Синдром дизостоза энхондрального - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Терещенко В.А., Стасова Ю.В.
Научный руководитель: к.м.н., доцент Нечаев В. Н.
ГБОУ ВПО Саратовский ГМУ им. В.И. Разумовского Минздрава РФ Кафедра госпитальной педиатрии и неонатологии
Резюме
В представленной статье описывается клинический опыт ведения пациента с врожденным заболеванием (мандибуло-фациальным дизостозом), возникающим в результате поражения структур, исходящих из первой жаберной дуги.
Ключевые слова
Статья
Мандибуло-фациальный дизостоз - это поражение структур, исходящих из первой жаберной дуги, наследуемый по аутосомно-доминантному типу с высокой (до 90%) пенетрантностью и различной экспрессивностью переменной, даже среди больных одной семьи. Может наблюдаться в двух или даже трех генерациях [1].
Этот синдром вызывается мутациями в TCOF1 гене (5q32-q33.1) или в POLR1C гене (6p21.1) и POLR1D гене (13q12.2), кодирующих РНК-полимеразы I и III подразделений. Он обусловлен дисплазией эмбрионального элемента первой жаберной дуги неизвестного происхождения [2]. Описывается, как врожденное нарушение развития костей черепа (чаще височной кости) и лица, характеризуется двусторонней симметричной ото-нижнечелюстной дисплазией без аномалий развития конечностей, он также связан с некоторыми дефектами головы и шеи [3].
Синдом Франческетти - редкое заболевание, встречающееся в 1 случае на 50 000 детей, рожденных живыми.
Дифференциальная диагностика проводится с синдромом глазо-ушно-позвоночной дисплазии Гольденхаара, при которой имеются эпибульбарные дермоиды, часто связанные с колобомой верхнего века, ушные отростки и предушные слюнные фистулы, нарушение строения наружного слухового прохода и тугоухость, шейный синостоз, увеличение количества грудных или поясничных позвонков, и другие аномалии позвоночника. Часто наблюдается высокое расщепленное небо, раздвоенный язык и аномалии зубов [4, 5].
Несмотря на наличие разнообразных аномалий, прогноз при синдроме Франческетти благоприятный. Этиологического лечения не существует, по возможности проводят пластические операции. Гибель пациентов чаще наступает от интеркуррентных заболеваний.
Под нашим наблюдением находилась новорожденная доношенная девочка Мария Г., родившаяся от 6 беременности, на фоне: отеков, вызванных беременностью, хронической внутриутробной гипоксии плода, фетоплацентарной недостаточности, вегето-сосудистой дистонии по гипертоническому типу, хронического гастрита. В 20 недель мать проходила лечение по поводу острого токсоплазмоза, отмечалось многоводие, в 34 недели у ребенка выявлен порок мочевыводящей системы (МВС). Роды 3 срочные в головном предлежании. Масса ребенка при рождении 2800 граммов, рост - 50 см., с оценкой по шкале Апгар 4, 5, 5 баллов.
Из родильного зала ребенок поступил в ОРИТН в тяжелом состоянии за счет явлений дыхательной недостаточности 3 степени, неврологической симптоматики, задержки внутриутробного развития, кожного геморрагического синдрома. С рождения до 20 суток жизни проводилась респираторная терапия, затем, в течение 7 дней дыхание через воздуховод, в последующем - спонтанное, лишь при нарастании дыхательной недостаточности периодически нуждалась в постановке воздуховода. Проводилось зондовое кормление.
Обращали на себя внимание множественные пороки развития: выраженная микрогения, гипоплазия скуловых костей, большой «клювовидный нос», короткий фильтр, глазной гипотеларизм, микрофтальмия, антимонголоидный разрез глаз, «птичье лицо», деформация ушных раковин (низко расположенные, большие уши), гипоплазия ногтевых пластинок (на кистях и стопах), широкая грудная клетка, сосковый гипертеларизм, выраженная бледность кожных покровов. В связи с выявлением пороков развития и более 5 стигм дисэмбриогенеза, ребенок консультирован генетиком. На основании фенотипических данных обследования методом синдромологического анализа был заподозрен синдром Франческетти: аутосомно-доминантное заболевание.
Пациентке было проведено кариотипирование, выявлен нормальный женский кариотип (46 хх).
