Синдром Бурневилля (Bourneville) - синонимы, авторы, клиника
Добавил пользователь Morpheus Обновлено: 22.01.2026
… является одним из наиболее тяжелых по течению и прогнозу среди факоматозов (нейрокожных синдромов).
.. чаще встречается, чем диагностируется, так как индивидуумы, имеющие факультативные неспецифические клинические признаки в большинстве случаев не учитываются, а больные с облигатными признаками и синдромами, не являющимися нозологическими формами, довольно часто курируются врачами разных специальностей.
… диагностика этого редкого заболевания не является сложной, если врач имеет четкие представления о его клинических проявлениях.
Туберозный склероз (далее - ТС) - это непрерывно прогрессирующее генетически детерминированное заболевание из группы факоматозов с широким спектром клинических проявлений вследствие образования доброкачественных опухолей (гамартом) в различных органах и тканях человеческого организма, включая головной мозг, глаза, кожу, сердце, почки, печень, легкие, желудочно-кишечный тракт, эндокринную и костную системы. Синонимы ТС - болезнь Бурневилля-Прингла, центральный нейрономатоз, нейрокожный синдром типа Бурневилля, EPILOIA (epilepsy, low intelligence, angiofibroma), синдром себорейной аденомы, судорог и умственной отсталости.
Справочная информация. Гамарто́ма (от др.-греч. ἁμάρτημα - «ошибка», «изъян» и -ωμα от ὄγκωμα - «опухоль») - узловое доброкачественное опухолевидное образование, представляющее собой тканевую аномалию развития. Гамартомы состоят из тех же тканевых компонентов, что и орган, где она расположена. При этом она отличается аномальным строением и степенью дифференцировки тканей. В тех случаях, когда в строении гамартомы преобладает какая-либо одна ткань, её называют по характеру этой ткани - хондроматозная, сосудистая и т. п. При обнаружении элементов разных тканей, напоминающих картину того или иного органа, используют термин «органоидная гамартома». Гамартомы являются доброкачественными новообразованиями. Крайне редко из них могут развиться злокачественные опухоли гамартобластомы.
ТС - аутосомно-доминантное генетически гетерогенное заболевание. Приблизительно от 10 до 30% случаев ТС обусловлено мутациями в гене TSC1 (ТС 1-го типа), локализованном на 9 хромосоме и кодирующим белок гамартин. Остальные случаи болезни обусловлены мутациями в гене TSC2 (ТС 2-го типа), локализованном на 16 хромосоме и кодирующим белок туберин (для генов ТС характерны высокая пенетрантность - до 100%, вариабельная экспрессивность и высокая частота возникновения новых [спонтанных - de novo] мутаций).Считается, что у пациентов с мутацией TSC1 заболевание течет более мягко, а мутации в гене TSC2 обусловливают более тяжелое развитие патологии. ТС относится к редким (орфанным) заболеваниям, его частота в популяции составляет 1 : 10 000 (у новорожденных - 1 : 6000).
Гены TSC1 и TSC2 в норме являются естественными генами-супрессорами опухолевого роста. Белковые продукты генов TSC1 и TSC2, гамартин и туберин, образуют гетеродимер, способный ингибировать опосредованный комплексом mTORС1 (mammalian Target of Rapamycin Complex 1) сигнальный каскад. Мутации в генах NCS1 и TSC2 приводят к потери функций гамартина и туберина, и, как следствие, к патологической активации киназы mTOR, которая является ключевым регулятором роста и пролиферации клеток, поэтому при заболевании развиваются множественные доброкачественные опухоли (гамартомы) в различных органах, включая головной мозг, глаза, кожу, сердце, почки, печень, легкие, желудочно-кишечный тракт, а также эндокринную и костную системы. Постепенно прогрессируя и увеличиваясь в размерах, они нарушают функции этих органов, иногда приводя к фатальным последствиям, чаще - к сокращению продолжительности жизни и инвалидизации пациентов.
