Синдром Гетчинсона (Hutchinson) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 02.02.2026
Синдром Гетчинсона-Гилфорда или прогерия (сенильный нанизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения. Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом. Этиология прогерии неясна. Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у больных. Однако исследования зарубежных ученых позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения. Характерен вид лица: с экзофтальмом, тонким клювовидным носом, большим мозговым и малым лицевым черепом, голос тонкий, имеются скелетные аномалии. Пубертат обычно не наступает, наружные гениталии гипоплазированы. Интеллект средний или выше среднего. Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Ключевые слова
Для цитирования:
For citation:
Синдром Гетчинсона-Гилфорда (Hutchinson-Gilford Progeria Syndrome), или прогерия (сенильный папизм) — исключительно редкое генетическое заболевание детей с клиническими чертами преждевременного старения.
Частота заболевания составляет 1 на 8 млн новорожденных (De Busk. 1972). К настоящему времени в мировой литературе описано около 70 пациентов с этим синдромом.
Этнология прогерии неясна.
Генетическая модель наследования неизвестна в связи с крайней редкостью встречаемости синдрома и отсутствием потомства у бальных. Однако исследования зарубежных ученых [2-4, 7] позволяют говорить о спорадической доминантной мутации как генетической основе данного синдрома. Отмечается влияние возраста отца как возможной причины новых мутаций. Так, средний возраст отцов составляет 35-37 лет.
Аутосомно-рецессивный тип наследования синдрома также обсуждается в литературе [7, 8, И]. Этот тип наследования был впервые предположен Gabr и соавт. в 1960 г. при описании двух моиозиготных сестер, а впервые сообщил о семейном случае прогерии Paterson в 1922 г., хотя описание двух больных братьев было неполным и без фотографий.
Клиническая картина прогерии представлена симптомами прогрессирующего преждевременного старения.
Дети рождаются нормальными, но к 1-му году жизни наблюдается выраженная задержка роста и массы тела. Конечный рост в среднем достигает 100 см. В первые годы жизни развивается тотальная алопеция. Кожа топкая, лоснящаяся, сухая, тугонатянутая (па кистях и стопах, наоборот, морщинистая). В нижней части живота и бедрах кожные изменения напоминают склеродермию. С возрастом появляются коричневые пигментные пятна. Потовые и сальные железы атрофируются.
Подкожный жировой слой полностью отсутствует, за исключением лобковой области. На черепе выражена подкожная венозная сеть. Нощи дистрофичные, влоть до аплазии. Зубы прорезываются с задержкой, аномально расположены, с ранним разрушением как молочных, так и постоянных зубов.
Характерен вид лица: с экзофтальмом, гонким клювовидным носом, большим мозговым и малым лицевым черепом. Голос тонкий.
Скелетные аномалии включают резорбцию ключицы с замещением фиброзной тканью, резорбцию конечных фаланг кистей (акроостеолиз), истончение длинных трубчатых костей и ребер, тугоподвижность сутавов пальцев, увеличение локтевых и коленных суставов. Часты асептические некрозы головки бедренной кости и вывих тазобедренного сустава.
Пубертат обычно не наступает, наружные гениталии ги- поплазированы.
Интеллект средний или выше среднего.
Для данного синдрома характерны распространенный атеросклероз с поражением коронарных и мезентериальных сосудов, аорты, сосудов головного мозга, с гиперлипидемией. Следует отметить, что заболевания, характерные для нормального процесса старения (катаракта, опухоли, сахарный диабет), встречаются при прогерии крайне редко.
Прогноз для жизни неблагоприятный: продолжительность жизни колеблется от 7 до 28 лет, в среднем составляя 12-13,5 года [1, 4, 5, 7, 10]. Основные причины летальных исходов - острый инфаркт миокарда, застойная сердечная недостаточность, инсульты. На аутопсии выявляются распространенный атеросклероз, гипоплазия гонад, иногда гипоплазия надпочечников и значительная гиперплазия тимуса, истончение коркового слоя костей.
Прогерия как модель преждевременного старения изучается в разных аспектах: метаболическом, гормональном, гистологическом, иммунологическом, молекулярном.
Гормональные исследования у детей [4] выявляют нормальную ночную секрецию соматотропного гормона гипофиза, но крайне низкий уровень инсулиноподобного фактора роста (ИФР-1) в плазме крови. Это позволяет предполагать наличие у данных больных бионеактивпого пула СТГ в крови, либо периферическую резистентность к эндогенному СТГ. либо выраженный дефицит питания. Трехмесячный период высококалорийного питания не увеличивает уровень ИФР-1 в крови, но ускоряет линейный рост.
Прогерия считается состоянием, связанным с ннсулиноре- зистентностыо умеренной степени. В 1983 г.[9] впервые была описана девочка, у которой в 2-легнсм возрасте уровень инсулина в крови натощак составлял 20-40 мкг/дл. В 4 года через 3 мес после удаления кисты яичника развилась гипергликемия натощак (до 250 мкг/дл) с высоким уровнем инсулина (более 2200 мкЕД/мл) и С-пептида (32,4 нг/мл) в крови. Уровень гемоглобина А1 достигал 10%. Связывание инсулина с рецепторами эритроциов было в пределах нормальных значений.
Иммунологический аспект в патогенезе прогерии впервые был выдвинут Walford в 1970 г. В 1973-1976 г. Singal и Goldstein выявили отсутствие либо резкое снижение экспрессии HLA культурой фибробластов кожи у детей с прогерией. Вместе с тем другие исследователи, анализируя экспрессию HLA у детей с прогерией и их здоровых родственников [2], нс выявили ни количественного, ни качественного дефицита в экспрессии HLA в фибробластах кожи. Различие в частоте встречаемости ряда HLA-антнгенов у больных прогерией и здоровых людей не дает право в настоящее время говорить о специфической ассоциации HLA и прогерии в связи с малым числом исследуемого материала.
Биохимическими исследованиями показано, что одним из биомаркеров старения является мочевая экскреция гиалуроновой кислоты. В норме у детей и подростков содержание ее составляет менее 1% от уровня общих гликозаминогликанов и увеличивается с возрастом до 5-6%. У детей с прогерией выявлено значительное повышение (до 10-20%) экскреции гиалуроновой кислоты с мочой по сравнению со здоровыми людьми [4]. Данное повышение не наблюдается ни при одном генетическом заболевании, кроме синдрома Вернера, или “прогерии взрослых” [4]. Считается, что гиалуроновая кислота является ключевым фактором антиангиогенеза в процессе созревания и старения.