Ребенок находился на зондовом питании, на третьи сутки жизни девочка была проконсультирована детским хирургом по поводу непроходимости носовых ходов. Поставлен диагноз: ВПР. Атрезия хоан.
Кроме того, дополнительно потребовался осмотр отоларинголога, проведена попытка зондирования носовых ходов зондом №4. Диагноз: Атрезия хоан слева, сужение справа. Было рекомендовано хирургическое лечение в плановом порядке.
На 23 сутки жизни, после стабилизации состояния, девочка была переведена в отделение патологии новорожденных и недоношенных детей для дальнейшего обследования и лечения.
По данным нейросонографии выявлена гипоплазия мозолистого тела, расширение межполушарной щели и субарахноидального пространства, дилатация 3 желудочка мозга.
После чего, девочка была осмотрена неврологом и выставлен диагноз: ВПР головного мозга: Микроцефалия? Гипоплазия мозолистого тела. Последствия гипоксического поражения нервной системы, синдром двигательных нарушений. Задержка психомоторного развития, ранний восстановительный период.
По результатам ДЭХО-КГ выявлена коарктация аорты, функционирующее овальное окно до 0,47 см, открытый артериальный проток - 0,4 см. Диагноз подтвержден кардиохирургом, было рекомендовано оперативное лечение в более позднем периоде.
После осмотра окулистом, у девочки выявлена микрофтальмия, а также атрофия дисков зрительных нервов обоих глаз.
По данным УЗИ брюшной полости обнаружена тазовая дистопия и гипоплазия левой почки. С четвертой недели жизни появились изменения в моче - лейкоцитурия до 25 в поле зрения, в динамике уровень лейкоцитов нарос до 40, появилась протеинурия. В посеве мочи на стерильность выявлен умеренный рост E. Coli, была назначена антибактериальная терапия. Девочка осмотрена детским урологом и, учитывая наличие врожденного порока развития МВС, заподозрено наличие пузырно-мочеточникового рефлюкса в гипоплазированную почку. Было рекомендовано: УЗИ до и после микции, продолжить проведение антибактериальной терапии, поставить постоянный катетер, контроль ОАМ 1 раз в 2 недели, а также наблюдение и обследование ребенка урологом в плановом порядке, после коррекции пороков развития.
Перед выпиской из отделения, ребенок повторно осмотрен генетиком и, с учетом выявленных пороков развития (атрезия хоан слева и сужение справа, сформировавшаяся микроцефалия, гипоплазия мозолистого тела, тазовая дистопия и гипоплазия левой почки, ВПС - коарктация аорты, ОАП, а также множественные стигмы дисэмбриогенеза), синдром Франческетти был подтвержден.
За время нахожнения в стационаре, девочке проводилось симптоматическое лечение, направленное на поддержание витальных функций и усиление адаптационных возможностей организма, антибактериальная, ноотропная, инфузионная терапия и парентеральное питание.
После проведенного лечения и объяснения маме особенностей ухода за ребенком, необходимости хирургической лечения по восстановлению проходимости носовых ходов, коррекции ВПС, необходимости динамического наблюдения узких специалистов, ребенок переведен ОДКБ.
В заключение следует сказать о том, что настоящее наблюдение представляет большой интерес с клинической точки зрения, поскольку крайне редко встречается в повседневной практике. Ранняя диагностика сложных генетических синдромов, к коим относится и описываемое нами клиническое наблюдение, представляет большие сложности. По нашему мнению, в подобных ситуациях оправдана постановка синдромологического диагноза с уточнением аномалий развития на основании анализа совокупности клинических данных, дополнительных методов обследования, с последующей хирургической коррекцией нарушенных функций.
Литература
1. Козлова С.И., Демикова Н.С., Блинникова О.Е. «Наследственные синдромы и медико-генетическое консультирование» - М.: Практика, 1996 год. (Kozlova S.I., Demikova N.S., Blinnikova O.E. «Hereditary syndromes and medico-genetic consultation» - Moscow: Practice, 1996).
2. Лазюк Г.И., Кручинский Г. В Кириллова, И. А. «Тератология человека» - М: Практика, 1991 год (Lazyuk G. I., Kruchinsky G. V, Kirillov I. A. "Teratologiya of the person" - Moscow: Practice, 1991).