Симптомы заболевания могут проявляться с рождения, однако чаще развиваются в первые годы жизни; процесс постепенно прогрессирует (особенно в период полового созревания) - имеет место возраст-зависимый характер [проявления] основных клинических симптомов. В клинической картине ТС доминируют симптомы со стороны ЦНС и кожные проявления.
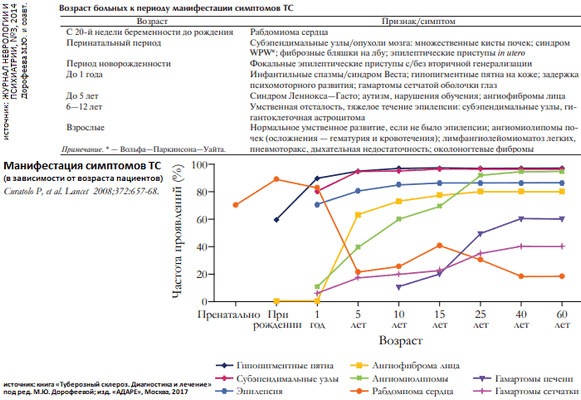
Симптомы со стороны ЦНС обнаруживаются более чем у 80% пациентов, а структурные изменения головного мозга, выявляемые по данным МРТ, - у 95%. ТС проявляется у пациентов с эпилепсией, нарушениями нервно-психического развития и расстройствами поведения. Эпилептические приступы отмечаются у 70 - 90% пациентов с ТС. У большинства пациентов приступы отмечаются на первом году жизни, но даже у взрослых есть риск развития приступов (12%). Инфантильные спазмы - самый распространенный тип приступов в дебюте эпилепсии, но у 54% пациентов развиваются различные типы приступов - простые и сложные фокальные, вторично-генерализованные. Около 30% пациентов страдают умственной отсталостью, половина больных имеют низкие показатели уровня интеллекта (IQ менее 70 баллов), при этом могут выявляться нарушения памяти, внимания, праксиса. К нейропсихиатрическим нарушениям, которые ассоциированы с ТС, относятся расстройства аутистического спектра (50%), синдром дефицита внимания/гиперактивности (30 - 50%), агрессивное поведение и вспышки ярости.
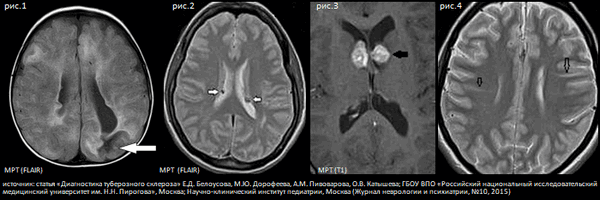
Описанная выше симптоматика ТС обусловлена структурными изменениями мозга, которые выявляются при нейро-радиологическом исследовании. К ним относятся корковые и подкорковые туберы [см. рис.1] (зоны дисплазии, в которых нарушено нормальное линейное строение коры и содержатся аномально большие клетки или «клетки-баллоны». Туберы обнаруживаются у 95 - 100% пациентов. Часто именно туберы являются причиной эпилепсии. Субэпендимальные узлы (см. рис.2) встречаются у 80% пациентов, по морфологическому строению они напоминают туберы и имеют характерную нейро-радиологическую картину. Одним из наиболее серьезных и жизнеугрожающих симптомов ТС являются субэпендимальные гигантоклеточные астроцитомы [см. рис.3] (СЭГА), которые встречаются примерно у 20% пациентов. Считается, что опухоль развивается из субэпендимальных узлов в первые 2 десятилетия жизни, но иногда она может обнаруживаться и у новорожденного ребенка. Как правило, СЭГА располагаются около отверстия Монро, растут медленно, но при росте вызывают развитие прогрессирующей гидроцефалии (проявляется рвотой, головной болью, снижением остроты зрения вплоть до слепоты, развитием спастического тетрапареза). Если пациент не будет прооперирован или не будет получать ингибитор m-TOR эверолимус, прогрессирующая гидроцефалия может привести к летальному исходу. Кроме того, у пациентов с ТС часто выявляются радиальные полосы белого вещества - радиальные миграционные тракты (см. рис.4), которые являются следствием нарушения нейрональной миграции, и кисты у задних рогов боковых желудочков. Могут встречаться и другие аномалии строения коры (фокальные дисплазии, шизэнцефалия, гетеротопии, частичная агенезия мозолистого тела).