Изучение культуры фибробластов кожи от пациентов с прогерией выявляет значительное снижение клеточного роста вследствие подавления митотической активности. С другой стороны, отмечается нормальное распределение типов коллагена в коже, характерное для детей, а именно, преобладание коллагена 3-го типа над коллагеном 1-го типа [8].
Имеются данные, что в основе прогерии лежит дефицит метаболизма витамина Е [7].
Ряд исследователей связывают прогерию с генетически обусловленной ошибкой в синтезе внутриклеточных белков. Так, показано, что эритроциты больных детей содержат повышенную термолабильную фракцию ферментов: глюкозо-6- фосфатдегидрогепазу и б-фосфоглюконатдегидрогепазу |6]. Другие работы не подтверждают эту концепцию [3].
Данные олене или детей с прогерией крайне малочисленны.
Патогенетически оправданными считаются терапия витамином Е для восполнения его дефицита (Лугез и МШап, 15974) и усиленное белковое питание.
Приводим описание собственного клинического случая.
Боль пая Н., 3 лет 9 мес., поступила в детское отделение ЭНЦ РАМН с жалобами на отставание в росте и массе, сниженный аппетит, облысение, резкую головную боль.
Раннее развитие: держит голову с 1 мес жизни, сидит с 6 .мес жизни, ходит с 1 года 2 мес, зубы появились в 1 год 1 мес, говорит с 1,5 лет. Грудное вскармливание - до 1 года 8 мес.
Перенесенные заболевания: дисплазия тазобедренный суставов (в 1 мес жизни), двусторонний врожденный вывих бедер (диагностирован в 6 мес жизни), стоматит, легкая форма (в 3 года).
Аллергологический анамнез не отягощен.
Наследственность по низкорослости не отягощена. Мать 24 лет, рост 165 см, родственники. со стороны матери: бабушка - рост 157 см, дедушка - 180 см, тетя - 168 см. Отец 26 лет, рост 176 см; родственники со стороны отца: бабушка - рост 157 см, дедушка - 170 см. Отягощена наследственность по сахарному диабету II типа, который имеется у бабушки со стороны отца и у прабабушки со стороны матери. Отягощена наследственность по бронхиальной астме, тяжелая форма которой имеется у прадедушки, со стороны матери и у двоюродной прабабушки со стороны матери.
Анамнез заболевания: с 2-месячного возраста замечены уплотнение кожных покровов и подкожной жировой клетчатки, лоснящаяся кожа на бедрах, животе, ягодицах, цианоз носогубного треугольника. В 3-месячном возрасте диагностирована легкая форма склеродермии. При исследовании биоптата кожи выявлен гиперкератоз эпителия и умеренный склероз дермы. Консультирована дерматологом: диагноз склеродермии был снят, больше данных, свидетельствующих о склеродерме. Получала лечение преднизолоновой мазыо в течение 3 мес с умеренным эффектом - блеск и плотность кожных покровов уменьшились. В возрасте 1 года появилась венозная сеть на голове. Отмечалась гипотрофия П-Ш степени. Невропатолог диагностировал перинатальную энцефалопатию, компенсированную гидроцефалию. В 1 год 7 мес девочка была впервые консультирована эндокринологом: диагностирована задержка физического развития смешанного генеза. Масса тела 7800 г, костный возраст соответствовал паспортному. В 1 год 10 мес начали выпадать полосы на голове. В 1 год 11 мес впервые консультирована генетиком, поставлен диагноз: “Синдром Гетчинсона—Гилфорда”. Девочка была обследована в стационаре по месту жительства: рост 73 см, масса 7900 г. На ЭКГ: метаболические изменения миокарда
Данные обследования в ЭНЦ РАМН. Хронологический возраст 3,9 года. Рост 81,2 см. Коэффициент стандартного отклонения (SDS роста) - 4,35. Масса тела 9,5 кг. Рост сидя 48,5 см, коэффициент “верхний ссгмент/пижпий сегмент" 1,48. Окружность головы 49 см.
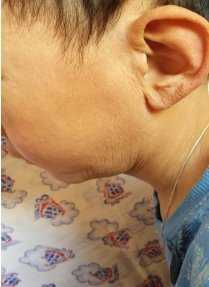
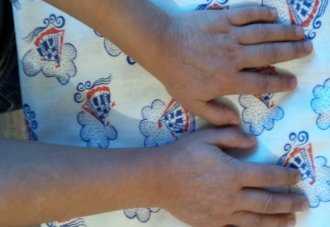
Отмечаются ярко выраженные черты синдрома Гетчинсона - Гилфорда: 1) крупная голова с диспропорционально большим мозговым черепом и малым лицевым. Вдавлен) гость височных костей; 2) выраженная венозная сеть на голове; 3) тотальная алопеция; 4) узкий, деформированный нос с истончением кожи на нем, цианоз носогубного треугольника; 5) истончение кожи на туловище, конечностях, морщинистость кожи на ладонях и ступнях. Склеродермоподобные изменения кожи на животе, спине и ягодицах - очаги депигментации диаметром 0,5-0,7 см, множественные, уплотненные. Депигментация сосков; 6) подкожная жировая клетчатка развита слабо, распределена равномерно; 7) гипоплазия ногтей кистей и стоп; утолщение межфаланговых суставов и концевых фаланг кистей и стоп; варусная девиация верхней трети предплечий, короткая шея (см. рисунок).
Область сердца визуально не изменена. Тоны сердца ясные, ритмичные. Часота сердечных сокращений 104 удара в минуту, АД 80/50 мм рт.ст. Дыхание везикулярное, хрипов нет. Зев чистый. Живот мягкий, безболезненный. Печень не увеличена, селезенка не пальпируется. Стул регулярный. Дизурических расстройств нет. Симптом Пастернацкого отрицательный с обеих сторон. Эндокринный статус: щитовидная железа не увеличена, симптомов нарушения функции нет. Симптомы гипокортицизма отсутствуют. Половой статус препубертатный: Ах 0, Р 0, Ма 0, Me 0.