3. Лорина Л.В. «Вопросы медицинской генетики» - Рязань: РязГМУ, 2011 год (Loreena L.V. "Questions of medical genetics" - Ryazan: RyazSMU, 2011).
4. Мутовин Г.Р. «Основы клинической генетики». - М.: Высшая школа, 2001 год (Mutovin G.R. "Fundamentals of clinical genetics". - Moscow: The higher school, 2001).
5. Шварц Ю.Г. «Избранные вопросы клинической генетики». - Саратов: СГМУ, 2011 год (Schwartz U.G. "The chosen questions of clinical genetics" - Saratov: SSMU, 2011).
Множественные дизостозы
Множественные дизостозы (хондродистрофия, липохондродистрофия, гаргоилизм) - группа врожденных заболеваний и синдромов аутосомнонаслелственного генеза, для которых общими признаками являются аномальная продукция, чрезмерное накопление и выделение одного или нескольких определенных мукополисахаридов, вследствие чего развиваются патологические изменения в хряще, фасциях, периосте, сухожилиях, сердечных клапанах, кровеносных сосудах, черепномозговых оболочках, роговице, печени, селезенке. Эти изменения заключаются в утолщении, гомогенизации, исчезновении ультраструктурных элементов, характерных для нормальных коллагеновых фибрилл. Остеоциты и хондроциты увеличены и вакуолизированы. Сердечные клапаны сморщены в результате рубцового процесса.
Описаны характерные для некоторых форм дизостозов клетки «гаргоилизма». Эти крупные клетки находят в периферических нервных узлах, в ЦНС, ядрах лейкоцитов. Глыбки вещества, окрашивающегося метахроматически, находят также в интиме аорты, легких, коронарных артериях, в клетках ретикулоэндотелиальной системы печени и селезенки, лимфатических узлах и в лимфоцитах.
Симптомы Множественных дизостозов:
Описано 6 синдромов, которые иногда внешне различить трудно, так как некоторые клинические признаки встречаются одновременно при нескольких вариантах поражений. Поэтому за последнее время стали пользоваться классификацией этой группы заболеваний по биохимическим признакам.
Синдромом Пфаундлера - Гурлер (гаргоилизм термин, получивший название по головке уродца - скульптурного украшения водосточной трубы католических костелов и соборов: изо рта уродца льется сточная вода, этим объясняется и другой термин синдрома- «человек, выплевывающий воду»). Больные выделяют с мочой большое количество хондроитинсульфата В и гиперинсульфата.
У больных нарушен энхондральный рост кости, так как хрящевые клетки в ядрах окостенения расположены беспорядочно. Укорочены прежде всего проксимальные отделы конечностей - бедра и плечи, в меньшей степени - голени, предплечья, плюсневые и пястные кости.
Характерна кисть больного: пальцы укорочены и одинаковы по размеру (изодактилия), расходятся веерообразно, напоминая трезубец.
Кости позвоночника, ключицы не изменены. Внешний вид больного также очень характерен: карликовый рост за счет непропорционально коротких конечностей при нормальных размерах туловища, череп гидроцефала с крутым лбом, плоским лицом, запавшим корнем носа, толстыми губками. Все больные гаргоилизмом похожи друг на друга: короткая шея, голова как бы сидит на туловище, отмечается кифоз в месте перехода грудного отдела позвоночника в поясничный, в нижнепоясничном отделе отмечается выраженный лордоз, в связи с чем живот выпячивается кпереди, а ягодицы кзади. Психика нормальная.
При рентгенологическом исследовании трубчатые кости утолщены за счет кортикального слоя, очень плотны, иногда дугообразно изогнуты. Места прикрепления мышц и сухожилий (бугры, бугристости, вертелы) резко утолщены и деформированы. Ядра окостенения имеют бахромчатый вид, деформированы, в суставах выявляются признаки вторичного остеоартроза - уплощение, извилистость, неровность контуров сочленяющихся поверхностей.
Синдром Хантера. Тяжесть клинических проявлений при этом синдроме мягче, чем при болезни Пфаундлера - Гурлер. В частности, отсутствует помутнение роговицы. Продолжительность жизни таких больных больше, чем больных гаргоилизмом (с мочой больные выделяют повышенное количество мукополисахаридов).