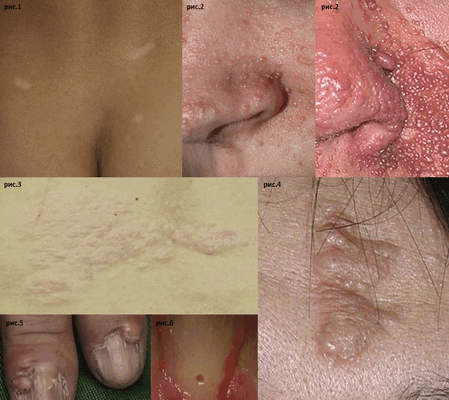
Самым очевидным и ранним кожным симптомом являются гипопигментные пятна (см. рис.1), которые могут присутствовать у новорожденного ребенка или обнаруживаться в младенчестве. Встречаются более чем у 90% пациентов с ТС в возрасте до 5 лет. Наиболее характерная форма пятен - овальная, похожая на лист ясеня, размером от 1 до 12 см. Их количество варьирует от 3 - 4 до 100 и более, с возрастом наблюдается тенденция к увеличению числа пятен. Другим достаточно распространенным симптомом ТС являются ангиофибромы лица (см. рис.2). Частота их встречаемости увеличивается с возрастом: от 8% у детей в возрасте до 2 лет, до 75% у детей в возрасте до 9 лет. Ангиофибромы располагаются симметрично с обеих сторон лица на щеках и носу по типу «крыльев бабочки»; первоначально это - папулы розового цвета, но со временем они приобретают красно-коричневую окраску. Также в первое - второе десятилетия жизни у 30 - 70% детей появляются участки «шагреневой кожи» (см. рис.3) - коллагеномы, как правило, расположенные в нижней части спины и внешне похожие на апельсиновую корку. Фиброзные бляшки (см. рис.4) часто появляются уже на первом году жизни, встречаются примерно у 25% больных, имеют бежевый цвет и шероховаты на ощупь, чаще всего локализуются на лбу. Околоногтевые фибромы [см. рис.5] (опухоли Коэнена) появляются на втором десятилетии жизни и встречаются в 17 - 52% случаев. Это папулы (узлы) красного или мясного цвета, растущие от ногтевого ложа или вокруг ногтевой пластинки рук и ног. Вышеописанные кожные симптомы относятся к так называемым первичным признакам. Кроме них у пациентов с ТС могут встречаться вторичные кожные симптомы. К ним относятся гипопигментные мелкие пятна по типу конфетти, фибромы ротовой полости (расположены на деснах, слизистой щеки и губах) и дефекты эмали зубов (см. рис.6). Последний симптом довольно часто встречается и при отсутствии ТС, но у пациентов с ТС его частота очень высокая - дефекты эмали можно найти практически у всех пациентов.
Обратите внимание! Фенотип пациента с ТС зависит от числа, локализации и размера гамартом. Возраст больного также играет важную роль, так как разные симптомы болезни проявляются в различные возрастные периоды. Большое количество клинических признаков ТС, вариабельность фенотипа и тот факт, что манифестация признаков зависит от возраста пациента, затрудняют диагностику заболевания.
[ 1 ] Полный осмотр кожных покровов. Гипопигментные пятна, как правило, хорошо видны невооруженным глазом, хотя в отдельных случаях заметить их невозможно. При подозрении на ТС для визуализации гипопигментных пятен рекомендуется применение лампы Вуда, которая излучает пучок света с длиной волны 360 нм, селективно абсорбирующегося меланиновыми клетками кожи. Под светом этой лампы здоровая кожа выглядит тусклой, а участки кожи, имеющие дефицит меланина, ярко светятся.