Гормональное исследование крови: кортизол 321,4 нмоль/л, общий трийодтиронин 2,48 нмоль/л, общий тироксин 100,0 нмоль/л, ТТГ 2,10 мкЕД/л, пролактин 248,0 мкЕД/л, ЛГ 1,60 ЕД/л, ФСГ 5,50 ЕД/о, 17-оксипрогестерон 1,5 нг/мл. Соматотропный гормон на фоне стимуляционной пробы с клофелином: 0 мин - 1,7 нг/мл, 30 мин - 1,5 нг/мл, 60 мин - 2,3 нг/мл, 120 мин - 71,0 нг/мл, 150 мин - 29,3 нг/мл.
Гормональное исследование мочи: свободный кортизол 1115,0 нмоль/л на 1 г креатинина (креатинин 0,24 г/л). Дс- гидроэпиандростерон-сульфат — следы.
Показатели гуморального аутоиммунитета: у больного ребенка не выявлено антител пи к тиреоглобулину человека, ни к микросомальному антигену тиреоцигов, ни к поверхностным антигенам клеток аденогипофиза и клеток коры надпочечников крысы.
Вместе с тем у матери ребенка обнаружены антитела к поверхностным антигенам клеток аденогипофиза крысы, а у бабушки по материнской линии - слабо положительная реакция па наличие антител к микросомальному антигену тиреоцигов при отсутствии антител к другим изучаемым антигенам.
Рентгенографическое исследование: на рентгенограмме черепа и кистей структура костей не изменена. Форма и размеры турецкого седла обычные. Сосудистый рисунок костей свода усилен. Дифференцирование скелета соответствует 12- 15 мес. Ногтевые фаланги деформированы, треугольной формы. На рентгенограмме стоп отмечается небольшое уплотнение стенок arteria dorsalis pedis.
Компьютерная томография головного мозга: на серии компьютерных томограмм изменения плотности мозговой ткани не выявлено. Желудочковая система не изменена. Хиазмальная цистерна расширена. Несколько расширена межполушарная щель. Данных, свидетельствующих об объемном процессе головного мозга, не выявлено.
ЭКГ: ЧСС 120 в минуту, ритм синусовый. Электроэнцефалограмма: на фоне умеренных диффузных изменений биоритмики с признаками диэнцефальной заинтересованности отмечаются признаки ирригации стволово-диэнцефальных структур с легким акцентом справа, в теменно-затылочной области. Эхоэнцефалограмма: эхо-пульсация неустойчиво усилена до 55%, смещения срединных структур нет, легкое расширение 111 желудочка (до 6 мм), вентрикулярный индекс умеренно выше нормы. Заключение: смещения срединных структур не выявлено. Расширение боковых желудочков.
Ультразвуковое исследование: щитовидная железа: типично расположена, контуры ровные, структура гомогенная. Правая доля 2,3x1,1хо,9 см, слева - 2,4x1,2x0,9 см, толщина перешейка 0,4 см. Объем щитовидной железы 2,55 мл. Надпочечники: нс увеличены. Органы малого таза: размеры матки и яичников соответствуют возрастной норме. Матка: 3,1x1,3x1,1 см, правый яичник: 1,6x1,2x0,8 см, левый яичник: 1,4x1,2x1,0 см. Печень: увеличена правая доля - 8,5 см, левая доля 2,6 см. Структура паренхимы гомогенная, внутри- печеночные протоки не расширены. Воротная вена не расширена. Желчный пузырь конкрементов не содержит, Почки: топография не изменена, размеры в пределах возрастной нормы. Структуры хорошо дифференцированы, без гидро- нефротических изменений и достоверных эхо-признаков дополнительных объемных включений. Паренхима гомогенна, толщина се соответствует возрастной норме.
Консультация окулиста: моторно-зрачковых нарушений не выявлено. Передний отрезок и среды без патологии. Глазное дно: диски бледно-розовые, сосуды умеренно расширены, полнокровны, извиты по всей протяженности сетчатки. Сетчатка на периферии разряжена, небольшие отеки. Заключение: повышение внутричерепного давления.
Консультация невропатолога: субкомпепсированпая гидроцефалия на фоне скудной церебральной симптоматики.
Консультация дерматолога: Alopecia totalis. Рекомендованы курсы лечения препаратами, улучшающими микроциркуляцию (трептал, троксевазип), поливитамины (Bj, В2, В(„ В15, А, Е); наружно — втирание с массажем головы геля актовегина, геля троксевазина, димексида, 01. Ricini.
Учитывая имеющиеся в литературе данные об эффективности применения гормона роста у детей с прогерией [4], для увеличения линейного роста девочке был назначен пробный 3-месячный курс лечения рекомбинантным гормоном роста человека “SAIZEN” (ARES-SERONO). Недельная доза составила 15 ЕД/м 2 , суточная доза — 1 ЕД, подкожно, ежедневно, 7 раз в неделю.
Лечение рекомендовано проводить под контролем гликемии и суточной глюкозурии.
Помимо гормона роста, девочке назначено лечение, рекомендованное дерматологом, и полноценное белковое питание.
Таким образом, представленный материал, основанный на данных анамнеза, жалоб, результатах клинического обследования и лабораторно-инструментальных исследований, подтверждает наличие у ребенка классического синдрома Гетчинсона-Гилфорда (прогерии).
Синдром прогерии Гетчинсона — Гилфорда (HGPS)
В статье рассматривается клинический случай синдрома прогерии Гетчинсона — Гилфорда. После краткого литературного обзора описывается клинический случай врожденной прогерии (синдром прогерии Гетчинсона — Гилфорда) у мальчика 5 лет. Приводимый случай представляет большой интерес и имеет практическое значение для дерматовенерологов, педиатров, генетиков, врачей общей практики, так как это заболевания встречается очень редко, в республиках Центральной Азии, в том числе в Узбекистане описывается впервые.
Ключевые слова: синдром прогерии, старение, этиология, патогенез, клиника, диагностика, дифференциальная диагностика, лечение.
The article deals with the clinical case of the Hetchinson-Gilford progeria syndrome. After a brief literature review, the clinical case of congenital progeria (Hetchinson-Gilford progeria syndrome) is described in a boy of 5 years. The presented case is of great interest and is of practical importance for dermatovenerologists, pediatricians, geneticists, general practitioners, since this is very rare, in the Central Asian republics, including Uzbekistan, for the first time.
Key words: progeria syndrome, aging, etiology, pathogenesis, clinic, diagnostics, differential diagnosis, treatment.