Синдром Санфилиппо характеризуется повышенным содержанием в моче гиперинсульфата. У больных выражены психические расстройства - деменция, идиотизм. Тяжесть соматических поражений меньшая, чем при описанных выше синдромах.
Синдром Моркио характеризуется повышенным выделением с мочой кератансульфата. Карликовость у таких больных зависит прежде всего от уплощения тел позвонков. Характерны такие признаки, как короткая шея, кифоз, вальгусная установка коленных суставов, плоскостопие, дефигурация коленных и локтевых суставов, ограничение подвижности, мышечная слабость.
Рентгенологически часто находят очаговую кальцификацию и уплощение головки бедра, расширение вертлужной впадины. Печень и селезенка имеют нормальные размеры. Психических расстройств нет.
Синдром Шиайа характеризуется повышенным содержанием в моче дерматансульфата. Первые признаки болезни развиваются в зрелом возрасте. Клинически выявляются дисплазия лица, низкий рост, атрофия межостных мышц, гиперэкстензия пястнофаланговых и сгибательные контрактуры межфаланговых суставов кистей, помутнение роговицы, коарктация аорты или недостаточность аортальных клапанов. Психика не нарушена.
Сидром Лами - Марото характеризуется повышенным выделением с мочой хондроитинсульфата В. Описаны следующие клинические симптомы: карликовый рост, аномалии стоп, варусная установка тазобедренных суставов, расширение промежутков между I и II пальцами стоп, изодактилия, торакальный сколиоз, дисплазия и гемангиомы ушных раковин, расщепленное небо.
К каким докторам следует обращаться если у Вас Множественные дизостозы:
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Множественных дизостозов, ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Энхондрома
Энхондрома - это доброкачественное новообразование, состоящее из хрящевой ткани. Локализуется внутрикостно, обычно в зоне диафизов и метадиафизов трубчатых костей. Чаще поражает мелкие трубчатые кости кистей и стоп, может быть как одиночной, так и множественной. Как правило, протекает бессимптомно, крупные энхондромы могут вызывать деформацию сегмента конечности. Иногда энхондрома осложняется патологическим переломом. Перерождение в злокачественную опухоль наблюдается редко. Диагноз подтверждают при помощи рентгенографии, КТ и биопсии. Лечение оперативное - удаление неоплазии.
МКБ-10


Общие сведения
Энхондрома (endo- внутри + chondros хрящ + -oma опухоль) - доброкачественная опухоль, представляет собой участок зрелого гиалинового хряща гетеротопической локализации (расположенный в анатомических зонах, где в норме хрящевая ткань отсутствует). Обычно локализуется в костях, однако описаны случаи, когда энхондромы обнаруживали в легких, подкожной жировой клетчатке, яичниках, молочных железах, тканях мозга и т. д. Энхондромы составляют около 10% от общего количества новообразований костной и хрящевой ткани.
Более чем в половине случаев энхондромы диагностируются у лиц моложе 40 лет. Пик заболеваемости приходится на возраст 11-16 лет. Энхондромы могут быть одиночными или множественными. Множественные энхондромы наблюдаются при болезни Олье и синдроме Маффуччи. По своей структуре множественные образования практически аналогичны одиночным хондромам, но отличаются от них большей упорядоченностью структуры. Несмотря на медленный рост и невысокую склонность к малигнизации, энхондромы рассматриваются как потенциально злокачественные новообразования, поэтому врачи-онкологи и травматологи обычно рекомендуют их хирургическое удаление.

Причины энхондромы
Опухоли развиваются из участков хряща, расположенных в местах, где в норме хрящевая ткань отсутствует. Исследователи выяснили, что гетеротопическая локализация хряща является следствием нарушения процесса окостенения во внутриутробном периоде и в первые годы жизни ребенка. Непосредственная причина нарушения окостенения пока не выяснена, однако установлено, что эта патология не связана с воздействием вредных веществ или радиации. Предполагается, что рост энхондромы из гетеротопически расположенного участка хряща провоцируется травматическими поражениями и воспалительными процессами в костной ткани.