[ 2 ] МРТ головного мозга - позволяет выявить туберы, СЭГА, субэпендимальные узлы и нарушения в белом веществе головного мозга. При подозрении на кальцификацию туберов проводится КТ головного мозга.
[ 3 ] Электроэнцефалография (ЭЭГ) - обязательно проводится при наличии эпилептических приступов или при пренатальном выявлении рабдомиомы сердца у новорожденных с целью своевременно обнаружить эпилептиформную активность.
[ 4 ] Ультразвуковое исследование органов брюшной полости и почек - проводится для исключения АМЛ, поликистоза почек, а также гамартом печени и поджелудочной железы. В сомнительных случаях выполняется МРТ почек.
[ 5 ] Электрокардиография и эхокардиография - проводятся для обнаружения рабдомиом сердца и нарушений сердечного ритма, обусловленных их наличием;
[ 6 ] Нейропсихологическое тестирование - для оценки речевого и интеллектуального развития ребенка.
[ 7 ] Молекулярная диагностика - генетическое тестирование позволяет определить конкретную мутацию, которая привела к развитию болезни. ДНК-исследование подтверждает диагноз ТС у пациента и его родителей и позволяет определить спорадический или наследственный характер заболевания. Если характерные мутации не обнаружены, а клинических проявлений недостаточно для постановки достоверного диагноза ТС, показано ежегодное динамическое наблюдение с целью выявления возможных симптомов в дальнейшем.
Диагностические критерии ТС: [ 1 ] несомненный диагноз ТС устанавливается на основании наличия 2 первичных признаков или 1 первичного и 2 вторичных признаков; [ 2 ] возможный диагноз - на основании наличия 1 первичного признака или 1 первичного и 1 вторичного признаков, или 2 (и более) вторичных признаков. Ниже приведены первичные и вторичные признаки (2012 TSC Clinical Consensus Conference). [ . ] Подтвержденная патогенная мутация TSC1 или TSC2 является главнейшим критерием, достаточным для постановки диагноза ТС.
Первичные (большие) признаки :
[1] ангиофибромы лица (не менее 3) или фиброзные бляшки на лбу;
[2] гипопигментные пятна (не менее 3 и не менее 5 мм в диаметре);
[3] нетравматические околоногтевые фибромы (не менее 2);
[4] участок «шагреневой кожи»;
[5] множественные гамартомы сетчатки;
[6] корковые дисплазии (не менее 3): корковые туберы и миграционные тракты в белом веществе головного мозга;
[7] субэпендимальные узлы (не менее 2);
[8] субэпендимальная гигантоклеточная астроцитома;
[9] рабдомиомы сердца множественные или одиночные;
[10] лимфангиолейомиоматоз легких;
[11] множественные ангиомиолипомы почек (не менее 2).
Вторичные (малые) признаки :
[1] многочисленные углубления в эмали зубов (не менее 3);
[2] фибромы в полости рта (не менее 2);
[3] гамартомы внутренних органов;
[4] ахроматический участок сетчатой оболочки глаза;
[5] пятна типа конфетти на коже;
[6] множественные кисты почек.
В мировой практике профилактика ТС сводится к его пренатальной диагностике. При спорадическом ТС риск повторного рождения больного ребенка составляет 2%, при наследственном - 50%. Поэтому при подозрении на наследственный характер ТС (при наличии подозрения или подтвержденного диагноза ТС у будущей матери, отца или родственников) генетическую диагностику ТС необходимо проводить еще при планировании беременности. Подтвержденный наследственный тип ТС является основанием для проведения инвазивной пренатальной диагностики. При подозрении на ТС у плода рекомендуется проведение эхокардиографии на сроках 20 - 24 недели беременности для исключения рабдомиомы сердца.