Старение —это физиологический, нормальный непрерывный процесс, который является неотъемлемой, составной частью жизни. Цицерон в своем произведение «De senectute» («О старости») упомянул эту проблему: «…всегда существовала необходимость в каком‑то завершении, и когда время наступает, нам приходится, подобно плодам деревьев или плодам земли, в известной мере увядать и опадать» [1].
На протяжении всей жизни в организме постоянно происходят изменения, без которых его существование немыслимо. Считается, что от рождения и до определенного возраста человек растет и развивается, но только в 25 лет обызвествляются последние хрящи в эпифизах трубчатых костей, и рост затормаживается, поэтому этот возрастной период считается переломным моментом, с которого начинается движение к старости [8].
При некоторых заболеваниях наступает ускорение темпа старения, и пациент выглядит значительно старше своих сверстников. Особое место по раннему проявлению занимают синдромы преждевременной старости наследственной природы, представляющие явную патологию. Это так называемые прогерии.
Прогерия (греч. progērōs преждевременно состарившийся) — комплексная мезоэктодермальная дисплазия [3]. Различают две формы прогерии: детская — Hutchinson — Gilford Progeria Syndrome (HGPS); прогерия взрослых — синдром Вернера [10].
В 1886 г. английский врач Dr. Jonathan Hutchinson описал преждевременное старение у 6-летнего ребенка, оно проявлялось атрофией кожи и её придатков [15]. Dr. Gilford ввел термин «прогерия», который изучил клинико-морфологические особенности этой патологии [12, 13].
Вызван HGPS спорадической аутосомно-доминантной мутацией в гене LMNA, который осуществляет синтез белка Lamin A, являющегося основой клеточного ядра и носящего название прогерин. Он способствует нестабильности ядер, приводя к ускорению процесса старения и развитию прогерии [11].
На первом или втором году жизни появляются первые признаки заболевания. Характерный внешний вид: низкий рост, относительно большую голову и уменьшенную лицевую часть черепа, тонкий клювовидный нос, оттопыренные уши, микрогнатию, экзофтальм. Кроме того, наблюдается уменьшение подкожной жировой клетчатки, гиперпигментация, истончение кожи, которая становится сухой, морщинистой, местами склеродермоподобной. Ногтевые пластинки дистрофичные, могут полностью отсутствовать. Отмечается преждевременное поседение волос, тотальная алопеция. У них брови и ресницы редкие, тонкие. Больные отстают в половом развитии. Интеллект не страдает. В крови повышен уровень холестерина и липопротеидов. Имеет место изменение коллагеновых волокон: дезорганизация, утолщение, снижение растворимости. Продолжительность жизни от 7 до 27 лет. Большинство больных погибают от атеросклеротических осложнений и злокачественных новообразований. На аутопсии выявляются генерализованный атеросклероз, фиброз миокарда; отложение жироподобного вещества в мозге, коре надпочечников, почках, печени, половых железах; истончение коркового слоя в костях [6,10].
На сегодняшний день патогенетического лечения и профилактики прогерии не существует. Но фондом Progeria Research Foundation проведен ряд клинических испытаний. Согласно этим исследованиям, препарат Lonafarnib типа фарнезилтрансферазы ингибитор (ФТИ), изначально разработанный для лечения рака, оказался эффективным в отношении прогерии. У каждого больного прогерией наблюдалось улучшение по одному из четырех параметров: повышение массы тела, улучшение слуха, улучшение костной структуры и, самое главное, повышение гибкости кровеносных сосудов [14]. Симптоматическая терапия включает антиоксидантные препараты, витамин Е и другие лекарственные средства. Больным рекомендуется избегать воздействия травмирующих факторов на кожу [9].
Дифференциальная диагностика детской прогерии проводится с синдромом Коккейна, для которого характерно: непропорциональная карликовость, диффузный пигментный ретинит с обесцвечиванием диска зрительного нерва, глухота, атаксический тремор, повышенная чувствительность к ультрафиолетовым лучам и снижение интеллекта.
Синдром Гетчинсона — Гилфорда следует также отличать от синдрома Ульриха —Фремерей — Доны — врожденного симптомокомплекса, проявляющегося треугольной формой черепа с расходящимися швами, катарактой, гипотрихозом с очаговой алопецией вдоль костных швов.
По мнению Беренбейн Б. А. дифференциальный диагноз с синдромом Ханхарта (гипофизарно-церебральный нанизм) базируется на наличии ранних геродермических изменений на лице, которое становится морщинистым, пропорциональной карликовости в сочетании с гипогенитализмом, иногда гипотиреозом. В крови снижена щелочная фосфатаза, отмечается анемия и лимфоцитопения, в моче — снижение уровня тирео- и гонадотропных гормонов. При cиндроме Ротмунда-Томпсона отсутствуют карликовость, евнухоидизм и патология сердечно- сосудистой системы [3].
В 1904 г. Вернером был описан прогерия взрослых (син.: синдром Вернера) В 20 % случаев отмечались кровнородственные браки. Она наследуется по аутосомно-рецессивному типу, при этом имеет место сцепление с группой маркеров 8‑й хромосомы [9]. Есть сведения о нарушении метаболизма соединительной ткани при этом синдроме, что подтверждается изменениями пролиферативной активности фибробластов и наличием дефекта синтеза гликозаминогликанов. Повышенная гиалинизация базальных мембран вызывает сдавление семявыводящих канальцев у мужчин, патологию овариальной системы у женщин, что приводит к бесплодию. По этой же причине нарушается функция печени, почек, желез внутренней секреции. Гистологическая картина: атрофия эпидермиса, дермы, скопление меланоцитов в базальном слое эпидермиса, гиалинизация и атеросклероз сосудов. Манифестирует заболевание в возрасте 14-18 лет. Носит прогредиентное течение [5]. Ранними признаками данной патологии являются преждевременное старение волос и прогрессирующая алопеция. Больные низкого роста с маскообразным лицом, клювовидным носом и выступающим подбородком. Кожа истончена, блестящая, натянутая, особенно в области голеней, где отчетливо видна сеть кровеносных сосудов. Подкожно-жировая клетчатка и мышцы атрофированы, в силу чего конечности непропорционально тонкие. Отмечается нарушение пигментации (де- и гиперпигментация). В ряде случаев диагностируют ювенильную катаракту, нарушения эндокринной системы (гипогонадизм, сахарный диабет), атеросклероз, остеопороз, инфаркт миокарда. А также, встречается ладонно-подошвенная кератодермия, дистрофические изменения ногтевых пластинок. Иногда обнаруживаются дефекты интеллекта [9]. Есть высокий риск развития меланомы, остеосаркомы, рака щитовидной железы и других видов опухолей. Имеются данные сочетания синдрома Вернера с псориатической эритродермией и несращением твердого неба [4,5,2]. Больные умирают от злокачественных новообразований или сердечно-сосудистой патологии после 40 лет. Лечение симптоматическое [9].