Патогенез
Опухоль представляет собой зрелый гиалиновый хрящ с утраченной структурой. В отличие от нормального хряща клетки энхондромы расположены хаотично, их размер и форма могут сильно различаться. Неоплазия покрыта надхрящницей и имеет дольчатое строение. Внутри обычно образуются небольшие очаги окостенения. Характерны дистрофические изменения ткани, проявляющиеся разжижением межклеточного вещества и формированием кист. При малигнизации клетки опухоли становятся более крупными, увеличивается количество клеток с двумя ядрами.
«Излюбленная» локализация энхондром - короткие трубчатые кости стоп и кистей. Возможно также поражение плечевой и бедренной кости. В области других длинных костей единичные энхондромы встречаются редко. Иногда поражаются плоские кости: кости таза, лопатка и т. д. При множественном хондроматозе (болезни Олье) опухоли могут выявляться в области одной половины тела (правой или левой) или в области одной конечности. Реже патологический процесс распространяется на обе нижние конечности.
Симптомы энхондромы
Клиническая симптоматика обычно скудная. Небольшие опухоли протекают бессимптомно и становятся случайной находкой при проведении рентгенологического обследования по другим поводам. При крупных энхондромах возникают деформации пораженного сегмента. Пальпаторно опухоль определяется как плотное безболезненное образование. Неприятные симптомы появляются при сдавлении соседних анатомических образований (сосудов, нервов). Крупные неоплазии, расположенные недалеко от суставов, могут провоцировать артралгии, ограничение движений и синовиты.
Хрящевая ткань не такая плотная и прочная, как кость. Она не приспособлена к высоким статическим и динамическим нагрузкам. Если энхондрома занимает весь поперечник кости или его значительную часть, прочность кости в этом месте резко снижается. В таких условиях даже небольшой травмы становится достаточно для возникновения патологического перелома. Переломы сопровождаются болью, патологической подвижностью, крепитацией и деформацией конечности. В отличие от обычных переломов данные об интенсивном травматическом воздействии в анамнезе отсутствуют.
Диагностика
Диагностика энхондром, как правило, несложна. На рентгенограммах длинных трубчатых костей выявляется центрально расположенное облаковидное просветление. В зоне просветления могут обнаруживаться более темные участки - очаги кальцификации. Опухоли в области коротких трубчатых костей обычно выглядят однородными и занимают большую часть поперечника или весь поперечник кости. Кортикальный слой не нарушен. На КТ кости определяется аналогичная картина, преимуществом компьютерной томографии является возможность более подробно рассмотреть структуру энхондромы.
При подозрении на озлокачествление опухоли выполняют биопсию хряща. О малигнизации свидетельствуют увеличенные хрящевые клетки округлой или неправильной формы и наличие большого количества многоядерных клеток. Для повышения достоверности диагностики забор материала всегда производят в нескольких участках, поскольку на начальных стадиях озлокачествления зоны нормальных клеток в энхондроме чередуются с зонами перерождения. Дифференциальный диагноз энхондромы проводят с гигантоклеточной костной опухолью, фиброзной дисплазией, костной кистой и хондросаркомой.
Лечение энхондромы
Тактика лечения зависит от локализации новообразования. Опухоли коротких трубчатых костей не склонны к малигнизации, поэтому при таком расположении энхондромы возможно динамическое наблюдение, включающее в себя регулярные осмотры и повторную рентгенографию пораженного сегмента. Из-за потенциальной опасности озлокачествления энхондром длинных трубчатых и плоских костей специалисты в области клинической онкологии обычно предлагают пациентам удалять опухоль сразу после постановки диагноза.
Растущие энхондромы коротких трубчатых костей иссекают в пределах здоровых тканей. При образовании крупных дефектов используют гомо- или аллотрансплантаты. При опухолях плоских и длинных трубчатых костей необходимо расширенное хирургическое вмешательство - сегментарная резекция кости с последующим замещением дефекта.
Прогноз и профилактика
При своевременном полном иссечении новообразования прогноз обычно благоприятный, наблюдается полное выздоровление. Рецидивы отмечаются достаточно редко. Вероятность озлокачествления первичной неоплазии невысока. Рецидивные энхондромы заслуживают особого внимания из-за высокой вероятности малигнизации. В подобных случаях проводятся абластичные (расширенные) резекции. Профилактические мероприятия не разработаны.