Подробнее о ТС в следующих источниках:
статья «Рекомендации по диагностике и лечению туберозного склероза» М.Ю. Дорофеева, Е.Д. Белоусова, А.М. Пивоварова; ФГБУ «Московский НИИ педиатрии и детской хирургии», Москва (Журнал неврологии и психиатрии, №3, 2014) [читать];
статья «Диагностика туберозного склероза» Е.Д. Белоусова, М.Ю. Дорофеева, А.М. Пивоварова, О.В. Катышева; ГБОУ ВПО «Российский национальный исследовательский медицинский университет имени Н. Н. Пирогова», Москва; Научно-клинический институт педиатрии, Москва (Журнал неврологии и психиатрии, №10, 2015) [читать];
книга «Туберозный склероз. Диагностика и лечение» под ред. М.Ю. Дорофеевой; изд. «АДАРЕ», Москва, 2017 [читать]
статья «Лечение эпилепсии при туберозном склерозе» Белоусова Е.Д., Дорофеева М.Ю., Охапкина Т.Г. ГБОУ ВПО «РНИМУ им. Н.Н. Пирогова» Минздрава России (журнал «Эпилепсия и пароксизмальные состояния» №2, 2016) [читать];
статья «Хирургическое лечение эпилепсии у пациентов с туберозным склерозом» Hans Holthausen, Tom Pieper, Hans Eitel, Manfred Kudernatsch; Neuropediatric Clinic and Clinic for Neurorehabiltation, Epilepsy Center for Children and Adolescents; Neurosurgery Clinic and Clinic for Epilepsy Surgery; Schoen-Klinik Vogtareuth (Русский журнал детской неврологии, №1, 2015) [читать]
Болезнь Бурневилля-Прингла: доброкачественная ошибка

Доброкачественность опухоли еще не означает ее безопасность. Существует генетическое заболевание, при котором многочисленные доброкачественные опухоли поражают разные органы и ткани, приводя к тяжелым патологиям. Речь идет о туберозном склерозе, или болезни Бурневилля-Прингла. MedAboutMe выяснял, что это за болезнь и чем опасны доброкачественные опухоли, когда их слишком много.
Факоматозы и гамартомы

Туберозный склероз входит в многочисленную группу болезней под названием «факоматозы», от греческого слова «факос» — пятно. Такие патологии являются результатом поражений кожи и тех тканей, которые имеют общее с ней происхождение: нервных тканей, клеток сетчатки, а также некоторых внутренних органов. Все они образуются из одного слоя зародышевых клеток.
Чаще всего факоматозы развиваются в результате мутации одного-двух генов. Но даже такого небольшого генетического дефекта, который начинает проявлять себя еще во время внутриутробного развития, достаточно, чтобы в значительной степени искалечить организм человека.
Для факоматозов характерно разрастание различных слоев кожи с образованием доброкачественных опухолей — телеангиэктазий, фибром, ангиом, аденом и др. Часто наблюдаются также нарушения пигментации кожи — образуются характерные пятна обесцвеченной кожи или пятна цвета «кофе с молоком». Например, в число известных и довольно распространенных факоматозов входит альбинизм.
История открытия болезни «туберозный склероз»
Впервые специфические множественные доброкачественные разрастания тканей были обнаружены и описаны в 1862 году Ф.Д. Реклингхаузеном. В 1880 году французский невролог, психиатр и депутат парламента Дезире Бурневилль объявил их отдельным заболеванием. А в 1890 году шотландский физиолог и врач Дж. Дж. Прингл в ходе исследования тканей мозга ребёнка с аналогичным заболеванием обнаружил клубневидные склерозированные разрастания нервной ткани, которые назвал «туберами» («тубер» — клубень). Он же и дал болезни ее нынешнее название — туберозный склероз.
Сегодня говорят о целой группе патологий, которые объединяются под названием «туберозный склероз» или «болезнь Бурневилля-Прингла». Болезнь имеет множество синонимов: эпилойя, центральный нейрономатоз, узелковый склероз, нейрокожный синдром типа Бурневилля, синдром себорейной аденомы, а также аденома сальных желез, и др.
Туберозный склероз — редкое (орфанное) заболевание. Заболевамость им в разных странах колеблется от 1:6000 населения до 1:15400.