Наши наблюдения: Больной Т., 2010 г.р. с Каракалпакстанской автономной республики обратился с матерью в ТашОблКВД 15.01.16 года с жалобами на морщинистую кожу, поредение ресниц, низкий рост.
Anamnesis morbi: со слов матери мальчик до двух лет рос и развивался нормально, почти не болел, прививки получал по календарю. Со стороны кожи и видимых слизистых заметных изменений не замечала. С трех лет начал отставать от своих сверстников. Когда мальчику исполнилось 4,5 лет, сделали обрезание, спустя несколько недель по всему телу стали появляться угревидные высыпания, через 10-15 дней высыпные элементы исчезли и потом вновь появились. Обратились в поликлинику по место жительству, там назначили внутрь ампицилин, пиковит, наружно левомицетиновую мазь. Высыпания исчезли. В декабре 2015 года (когда мальчику исполнялось 5 лет) опять стали появляться на гладкой коже лица и рук “угревидные” высыпные элементы. В январе 2016 г. вся кожа покрылась аналогичными высыпаниями, больной был госпитализирован в Республиканский КВД в г. Нукус, от проводимого лечения эффекта не было и выписан домой. Дома поднялась температура тела, больного осмотрела педиатр и отправила в инфекционную больницу, там назначили амбулаторное лечение. После лечения высыпные элементы исчезли, температура нормализовалась (каким диагнозом и какими препаратами лечились не помнит). На местах, где были высыпания, остались следы. Весной кожно-патологический процесс обострился, в связи чем родители мальчика отвезли табибу. Табиб дал смесь трав для заваривания и приема внутрь. В процессе лечения высыпные элементы исчезли, но кожа стала сухая, морщинистая, дряблая. В связи с чем родители больного 15.01.16 года обратились на кафедру дерматовенерологии ТашПМИ.
Anamnesis vitae: Ребенок от 4 беременности по счету, роды 3, второй ребенок в семье. Беременность протекала с легким токсикозом, на 7 месяце беременности по показаниям положили на сохранение. Роды в срок путем Кесарево сечения, без осложнений. Ребенок до года находился на естественном вскармливании. Со слов родителей мальчик до 2-х лет психически и физически развивался соответственно по возрасту. Перенесенные заболевания: паховая грыжа, ОРВИ. Брак не является близкородственным. Мать отрицает в роду близкородственные браки.
Status praesens: Общее состояние больного удовлетворительное, сознание ясное, положение активное. Астенического телосложения. Тургор кожи снижен. Периферические лимфатические узлы не увеличены, безболезненные. В легких прослушивается везикулярное дыхание, сердечные тоны ясные, ритмичные. Язык обложен беловатым налетом, отмечается дистрофия и искривление зубов особенно жевательных. Живот мягкий, безболезненный при пальпации. Селезенка и печень не увеличены. На вопросы отвечает неохотно, интеллект снижен. Сон сохранен, аппетит снижен.
Status loсalis: Кожа по всему телу истонченная, морщинистая, дряблая, отмечается снижение тургора и эластичности (Рис.1,2,3) Отмечается уменьшение подкожно-жировой клетчатки. Эти изменения большее всего выражены на гладкой коже лица, особенно вокруг рта, носа, щек. Ушные раковины увеличены по размеру, кожа тоже дряблая особенно в области макушки (2). В области голеней кожа истончена, блестящая, натянута, местами отчетливо видна сеть кровеносных сосудов.
Брови, ресницы и волосы в боковых поверхностях волосистой части головы, редкие, тонкие. Ногтевые пластинки дистрофичные на поверхности некоторых ногтевых пластинок имеются поперечные линии, местами мелкие белые пятни. Субъективные ощущения: отсутствуют.
Лабораторные исследования: Общий анализ крови: гемоглобин-105 г/л, эритроциты — 3.6, цветной показатель — 0.88, лейкоциты — 5.2, моноциты — 9.0, СОЭ — 14. Показатели общего анализа мочи и кала в пределах нормы. Биохимический анализ крови: глюкоза 4,1 моль/л, ревматоидный фактор отрицательный. Больной проконсультирован у эндокринолога (проверено гормоны щитовидной железы и надпочечника) патология не выявлено. Больной обследован в Республиканском центре Скрининга: Заключение: “Наследственно-генетическое заболевание прогерия”.
УЗИ почек: эхопризнаки МКД почек. Незначительная дилятация ЧЛС справа. Каликоэктазия справа. УЗИ брюшной полости: эхопризнаки деформированной Ж. П., незначительная спленомегалия, печен без эхопатологии. Рентгенография кистей рук (в прямой проекции): Зоны роста открыты. Крупнопетлистый тип строения костей. Укорочение средней фаланги 5 пальца.
На основании анамнеза заболевания, течения, клиники, данных проведенных анализов и заключении смежных специалистов установлен диагноз: Врожденная прогерия (Синдром прогерии Гетчинсона Гилфорда).
Рассмотренный клинический случай представляет большой интерес и имеет практическое значение для дерматовенерологов, педиатров, генетиков и врачей общей практики, так как это заболевания встречается редко, в республиках Центральной Азии, в том числе в Узбекистане описывается впервые.
- Айрапетов С. Г. Здоровье, эмоции, красота. М.: Молодая гвардия, 1977; 11 c. 1.
- Балявичене Г. Р. Синдром Вернера в сочетании с частичным несращением твердого неба. Вестник дерматологии и венерологии 1980; (6): 55-58. 19.
- Беренбейн Б. А., Студницин А. А. Дифференциальная диагностика кожных болезней. М.: Медицина, 1989; 538 с. 4.
- Бутов Ю. С., Пономарев Б. А., Сасиков Б. М. Сочетание псориатической эритродермии и синдрома Вернера у одного больного. Вестник дерматологии и венерологии 1977; (3): 68-70. 18.