ДИЗОСТОЗ
Дизостоз (dysostosis; греч. dys- + osteon кость + osis) — нарушение развития костей, лежащее в основе врожденных наследственных семейных заболеваний костной системы. Чаще всего возникают аномалии развития костей черепа в сочетании с другими симптомами, однако встречаются множественные и генерализованные поражения костей скелета. Термин «Дизостоз» применяют к генерализованным поражениям скелета — хондродистрофии (см.), гаргоилизму (см.), остеогенезу несовершенному (см.) и т. д.
Важнейшие разновидности Дизостозов: ключично-черепной, черепно-лицевой, челюстно-лицевой и челюстно-черепной.
Содержание
Ключично-черепной Дизостоз
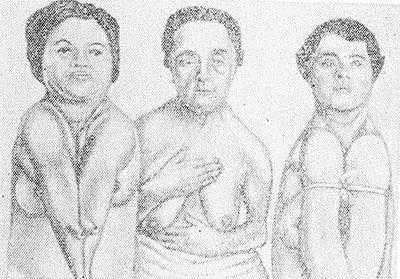
Рис. 1. Ключично-черепной Дизостоз (синдром Шейтхауэра—Мари—Сентона) у трех сестер. Отсутствие ключиц у двух сестер приводит к полному соприкосновению плеч.
Ключично-черепной Дизостоз (синдром Шейтхауэра — Мари — Сентона) характеризуется гипоплазией покровных костей черепа в сочетании с полным или частичным недоразвитием одной или обеих ключиц, т. е. нарушением развития так наз. мембранозных костей. Для Д. этого вида характерно незаращение или позднее заращение черепных швов и родничков, брахицефалия (см.) с преобладанием расширения свода черепа в латеральных направлениях, выдающийся лоб, гипоплазия лицевых костей, гл. обр. верхней челюсти, обусловливающая псевдопрогению (кажущееся увеличение нижней челюсти). Нарушение развития челюстей сопровождается запаздыванием прорезывания зубов. Отсутствие ключиц или частичное недоразвитие их с дефектом внутренних, средних или наружных частей ведет к увеличению подвижности плечевого пояса, а при полном отсутствии их — к полному соприкосновению плеч (рис. 1).
Описанные изменения часто сопровождаются деформациями позвоночника, костей верхних и нижних конечностей, стоп, тазовых костей. Аномалия наследуется по рецессивному и доминантному типу, может быть семейной.
При ключично-черепном дизостозе рентгенологически выявляются многочисленные изменения со стороны скелета, однако наиболее характерны изменения ключиц и костей черепа. Дефекты ключиц чаще симметричны и могут быть разных размеров: от небольших до полного отсутствия ключиц. Чаще же всего отсутствует акромиальный конец ключицы. Свободный конец оставшейся части закруглен, покрыт замыкающей костной пластинкой и связан плотным фиброзным тяжем с акромиальным отростком лопатки. По ходу фиброзного тяжа иногда обнаруживаются костные включения.
При рентгенологическом исследовании черепа определяется брахицефалия: мозговой череп увеличен в поперечнике и уменьшен в передне-заднем размере. Основание черепа укорочено в поперечном направлении и несколько удлинено в продольном. Кости свода, особенно лобная, истончены и как бы раздуты, значительно выдаваясь в стороны. Передний родничок остается незаращенным. В местах перекреста швов могут наблюдаться и дополнительные роднички или дополнительные костные включения в самих швах. Кости лицевого черепа малы, верхнечелюстные пазухи недоразвиты. Размеры нижней челюсти не изменены. Обнаруживаются аномалии прикуса, расположения, формы и сроков прорезывания зубов.
При исследовании скелета туловища и конечностей могут быть обнаружены отклонения в развитии ряда костей: уменьшенные размеры лопаток, крестца, костей таза с отсутствием слияния между собой лобковых, седалищных и подвздошных костей и недоразвитием лобкового симфиза; недоразвитие проксимальных отделов бедер с варусной деформацией их; укорочение или отсутствие ногтевых бугристостей у концевых фаланг пальцев кистей и стоп; незаращение дужек позвонков.
При множественном поражении скелета наличие характерных изменений ключиц делает рентгенологический диагноз достоверным.