Генетика туберозного склероза

Сегодня известны гены, отвечающие за развитие туберозного склероза. Это TSC1, расположенный на 9-й хромосоме, и TSC2, который находится на 16-й хромосоме. В норме эти гены выполняют функции опухолевых супрессоров, то есть подавляют развитие опухолей. Первый из них кодирует белок гамартин, а второй — белок тубертин. Вместе они образуют димер, который блокирует патологический рост и размножение клеток. Мутация, напротив, активизирует так называемый сигнальный путь mTOR, что и приводит к образованию опухолей.

По последним данным, мутация гена TSC1 также приводит к нарушению миелинизации нейронов, то есть к сбою в образовании специального миелинового покрытия на поверхности нервных клеток.
Мутация каждого из этих двух генов ответственна за половину случаев передачи заболевания по наследству — это примерно треть всех случаев болезни Бурневилля-Прингла. Две трети случаев (73%) — результат новых случайных мутаций.
Больные туберозным склерозом редко обзаводятся детьми, поэтому наследственная передача мутации встречается нечасто, хотя она является аутосомно-доминантной (то есть, достаточно одного больного родителя, чтобы поврежденные гены проявились у его ребёнка).
Симптомы болезни Бурневилля-Прингла

Из-за того, что генетическая мутация оказывает влияние на организм еще на эмбриональном уровне, развиваются полисистемные поражения кожи, нервной системы, зрения, внутренних органов, эндокринной системы и костной ткани. Лечением и наблюдением таких пациентов занимаются сразу несколько разных специалистов.
Болезнь проявляет себя на первом году жизни следующими основными симптомами:
- Поражения кожи.
- Опухолевидные образования в разных органах и тканях.
- Нарушения интеллекта.
- Судорожные приступы.
Поражения кожи могут проявляться в виде гипопигментированных пятен по всему телу (окрашенных как «кофе с молоком» и в форме «кленовых листьев»), кожных утолщенных образований в виде «шагреневой кожи». Часто наблюдается изменение пигментации ресниц и волос — появляются белые пряди или целые участки, лишенные пигмента. Нарушение выработки меланина может также наблюдаться на радужной оболочке глаз.
Как было сказано выше, главным симптомом и причиной тяжелых поражений органов и тканей являются многочисленные доброкачественнее опухоли. При туберозном склерозе образуются:
- гамартомы — опухоли, развивающиеся из эмбриональной ткани. Такие гамартозные образования формируются на сетчатке глаза в тканях сердца и почек, а также в области прямой кишки в виде полипов;
- ангиофибромы на лице — один из характерных признаков болезни, который представляет собой многочисленные шаровидные образования (аденомы сальных желез Прингла) по обеим сторонам крыльев носа, в области век;
- опухоли Кенена — околоногтевые фибромы, опухоли, образующиеся из тканей, окружающих ногти. Чаще поражаются пальцы на ногах;
- фибромы десен, тканей шеи, рук и ног, а также туловища.
Поражения интеллекта проявляются в виде умственной отсталости (у половины таких пациентов), нарушениях поведения. Лишь в редких случаях интеллект остается сохранным. У трети детей с туберозным склерозом в возрасте до 5 лет диагностируется аутизм.
Недавно американские ученые из детской клиники Цинциннати предложили использовать метод диагностики аутизма в группах высокого риска для выявления пациентов с туберозным склерозом. Предполагается, что таким образом можно будет ставить диагноз на самых ранних стадиях, а значит, раньше начать лечение.
Судорожные приступы — один из первых симптомов заболевания. Они отмечаются у 80-92% детей, страдающих от туберозного склероза, и развиваются в первые три года жизни у 80% пациентов. Нередко при этом также происходит нарушение цикла «сон-бодрствование».
Не все перечисленные симптомы встречаются одновременно. Для диагностики обычно достаточно комбинации нескольких признаков.
Можно ли вылечиться от туберозного склероза?
Нет. Никакой возможности излечить эту патологию на данный момент не существует. Поэтому применяется только симптоматическое лечение.