- Каламкарян А. А., Мордовцев В. Н., Трофимова Л. Я. Клиническая дерматология: редкие и атипичные дерматозы. Ер.: Айастан, 1989; 567 с. 16.
- Козлова С. И., Демикова Н. С., Семанова Е. и др. Наследственные синдромы и медико-генетическое консультирование. М.: Практика, 1996; 230 с. 11.
- Лазебник Л. Б., Вёрткин А. Л., Конев Ю. В. и др. Старение: профессиональный врачебный подход. М.: Эксмо, 2014; 7 с. 3.
- Махотин Ю. В., Карева О. В., Лосева Т. Н. Книга о здоровье. М.: Медицина, 1988; 417 с. 2.
- Суколин И. Г. Клиника наследственных дерматозов. Атлас — справочник. М.: БИНОМ, 2013; 96 с. 14.
- Федорова Е. В. и др. О врожденной прогерии. Педиатрия 1980; 4: 66. 5.
- Burtner R. C., Kennedy B. K. Progeria syndromes and ageing: what is the connection? Nature review. Molecular cell biology 2010; (11): 567-578. 9.
- Gilford H. On a condition of mixed premature and immature development. Medico-Chirurgical Transactions 1897; (80): 17-45. 7.
- Gilford H. Progeria: a form of senilism. Practitioner 1904; (73): 188-217. 8.
- Gordon L. B., Kleinman M. E., Miller D. T., et al. Clinical Trial of a Farnesyl transferase Inhibitor in Children with Hutchinson-Gilford Progeria Syndrome. Proceedings of the National Academy of Sciences 2012; 109 (41). 13.
- Hutchinson J. A case of congenital absence of hair with atrophic condition of the skin and its appendages. Lancet 1886; (1): 923. 6.
Основные термины (генерируются автоматически): HGPS, Синдром, больной, LMNA, гладкая кожа лица, общая практика, подкожно-жировая клетчатка, практическое значение, Центральная Азия, щитовидная железа.
Болезнь прогерия, или Синдром Хатчинсона-Гилфорда

Прогерия, также известная как синдром Хатчинсона-Гилфорда, является чрезвычайно редким, прогрессирующим генетическим заболеванием, которое заставляет детей быстро стареть, начиная с первых двух лет их жизни.
Дети с этим расстройством при рождении ничем не отличаются внешне от других младенцев, однако в течение первого года начинаются проявляться первые признаки - замедленный рост и выпадение волос.
Средняя продолжительность жизни ребенка с прогерией составляет около 13 лет, однако в некоторых случаях она достигает 20 лет.
К сожалению, на данном этапе лекарства от синдрома Хатчинсона-Гилфорда нет, однако исследования продолжаются.
Симптомы синдрома Хатчинсона-Гилфорда
Обычно в течение первого года жизни рост ребенка с прогерией заметно замедляется, но двигательное развитие и интеллект остаются в норме.
Признаки этого прогрессирующего расстройства включают в себя специфический внешний вид:
- замедленный рост и вес ниже среднего;
- узкое лицо, маленькая нижняя челюсть, тонкие губы и клювастый нос;
- голова непропорционально большая для лица;
- выпуклые глаза и неполное смыкание век;
- выпадение волос, включая ресницы и брови;
- истонченная, пятнистая, морщинистая кожа;
- видимые вены;
- высокий голос.
Признаки и симптомы также включают проблемы со здоровьем:
- Тяжелое прогрессирующее заболевание сердца и кровеносных сосудов (сердечно-сосудистые заболевания);
- Отвердение и стягивание кожи на туловище и конечностях (по аналогии со склеродермией);
- Замедленное и неправильное формирование зубов;
- Частичная потеря слуха;
- Потеря жира под кожей и мышечной массы;
- Скелетные аномалии и хрупкие кости;
- Жесткие суставы;
- Вывих бедра;
- Резистентность к инсулину.
Когда следует обращаться к врачу?
Прогерия обычно обнаруживается в младенчестве или раннем детстве, часто при регулярных осмотрах, когда у ребенка впервые проявляются характерные признаки преждевременного старения.
Причины прогерии
Причина синдрома Хатчинсона-Гилфорда - мутация гена.
Ген, известный как ламин А/C (LMNA), создает белок, необходимый для удержания ядра клетки. Когда этот ген имеет дефект (мутацию), образуется аномальная форма белка ламина, называемого прогерином, и делает клетки нестабильными. Это, по-видимому, приводит к процессу старения.
В отличие от многих генетических мутаций, cиндром Хатчинсона-Гилфорда редко передается по наследству.
Другие подобные синдромы
Есть и другие прогероидные синдромы, которые передаются по наследству и вызывают быстрое старение, сокращая продолжительность жизни:
- Синдром Видемана-Раутенштрауха, также известный как неонатальный прогероидный синдром, начинается в утробе матери, причем признаки и симптомы старения проявляются при рождении.
- Синдром Вернера, также известный как взрослая прогерия, начинается в подростковом возрасте или в раннем взрослом возрасте, вызывая преждевременное старение и состояния, типичные для старости, такие как катаракта и диабет.
Фактор риска
Нет никаких известных факторов, таких как образ жизни или экологические проблемы, которые увеличивают риск возникновения прогерии или рождения ребенка с синдромом Хатчинсона-Гилфорда.
Для родителей, у которых был один ребенок с прогерией, вероятность рождения второго ребенка с синдромом Хатчинсона-Гилфорда составляет 2-3%.
Осложнения, вызванные прогерией
У детей с прогерией обычно развивается тяжелое затвердение артерий (атеросклероз). Это состояние, при котором стенки артерий - кровеносных сосудов, несущих питательные вещества и кислород от сердца к остальной части тела, - застывают и уплотняются, часто ограничивая кровоток.
Большинство детей с прогерией умирают от осложнений, связанных с атеросклерозом, в том числе:
- проблем с кровеносными сосудами, которые снабжают сердце (сердечно-сосудистые проблемы), приводящих к сердечному приступу и застойной сердечной недостаточности;
- проблем с кровеносными сосудами, которые снабжают мозг (цереброваскулярные проблемы), приводящих к инсульту;
- других проблем со здоровьем, часто связанных со старением - таких как артрит, катаракта.