Черепно-лицевой Дизостоз
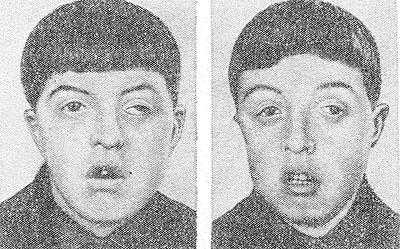
Рис. 2. Однояйцовые близнецы 13 лет с черепно-лицевым дизостозом (синдром Крузона). Характерны широко расставленные глаза, выражено косоглазие, гипоплазия верхней челюсти.
Черепно-лицевой Дизостоз (синдром Крузона, гипертелоризм) — недоразвитие костей черепа, мозга и верхней челюсти в сочетании с преждевременным закрытием черепных швов, экзофтальмом (см.), косоглазием (см.), нистагмом (см.), расстройством зрения. Лоб в области переносицы бугрист, глаза широко расставлены (рис. 2), нос своеобразной крючковидной формы («клюв попугая»), гипоплазия верхней челюсти, псевдопрогения; в резко выраженных случаях наблюдается снижение умственного развития. Наследуется по доминантному типу.
Рентгенологически выявляются изменения черепа. На первый план выступает характерная деконфигурация головы и нарушение нормальных соотношений между мозговым и лицевым черепом: первый уменьшен в размерах, имеет почти шаровидную форму, швы заращены, усилены пальцевые вдавления. Кости свода черепа истончены, несколько выпячиваются кнаружи в области переднего родничка. Основание черепа укорочено и углублено, область турецкого седла сужена, глазницы уплощены.
Кости лицевого черепа малы: верхняя челюсть и носовые кости недоразвиты, нижняя челюсть значительно выдается вперед, в силу чего образуется резкий прогиб носа внутрь.
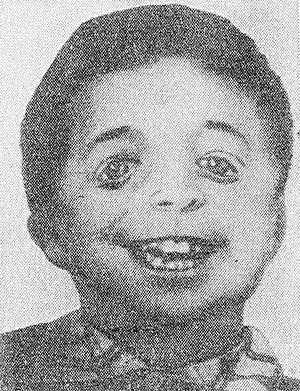
Рис. 3. Ребенок с челюстно-лицевым дизостозом. Характерны широкие косо расположенные глазные щели, нарушение развития зубов.
Челюстно-лицевой Дизостоз
Челюстно-лицевой Дизостоз (синдром Берри—Франческетти, синдром Франческетти—Цвалена) — гипоплазия гл. обр. нижней челюсти и скуловых костей, макростомия (своеобразное «рыбье» или «птичье» лицо), широкие косо расположенные глазные щели (рис. 3), с вывороченными и скошенными книзу веками и колобомами в наружных отделах, слепые фистулы от углов рта к ушам, языковидное оволосение щек, нарушения развития зубов, деформация ушных раковин, иногда среднего и внутреннего уха с развитием глухоты, устранимой операцией. В противоположность синдромам Крузона и Апера (см. Апера синдром) определяется сильное развитие лобных пазух. Встречается деформация грудной клетки и позвоночника. Наследуется по доминантному типу.
Челюстно-черепной Дизостоз
Челюстно-черепной Дизостоз (синдром Петерс — Хевельса) — гипоплазия верхней челюсти, скуловых дуг, открытый прикус, прогения (выстояние нижней челюсти), укорочение переднего отдела основания черепа. Аномалия наследуется по доминантному типу.
Существуют другие формы черепных Д.: синдромы Гегенхара, Робена, Франсуа и др. Внешний вид больных с различными формами Д. характерен. Д. сохраняется всю жизнь, не поддается оперативной коррекции, почти не требует дифференциальной диагностики с другими заболеваниями. В сомнительных случаях важным диагностическим методом является рентгенологическое исследование.
Различают так наз. неполные типы перечисленных Д., когда имеют место не все характеризующие их симптомы. Отдельные признаки могут комбинироваться в различных сочетаниях, составляя как бы промежуточные типы Д.
Прогноз для жизни благоприятный.