В список препаратов, которые назначаются таким пациентам, входят антиконвульсанты и другие лекарства, снижающие частоту пароксизмальных спазмов. Им также предписывается кетогенная диета (питание с резким ограничением количества углеводов и заменой их на жиры). Тяжелые поражения кожи лечатся при помощи лазерной терапии и методов электрокоагуляции. Опухоли головного мозга, угрожающие жизни пациента, удаляются хирургическим путем. Назначается медикаментозная терапия поликистоза почек, при крайней необходимости (рак почки) проводится нефрэктомия — удаление почки. Такие пациенты также проходят лечение у психиатров и являются участниками специальных программ социальной адаптации.
В России сегодня проживает примерно 7 тысяч человек с диагнозом «туберозный склероз». Они получают инвалидность еще в детстве и нуждаются в постоянном уходе, так как в связи с умственной отсталостью и повышенным риском развития эпилептических приступов не в состоянии жить самостоятельно. Тем из них, кому может помочь эверолимус, государство оплачивает терапию.
Болезнь Бурневилля

Туберозный склероз (болезнь Бурневилля) — редкое генетическое заболевание, при котором во множестве органов и тканей образуются доброкачественные опухоли. Полисистемный характер нарушений порождает широкий спектр симптомов — повреждения мозга могут вызвать эпилепсию, снижение интеллекта; поражаются внутренние органы — почки, сердце, легкие; характерные новообразования кожи лица и глазного дна могут быть использованы при начальной диагностике.
Первое слово в названии болезни происходит от латинского tuber — нарост, опухоль, и описывает «туберсы» — характерные новообразования в коре мозга больных, обычно на границе серого и белого вещества. Туберсы были впервые описаны французским неврологом Бурневиллем, поэтому иногда болезнь называют его именем.
Были установлены два локуса, связанные с заболеванием, а позднее описаны расположенные в них гены TSC1 и TSC2, кодирующие белки гамартин и туберин. Оба гена принадлежат к числу генов-супрессоров опухолей, в норме не позволяющих развиваться патологиям, ограничивающих избыточный рост тканей.
Содержание
История заболевания
Эпидемиология
Этиология
Патогенез
Клиника заболевания
Диагностика
Лечение
Прогноз
Примечания
Ссылки
Wikimedia Foundation . 2010 .
Полезное
Смотреть что такое "Болезнь Бурневилля" в других словарях:
Болезнь Бурневилля-Прингля — См. Склероз туберозный … Энциклопедический словарь по психологии и педагогике
БУРНЕВИЛЛЯ СИНДРОМ — (Бурневилля - Прингла болезнь, описана французским неврологом D. M. Bourneville, 1840-1909, британским дерматологом J. J. Pringle, 1855-1922; синонимы - эпилойя, туберозный склероз) - наследственное заболевание из группы факоматозов (греч. phakos … Энциклопедический словарь по психологии и педагогике
Бурневилля болезнь — (D. M. Bourneville, 1840 1909. франц. врач) см. Склероз туберозный … Большой медицинский словарь
Бурневилля-Прингла болезнь — (D. M. Boumeville, 1840 1909, франц. врач: J. J. Pringle, 1855 1922, англ. врач) см. Склероз туберозный … Большой медицинский словарь
Список наследственных заболеваний — Список генетических заболеваний Основные статьи: наследственные заболевания, Наследственные болезни обмена веществ, Ферментопатия. В большинстве случаев приведен также код, указывающий на тип мутации и связанные с ней хромосомы. См. также система … Википедия
склероз туберозный — (sclerosis tuberosa; син.: Бурневилля болезнь, Бурневилля Прингла болезнь, эпилойя) наследственная болезнь из группы факоматозов, характеризующаяся сочетанием судорожных припадков, слабоумия и поражений кожи (фибромы, аденомы сальных желез,… … Большой медицинский словарь
