма (дыхание, пищеварение, сердцебиение).
Дети с выявленным заболеванием в раннем возрасте живут около 3-х лет, у подростков (позднее проявление патологии Альперса) срок жизни не спрогнозирован из-за того, что эта форма болезни почти не возникает.
Рекомендация врачей при планировании беременности сдавать кровь обоих родителей на предрасположенность к генетическим заболеваниям основана на том, что раннее выявление дефекта генов у матери и отца возможно. Чтобы рождение младенца с синдромом Альперса не стало неожиданностью, не стоит пренебрегать лабораторным тестированием. Своевременное выявление генных отклонений поможет избежать тяжелой трагедии - болезни и ранней смерти ребенка.
Читайте также:
- Изменения анализов крови при заболеваниях почек
- Рентгеновский снимок с консолидацией легочной ткани в язычковых сегментах: описание, заключение
- Глубокий прикус: причины, симптомы и лечение
- Гипертрофия правого желудочка при тетраде и пентаде Фалло. Гипертрофия при пороках сердца Фалло
- Нормальный сон. Нормальная продолжительность сна и бессоница