Постановка диагноза синдрома Хатчинсона-Гилфорда
Врачи могут заподозрить прогерию на основании признаков и симптомов, характерных для этого синдрома. Для подтверждения диагноза используется генетический тест на мутации LMNA.
Тщательное физическое обследование ребенка включает в себя:
- измерение роста и веса;
- построение измерений на графике кривой нормального роста;
- проверку слуха и зрения;
- измерение жизненно важных показателей, включая кровяное давление;
- поиск видимых признаков и симптомов, характерных для прогерии.
Лечение прогерии
Нет лекарства от прогерии, но регулярное наблюдение за состоянием сердца и кровеносных сосудов (сердечно-сосудистых заболеваний) помогает в облегчении симптомов. Во время медицинских визитов измеряются вес и рост ребенка с последующим нанесением на график нормальных значений.
Дополнительные регулярные обследования, включая электрокардиограммы и осмотры зубов, зрения и слуха, могут быть рекомендованы врачом для проверки наличия изменений. Некоторые методы могут облегчить или отсрочить некоторые признаки и симптомы.
Лечение зависит от состояния и симптомовребенка. Они могут включать в себя:
- Аспирин в малых дозах. Суточная доза может помочь предотвратить сердечные приступы и инсульт.
- Другие лекарственные препараты. В зависимости от состояния врач может назначить другие лекарства, такие как статины для снижения уровня холестерина, лекарства для снижения артериального давления, антикоагулянты для предотвращения образования тромбов и лекарства для лечения головных болей и судорог.
- Физиотерапия и трудотерапия. Эти методы лечения помогают при тугоподвижности суставов и проблемах с бедрами.
- Питание. Питательные, высококалорийные продукты и добавки необходимы для поддержания полноценного питания.
- Стоматологическая помощь. Проблемы с зубами часто встречаются при прогерии.
Потенциал дальнейшего лечения прогерии
Современные исследования направлены на понимание прогерии и выявление новых вариантов лечения.
Некоторые области исследований включают в себя:
- Изучение генов и течения болезни, чтобы понять, как она прогрессирует. Это может помочь выявить новые методы лечения.
- Изучение способов профилактики заболеваний сердца и сосудов. Проведение клинических испытаний с использованием препаратов, известных как ингибиторы фарнезилтрансферазы (ФТИ), таких как лонафарниб, которые были разработаны для лечения рака, но могут быть эффективны для лечения прогерии, помогая с увеличением веса и повышением гибкости кровеносных сосудов.
Образ жизни при прогерии
Вот несколько шагов, которые можно предпринять, чтобы помочь ребенку с прогерией:
Прогерия
Прогерия - редкое генетическое заболевание, характеризующееся преждевременным старением организма, соответствующими изменениями внутренних органов. Проявляется гиперпигментацией, истончением и утратой эластичности кожи, поседением и выпадением волос, увеличением размеров черепа, уменьшением его лицевой части, экзофтальмом, развитием атеросклероза сосудов, инфаркта и фиброза миокарда, остеопороза, сахарного диабета, формированием злокачественных опухолей. Диагностика основана на сборе клинических данных, лабораторном исследовании ДНК. Лечение направлено на устранение симптомов заболевания: профилактику и терапию атеросклероза, инфаркта миокарда, диабета, остеопороза, рака.
МКБ-10
Общие сведения
Термин «прогерия» в переводе с древнегреческого языка означает «преждевременное старение». Детская форма прогерии называется синдромом Хатчинсона-Гилфорда согласно фамилиям исследователей, которые впервые независимо друг от друга описали данную патологию: британский врач Дж. Хатчинсон - в 1889 году, его соотечественник Х. Гилфорд - в 1897 году. Взрослая форма носит название «синдром Вернера», так как в 1904 году немецкий врач О. Вернер первым официально наблюдал симптомы старения у подростков 14-19 лет. Оба варианта болезни являются крайне редкими. Распространенность детской прогерии составляет 1 случай на 7-8 млн. человек, с момента начала исследований патологии выявлено около 150 больных детей. Взрослая прогерия встречается чаще, средние эпидемиологические показатели - 1 больной на 100 тыс. населения.
Причины прогерии
В основе заболевания лежит генетическая мутация. Причиной детского типа прогерии является дефект гена LMNA, который кодирует особый белок ламин A, выстраивающий оболочку ядра клетки. Как правило, мутация происходит спорадически - во время созревания половых клеток у родителей или при зачатии. Однако исследователи выявили несколько случаев патологии у родных братьев и сестер, часть из которых входила в семьи с близкородственными браками. Это свидетельствует о возможности наследования мутации по аутосомно-рецессивному типу (родители являются носителями дефектного гена, но не болеют и способны к деторождению).
Прогерия взрослых - наследственное заболевание. Оно развивается при наличии дефекта в гене RECQL2, который ответственен за производство белка WRN, поддерживающего стабильность генома, целостность и структуру ДНК. Передача мутации происходит аутосомно-рецессивным способом, болезнь проявляется у людей, получивших два дефектных гена в одной аллели (от матери и от отца).
Патогенез
При детском варианте прогерии генетический дефект приводит к нарушению синтеза структурно правильного белка ламина А, образующего внутренний каркас ядерной оболочки. В результате мутации налаживается производство укороченного белка-предшественника преламина А, а строение созревшего конечного белка отличается от нормального. Молекулы белка встраиваются в ядерную мембрану, изменяют ее форму и повышают хрупкость. Ядро не может сохранять свою целостность, клетка гибнет раньше времени.
При болезни Вернера возникает недостаточность WRN-белка, определяется генетически детерминированная нестабильность хромосом. Их структура очень часто изменяется спонтанно или под воздействием некоторых факторов. В итоге ухудшается способность клеток к делению, пролиферативный потенциал становится в 3-5 раз меньше, чем должен быть в норме. В 10 раз увеличивается частота спонтанных мутаций, аномально укорачиваются теломеры - концевые участки хромосом, защищающие гены от повреждений.
Симптомы прогерии
Прогерия Хатчинсона дебютирует после периода нормального развития в возрасте от 6 месяцев до 2 лет. Изменяется внешность детей: замедляется рост, увеличиваются размеры черепа, но лицевая часть остается маленькой, нижняя челюсть - недоразвитой. Формируется клювовидный тонкий нос и экзофтальм (выпячивание глаз). Тотально выпадают волосы - на голове и теле, ресницы и брови. При алопеции разрушаются волосяные фолликулы, поэтому дальнейший рост волос становится невозможным. На волосистой части головы выбухают вены. Дерма и подкожная клетчатка подвергаются атрофическим изменениям: кожа истончается, иссушается, покрывается морщинами. Развивается липодистрофия - заметное снижение количества подкожного жира. Относительно сохранным он остается на щеках и лобке.