Склеро́з туберо́зный — (sclerosis tuberosa; син.: Бурневилля болезнь, Бурневилля Прингла болезнь, эпилойя) наследственная болезнь из группы факоматозов, характеризующаяся сочетанием судорожных припадков, слабоумия и поражений кожи (фибромы, аденомы сальных желез,… … Медицинская энциклопедия
Список эпизодов телесериала «Доктор Хаус» — Основная статья: Доктор Хаус … Википедия
ОЛИГОФРЕНИЯ — - группа различных по этиологии и патогенезу заболеваний, основным проявлением которых служит врожденное или приобретенное в первые 3 года жизни слабоумие и затруднение социальной адаптации. Причиной олигофрении могут быть хромосомные аномалии,… … Энциклопедический словарь по психологии и педагогике
БУРНЕВИЛЛЯ СИНДРОМ
БУРНЕВИЛЛЯ СИНДРОМ (Бурневилля - Прингла болезнь, описана французским неврологом D. M. Bourneville, 1840-1909, британским дерматологом J. J. Pringle, 1855-1922; синонимы - эпилойя, туберозный склероз) - наследственное заболевание из группы факоматозов (греч. phakos - чечевица, родимое пятно): системных дисплазий, характеризующихся комбинированными опухолевидными пороками развития кожи, нервной системы, глаз и др. Клинически проявляется триадой признаков: большие и малые судорожные приступы, кивки («салаамовы» судороги); умственная отсталость разной степени; поражения кожи - ангиофиброма щек в форме «бабочки»; околоногтевые и подногтевые подкожные фибромы; мягкие, плоские, слегка выступающие соединительнотканные невусы типа «шагреневой кожи» в пояснично-крестцовой области; телеангиэктазии и др. Встречаются опухолевидные изменения сетчатки, опухоли внутренних органов. Рентгенография и компьютерная томография черепа выявляют внутримозговые кальцификаты. Наследуется аутосомно-доминантно; 85 % случаев - спорадические, как результат новых мутаций. Лечение симптоматическое: противосудорожные средства, удаление кожных образований - криодеструкция снегом угольной кислоты, жидким азотом; дермабразия; диатермокоагуляция и др.; диспансерное наблюдение у невролога, офтальмолога, онколога и др.
D. M. Bourneville. Sclerose tubereuse des circonvolution cerebrales: Idiotie et epilepsie hemiplegique. Archives de neurologie, Paris, 1880; 1: 81-9l.
J. J. Pringle. A case of congenital adenoma sebaceum. British Journal of Dermatology, Oxford, 1890; 2: 1-14.
Энциклопедический словарь по психологии и педагогике . 2013 .
Смотреть что такое "БУРНЕВИЛЛЯ СИНДРОМ" в других словарях:
Туберозный склероз — Перва … Википедия
Мезенхимальные опухоли — Опухоли мягких тканей (за исключением нейрогенных новообразований) и специфические опухоли костей в онкоморфологии объединяются понятием мезенхимальные опухоли [mesenchymal tumors]. К опухолям мягких тканей [soft tissue tumors] относятся опухоли… … Википедия
Бриссо, Эдуард — Эдуард Бриссо Édouard Brissaud … Википедия
Склероз туберозный головного мозга — (Bournevill, 1880) - генетическая патология с аутосомно доминантным типом наследования, выражается системной патологией органов преимущественно эктодермального происхождения. Заболевание начинается обычно в молодом возрасте с появления… … Энциклопедический словарь по психологии и педагогике
Бурневилля болезнь
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
Смотреть что такое "Бурневилля болезнь" в других словарях:
Болезнь Бурневилля — Туберозный склероз Первая известная иллюстрация заболевания, из атласа кожных болезней французского врача Пьера Райе, 1835 год МКБ 10 Q85.1 МКБ 9 … Википедия
Читайте также:
- Вспомогательный аппарат глаза. Слезные железы. Мышцы глаза.
- Вестибуло-глазодвигательные тесты
- Доступ и ход операции резекции холангиокарциномы с наложением билатеральных гепатикоеюностом
- Лучевая диагностика оссификации задней продольной связки (ОЗПС)
- Факторы влияющие на венозный приток и сердечный выброс. Положительное давление в конце вдоха (ПДКВ)