Библиография: Алексеев В. А. Случай черепно-ключичного дизостоза, Вестн, рентгенол, и радиол., № 3, с. 80, 1974; Косинская Н. С. Нарушения развития костно-суставного аппарата, Л., 1966; Кручинский Г. В. Редкие врожденные синдромы лица и челюстей (в границах первой и второй жаберных дуг), Минск, 1974, библиогр.; Рейнберг С. А. Рентгенодиагностика заболеваний костей и суставов, кн. 1—2, М., 1964; Ромоданов А. П. и Лягценко Д. С. Черепно-лицевой Дизостоз, Журн, невропат, и психиат., т. 72, № 10, с. 1487, 1972, библиогр.; Fleischer-Peters A. Kiefermissbildungen bei Dysostose-Syndromen des Schadels, Dtsch, zahnarztl. Z., Bd 24, S. 932, 1969; Humangenetik, hrsg. v. P. E. Becker, Bd 2, S. 489, Stuttgart, 1964, Bibliogr.; Hylton R. P. a. Albright J. E. Cleidocranial dysostosis, J. oral Surg., v. 28, p. 682, 1970; Tessier P. The definite plastic surgical treatment of the severe facial deformities of craniofacial dysostosis, Plast. reconst. Surg., v. 48, p. 419, 1971.
Энхондрома причины, симптомы, методы лечения и профилактики
Энхондрома — доброкачественная костная опухоль, формирующая хрящевую ткань. Образуется внутри костей, преимущественно в области диафизов и метадиафизов. По мере своего роста вызывает деформацию костной ткани. В редких случаях возможно злокачественное перерождение новообразования. Рассмотрим основные причины новообразования, методы диагностики и лечения.

При таком заболевании гиалиновый хрящ появляется в нетипичных для него анатомических структурах. Наиболее распространенная локализация — крупные и мелкие трубчатые кости. Например, энхондрома бедренной кости характеризуется образованием доброкачественной опухоли в зоне тела (диафиза). Реже патологический процесс затрагивает другие органы, вроде легких или молочных желез.
Возможные факторы риска:
- неблагоприятная наследственность
- перенесенные травмы
- воспаление кости
Энхондрома большеберцовой кости, ребер или другой анатомической структуры может быть диагностирована в любом возрасте, но чаще всего опухолевый рост приходится на подростковый период. У взрослых пациентов старше 45 такую болезнь выявляют сравнительно редко.
Статью проверил
Дата публикации: 05 Мая 2022 года
Дата проверки: 11 Мая 2022 года
Дата обновления: 24 Октября 2022 года
Содержание статьи
Проявления патологии зависят от того, где локализуется новообразование. Симтоматика скудная. На ранних этапах явные симптомы обычно отсутствуют. Из-за медленного прогрессирования бессимптомное течение зачастую сохраняется на протяжении многих лет. Если патологический очаг расположен рядом с костным сочленением, повышается риск нарушения его двигательной функции с развитием болевого синдрома. Например, энхондрома плечевой кости иногда нарушает подвижность верхней конечности. Опухоль бедра негативно влияет на работу коленного сустава. Поражение малоберцовой кости подчас затрудняет ротацию стопы.
- деформация скелета
- ощущение плотного нароста при пальпации
- болезненность на фоне сдавливания сосудов и нервов
- склонность к переломам
У ряда пациентов заболевание впервые диагностируют после возникновения патологического перелома, не связанного с травматическим воздействием.
Как диагностировать
Врач расспрашивает пациента о симптомах и собирает анамнез для обнаружения факторов риска. Визуальный осмотр дает возможность выявить неспецифические признаки изменения структуры костной ткани или нарушения двигательной функции сустава. Требуется дифференциальная диагностика для исключения патологий с похожими симптомами: энхондроматоза, фиброзной дисплазии, экхондромы и других.
Рентгенография. Рентгенологическое исследование выявляет патологический очаг в пределах здоровой кости
Компьютерная или магнитно-резонансная томография. Врач получает описание костей и расположенных рядом мягких тканей.
Биопсия. Производится забор опухолевой ткани для уточнения ее типа.
МРТ — более точный метод визуализации, позволяющий исключить другие патологии и подготовиться к лечению. Рентгенологи в клиниках ЦМРТ ставят диагноз энхондрома по МРТ.
Читайте также:
- Анемический невус у ребенка
- Диагностика лепры. Принципы микробиологической диагностики проказы. Лечение лепры. Методы лечения проказы.
- Рефлекторные движения в акте жевания. Влияние зубов на произношение и речь
- Случай успешного лечения смешанного геморроя
- Образование губчатой кости у эмбриона. Возникновение эндохондральной кости плода