На туловище образуются склеродермоподобные очаги - уплотнения в нижней части живота, на ягодицах и бедрах. На открытых участках кожи появляются пигментные пятна. Ногти недоразвитые, ломкие, утолщенные, округло выпуклые - имеют форму «часовых стекол». К 2 годам развивается фиброз органов и околосуставных тканей. Пассивные движения в локтевых и коленных суставах ограничены (контрактуры), формируется характерное положение костей ног - «поза всадника». Скелет подвергается гипоплазии, дисплазии и дегенеративным изменениям. Молочные и постоянные зубы прорезываются с опозданием, частично отсутствуют, скученные, искривленные, подвержены кариесу. К 5-6 годам диагностируется атеросклероз сосудов, сердечные шумы, гипертрофия левого желудочка, помутнение хрусталика, резистентность к инсулину. Половые органы остаются недоразвитыми. Уровень умственного развития обычно выше, чем у сверстников.
Клинические проявления синдрома Вернера обнаруживаются с 14 до 18 лет. Подросток начинает отставать в росте, его волосы седеют и выпадают. К 20 годам пациенты лысеют. Кожа лица и конечностей бледнеет, истончается, натягивается. Под ней видна сеть кровеносных сосудов. Мышечная и жировая ткани атрофируются, руки и ноги становятся непропорционально тонкими, кожа над суставными выступами изъязвляется. К 30 годам появляется катаракта, голос слабеет и хрипнет, на ногах образуются язвы, на подошвах - мозоли, сосудистые звездочки, кератоз. Внешний вид пациентов специфичен: низкий рост, лунообразное лицо, выступающий подбородок, суженное ротовое отверстие, псевдоэкзофтальм.
Сальные и потовые железы атрофируются. Костно-суставные изменения включают метастатическую кальцификацию, явления генерализованного остеопороза, эрозивных остеоартритов, ограниченную подвижность и деформацию пальцев рук, сгибательные контрактуры, боли в конечностях, артрит и остеомиелит. Отмечаются атеросклеротические изменения сосудов, медленно прогрессирует катаракта, снижаются интеллектуальные способности. После 30 лет проявляются эндокринные заболевания - сахарный диабет, гипогонадизм, тиреопатии. У 5-10% больных обнаруживаются злокачественные опухоли различных органов, костей и кожи. Причиной смерти становится онкопатология или тяжелое сердечно-сосудистое заболевание.
Диагностика
Диагноз прогерии устанавливается на основании клинико-анамнестических данных. В зависимости от имеющейся симптоматики в диагностике могут принимать участие эндокринологи, неврологи, дерматологи, терапевты, врачи-генетики. Детскую и взрослую прогерию необходимо дифференцировать с системной склеродермией, пойкилодермией, синдромом Ротмунда-Томсона, синдромом Видемана-Раутенштрауха. К основным методам обследования больных относятся:
- Опрос. В ходе беседы врачи уточняют время появления симптомов, их выраженность, степень физической и социальной дезадаптации, наличие периода обычного развития (6-24 месяца при детской форме, 14-20 лет - при взрослой). Собирают данные семейного и генеалогического анамнеза, выявляя генетическую патологию или ее отсутствие в роду.
- Осмотр. Специалисты оценивают неврологический статус, пассивную и активную подвижность суставов, костные деформации, состояние кожи и подкожной клетчатки, сохранность зрения, интеллектуальных функций. Пациенты могут быть направлены на инструментальные исследования: рентгенографию костей, УЗИ и МР-томографии органов, ЭКГ, офтальмоскопию.
- Биогенетическое исследование. Выполняется анализ генетического материала методом секвенирования ДНК. Исследуются участки генов, дефекты в которых приводят к развитию прогерии. У пациентов с детской и взрослой формой болезни обнаруживаются мутации в гомозиготном и компаунд-гетерозиготном состоянии.
Лечение прогерии
Специфические методы терапии прогерии не разработаны. Медицинская помощь пациентам заключается в облегчении симптомов болезни. Проводится лечение атеросклероза, остеопороза, остеомиелита, катаракты, диабета, патологий сердца и онкологических заболеваний. Известен случай, когда состояние больного заметно улучшилось на несколько лет после операции трансплантации сердца.
Научные исследования ведутся в области генной инженерии - совершаются попытки выделить мутационный ген из ДНК и заменить его здоровым. Параллельно разрабатывается метод этиотропной терапии. Ученые из Швеции нашли способ нейтрализации дефектных молекул ламина A, которые проникают в мембрану клеточных ядер и способствуют гибели клетки. Положительные результаты получены на экспериментах с мышечной тканью крыс, на людях препарат пока не опробован. В США с 2012 года ведется изучение эффективности применения ингибитора фарнезилтрансферазы (лекарственного средства, разработанного для лечения рака). После терапии дети из экспериментальной группы чувствовали себя лучше, повысилась жесткость и эластичность артерий, плотность костей, снизились случаи транзиторных ишемических атак, сердечных патологий, утраты зрения и слуха. Продолжительность жизни увеличилась на 1,6-6 лет.
Прогноз и профилактика
1. Синдром прогерии Гетчинсона-Гилфорда (HGPS)/ Абдуллаев М. И., Абидова Ш. А. // Молодой ученый — 2018 — №19.
2. Наследственные синдромы и медико-генетическое консультирование/ Козлова С. И., Демикова Н. С., Семанова Е. и др. - 1996.
3. Hutchinson-Gilford progeria syndrome: A rare case report/ Subhash Kashyap, Vinay Shanker, Neeraj Sharma// Indian Dermatol Online Journal - 2014 - №5.
Синдром Гетчинсона-Гилфорда (прогерия)
Фофанова О.В. Синдром Гетчинсона-Гилфорда (прогерия). Проблемы Эндокринологии. 1995;41(4):24-26.
Fofanova O.V. Syndrome Hutchinson-Gilford (progeria). Problems of Endocrinology. 1995;41(4):24-26. (In Russ.)
Читайте также: