Синдром Кирле (Kyrle) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 01.02.2026
Главный военный клинический госпиталь внутренних войск МВД России, Балашиха, Московская область
Болезнь Кирле (кератоз фолликулярный и парафолликулярный, проникающий): клинический случай
Журнал: Клиническая дерматология и венерология. 2011;9(5): 25‑28
Карпов В.В. Болезнь Кирле (кератоз фолликулярный и парафолликулярный, проникающий): клинический случай. Клиническая дерматология и венерология. 2011;9(5):25‑28.
Karpov VV. Kyrle's disease (follicular and parafollicular, penetrating keratosis): a case report. Klinicheskaya Dermatologiya i Venerologiya. 2011;9(5):25‑28. (In Russ.).
Рассмотрены вопросы эпидемиологии, этиопатогенеза, клинические проявления, методы диагностики и лечения парафолликулярного проникающего кератоза (болезни Кирле). Представлено собственное клиническое наблюдение.
Кератоз фолликулярный (парафолликулярный кератоз, болезнь Кирле — БК) — редкое заболевание кожи из группы перфорирующих дерматозов, в основе которого лежит нарушение ороговения с интрадермальной инвагинацией эпидермиса [1] (синонимы: гиперкератоз фолликулярный и парафолликулярный, проникающий в кожу, гиперкератоз фолликулярный и парафолликулярный с пенетрацией в кожу, (hyperkeratosis follicularis et parafollicularis in cutem penetrans), проникающий гиперкератоз [2, 3]).
Заболевание в 1916 г. впервые описал австрийский дерматолог Дж. Кирле (J. Kyrle), наблюдавший этот дерматоз у больного сахарным диабетом [3—5].
Эпидемиология. Заболевание редкое. Возраст больных 20—70 лет. Женщины болеют чаще, дети — очень редко. Сопутствующие заболевания: часто — сахарный диабет и хроническая почечная недостаточность, реже — сердечная недостаточность, гипотиреоз, гиперлипопротеинемия II типа (повышение в крови уровня холестерина и триглицеридов) [1, 3—6].
Этиология и патогенез окончательно не изучены. Ранее предполагаемая роль вирусной или бактериальной инфекции, нарушение обмена витамина А в развитии этого дерматоза представляет лишь исторический интерес [4]. Определенное значение в развитии заболевания, помимо нарушения углеводного обмена, придается поражению печени (хронический гепатит) с развитием вторичной недостаточности витамина А [5].
Генетическая предрасположенность к развитию БК окончательно не доказана, хотя описаны случаи заболевания среди родственников в одной семье при кровном родстве (родственники первой степени) [1, 3—5].
По первоначальному определению Дж. Кирле [7], это заболевание, при которой атипичный клон кератиноцитов проникает через эпидермис в дерму. Полагают, что в основе патологического процесса лежит нарушение кератинизации, дифференцировки и ороговения кератиноцитов (образование дискератотических фокусов и ускорение процесса ороговения). Это ведет к образованию кератотических пробок с участками паракератоза. Ороговение начинается уже на границе эпидермиса и дермы. Скорость дифференцировки и кератинизации превышает скорость клеточной пролиферации, поэтому паракератотический конус частично проникает глубже, в нарушенный эпидермис, и обусловливает перфорацию его в дерму [1, 3—6].
Единой классификации фолликулярных кератозов нет. Среди самостоятельных нозологических форм выделяют папулезные, атрофирующие и вегетирующие фолликулярные кератозы [8]. Ряд авторов [4, 6] обращают внимание на сходство БК с лентикулярным стойким гиперкератозом Флегеля и рассматривают последний как вариант БК, хотя клинические проявления различаются. Другие авторы [5, 8] относят БК к вегетирующим фолликулярным кератозам, а кератоз фолликулярный стойкий (болезнь Флегеля) — к папулезным.
Рассматривается концепция реактивного перфоративного нарушения кератинизации на фоне болезней почек, печени, сахарного диабета. Появилось понятие — перфорирующие заболевания кожи, к нему относят группу заболеваний, при которых происходит элиминация измененных компонентов кожи через эпидермис (процесс, называемый трансэпидермальной элиминацией). Некоторые авторы оспоривают БК, как самостоятельную нозологическую единицу [7].
В Международной классификации болезней 10-го пересмотра в классе XII (болезни кожи и подкожной клетчатки) выделена рубрика L87 — трансэпидермальные прободные изменения, в которой БК отведена отдельная подрубрика L87.0.
Патоморфология весьма характерна и имеет диагностическую значимость. Углубления эпидермиса и расширенные устья волосяных фолликулов заполнены гиперкератотическими пробками. Под пробками в эпидермисе выражено разрастание зернистого слоя, а в местах отсутствия гипергранулеза — паракератоз, проникающий весь эпидермис до дермы. В дальнейшем происходит истончение эпидермиса и проникновение в дерму роговых масс с формированием в дерме воспалительных инфильтратов типа гранулем из лимфоцитов, лейкоцитов, гистиоцитов и гигантских клеток инородных тел. Наблюдается гибель сальных желез в области пораженных фолликулов, дистрофия коллагеновых волокон, гиперэластоз [1, 2, 5, 6].
Клиническая картина. Начало заболевания постепенное, новые высыпания появляются по мере исчезновения старых элементов. Характерны фолликулярные или парафолликулярные папулы сначала цвета здоровой кожи, а впоследствии сероватой или буровато-красной окраски, диаметром от булавочной головки до 1 см. В центре элементов — роговая пробка, при удалении которой образуется кратерообразное углубление. Папулы склонны к периферическому росту и слиянию с формированием сухих полициклических бляшек, покрытых наслоением чешуек, корочек. Консистенция плотная, поверхность неровная, бородавчатая. Свежие высыпания сопровождаются легким зудом (чаще у больных сахарным диабетом) или не беспокоят больного. Старые, более крупные очаги бывают болезненными, особенно при надавливании. В местах расчетов линейное расположение, что свидетельствует о возможности возникновения феномена Кебнера. Возможно присоединение вторичной инфекции. Локализация высыпаний — разгибательные поверхности конечностей, туловище, ягодицы. Слизистые оболочки не поражаются [1—6]. Высыпания в полости рта, на ладонях, подошвах, гениталиях встречаются редко [4].
Течение БК хроническое, рецидивирующее. Заболевание с трудом поддается лечению. Прогноз во многом зависит от основного заболевания.
Диагностика основывается на анамнезе, клинической и гистологической картине заболевания. Дифференциальную диагностику БК необходимо проводить с красным волосяным лишаем Девержи, фолликулярным дискератозом Дарье, порокератозом Мибелли, трансэпидермальными прободными изменениями кожи (перфорирующий ползучий/серпигинозный эластоз, реактивный перфорирующий коллагеноз), болезнью Флегеля [8].
Болезнь Флегеля (hyperkeratosis lenticularis perstans), так же как и БК, является редкой формой фолликулярного кератоза. Предполагают, что в основе данного заболевания лежит наследственно обусловленная (аутосомно-доминантный тип) патология ороговения, выражающаяся в нарушении синтеза кератина 55К, содержание которого резко снижено в очагах поражения при болезни Флегеля, в то время как в нормальной коже этот вид кератина доминирует. Гистологически выявляют истончение эпидермиса, фолликулярный ортокератоз с фокусами паракератоза, местами спонгиоз, в дерме выражен полосовидный лимфоцитарный (преимущественно Т-хелперный) инфильтрат. Придававшееся ранее большое значение в патогенезе дерматоза отсутствию или нарушению структуры телец Одланда [4] в настоящее время подвергается сомнению. Гранулы Одланда появляются в верхних рядах шиповатого слоя в кератиноцитах, занимают периферическое положение и выделяют путем экзоцитоза свое содержимое в межклеточное пространство, где оно принимает пластинчатое строение (межклеточный цемент) — компонент нормальной кератинизации. В отличие от БК этот дерматоз проявляется чаще в юношеском и среднем возрасте, характеризуется образованием диссеминированно расположенных более мелких (1-5 мм) и более поверхностно залегающих роговых папул (без тенденции образовывать бляшки). При БК нет нарушений кератина 55К [6].
Лечение: витамин А, аевит длительно, ретиноиды (неотигазон, этретинат, ацитретин из расчета 0,5 мг/кг в сутки). Наружно - кератолитические мази, 2% серносалициловая мазь, ретиноидный крем, 0,1% тигазоновый крем, 5% фторурациловый крем (на площадь не более 500 см одновременно), электро-, крио- и лазеродеструкция (углекислый лазер) отдельных очагов, ультрафиолетовое облучение. Эффект от лечения временный. После удаления роговой пробки остается атрофический, неравномерно пигментированный рубец; рецидив может возникнуть рядом. Необходима коррекция висцеральной патологии [3—6].
Приводим собственное клиническое наблюдение.
Больной Б., 57 лет, пенсионер (музыкант военного оркестра), поступил в отделение гнойной хирургии 13.09.10 на оперативное лечение по поводу остеомиелита левой стопы. Лечащим врачом назначена консультация дерматолога из-за высыпаний на коже.
Анамнез. Первые высыпания на коже грудной клетки спереди появились около 20 лет назад. Высыпания практически не беспокоили, редко отмечался незначительный зуд, больше «волновал» косметический аспект. Выставлялись диагнозы: дерматит, ксероз, кератоз. Эффекта от рекомендованных салициловой мази, кремов с ромашкой, чередой, приема витамина А внутрь не отмечалось, что объясняли «своеобразным строением кожи пациента». По словам пациента, наилучший эффект отмечал от применения циндола («болтушка хорошо подсушивает и слущивает пробочки» на коже). Полностью высыпания не исчезали, сезонность не отмечена.
С 1995 г. страдает сахарным диабетом 2-го типа, принимал перорально гипогликемические препараты (диабетон). В 2009 г. стационарно лечился по месту жительства по поводу синдрома диабетической стопы, гнойно-некротической раны на левой стопе; заживление вторичным натяжением. С лета 2010 г. высыпаний стало значительно больше, что пациент связывал с сильной жарой. В июле получил колотую рану левой стопы (наступил на гвоздь на даче), которая длительно не заживала, повысился уровень сахара крови. В связи с ухудшением состояния выраженного болевого синдрома пациент госпитализирован.
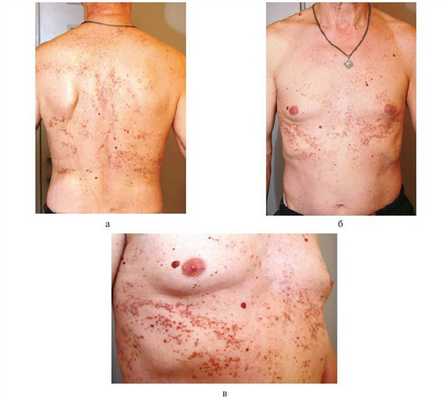
Кожный статус. На коже туловища — множественные фолликулярные и парафолликулярные папулы красно-бурого цвета, местами сгруппированные в мелкие бляшки, покрытые плотными чешуйками. В центре папул — роговые пробки. Там, где пробки отторглись, кратерообразные углубления, заполненные нанесенным ранее циндолом. На месте разрешившихся элементов — мелкие рубчики. При пальпации высыпания плотные, кожа напоминает «терку», влажная. Несколько внутридермальных невусов округлой куполообразной формы, мягкой консистенции, коричневого цвета (рисунок, а—в). Рисунок 1. Пациент Б., 57 лет: проявления БК. Пациент страдает сахарным диабетом, диабетической нефропатией. а—в — множество розовато-красного и сероватого цвета папул и узлов с роговыми пробками. В тех местах, где больной расчесывал кожу, элементы сыпи располагаются линейно (симптом Кебнера). Несколько внутридермальных невусов. Кожа головы, конечностей, видимые слизистые оболочки без высыпаний, физиологической окраски.
Результаты специальных обследований. Анализ крови клинический: гемоглобин — 124 г/л, эритроциты — 4,0·1012/л, лейкоциты — 6,7·109/л (при поступлении 12,8·109/л), нейтрофилы — 67%, сегментоядерные — 65%, палочкоядерные — 2%, эозинофилы — 3%, лимфоциты — 23%, моноциты — 9%, скорость оседания эритроцитов — 35 мм/ч (при поступлении 53 мм/ч). Титр АСТЛ-0 и ревматоидный фактор — не обнаружены. СРП —10 мг/л. Анализ мочи при поступлении: удельный вес — 1020, белок — 0,132‰, сахар — 0,3 мг/л, ацетон — нет, лейкоциты — 3—5 в поле зрения, эритроциты — 4—5 в поле зрения, цилиндры — 4 в препарате. При выписке анализ мочи в норме.
Рентгенография органов грудной клетки — без патологии. На рентгенограммах левой стопы - полная деструкция головки III плюсневой кости, частичное разрушение II плюсневого сустава. Электрокардиограмма — ритм синусовый, частота сердечных сокращений 76 в минуту; изменения в миокарде; феномен ранней реполяризации желудочков.
Эндокринолог. Сахарный диабет 2-го типа, тяжелое течение, инсулинопотребный, декомпенсированный. Диабетическая ангиопатия сосудов нижних конечностей. Диабетическая нефропатия. ХПН-0.
Офтальмолог. Пролиферативная диабетическая ретинопатия. Начальная катаракта правого глаза.
Хирург. Вялогранулирующие раны подошвенной поверхности левой стопы. Остеомиелит II—III плюсневых костей левой стопы.
Дерматолог. Кератоз фолликулярный и парафолликулярный, проникающий (БК). Внутридермальные невусы. Противопоказаний к оперативному лечению нет.
Больному 20.09.10 произведена трансметатарзальная резекция головки III плюсневой кости. Ампутация III пальца левой стопы. Получал антибактериальную терапию, хумулин, лантус; проводились перевязки с димексидом, мазями на водорастворимой основе, физиотерапия. Кожу туловища 1 раз в день обрабатывал циндолом.
Рекомендации при выписке. Наблюдение эндокринолога, хирурга по месту жительства; хумулин по 18 ЕД 3 раза в день перед основным приемом пищи, лантус в 22:00 по 16 ЕД. Перевязки остаточных ран с мазью бетадин. Лазерная коагуляция сетчатки после нормализации сахаров в отдельную госпитализацию, тогда же показаться дерматологу.
Каждый дерматолог встречается с непонятными больными. Особенно это касается редких дерматозов, с некоторыми из них врач может столкнуться лишь раз за свою практику. «Лучше один раз увидеть, чем сто раз услышать», — гласит русская народная мудрость. «Главный инструмент дерматолога — его глаза» [3].
То что у нашего пациента выраженные явления гиперкератоза туловища, было очевидно, но мы не встречались раньше ни с БК, ни с болезнью Флегеля. Только на основании анамнестических данных, характерной клинической картины и сведений, полученных из источников литературы (от предложенной биопсии кожи больной отказался), был выставлен окончательный диагноз. Наш пациент страдает сахарным диабетом, диабетической нефропатией. Если бы рекомендуемое обследование всех больных с гиперкератозами для выявления латентного диабета [4] было сделано раньше, возможно, и диагнозы были выставлены раньше. Цель данной публикации заключается именно в этом.
Синдром Криглера-Найяра
Синдром Криглера-Найяра - генетическое заболевание из класса ферментопатий, характеризующееся нарушением одного из звеньев процесса обезвреживания и выведения билирубина - конъюгации. Симптомами этого состояния являются желтуха печеночного генеза и тяжелые неврологические нарушения, которые могут привести к летальному исходу еще в младенческом возрасте. Диагностика синдрома Криглера-Найяра производится посредством биохимических проб и определения уровня неконъюгированного билирубина в плазме крови, а также молекулярно-генетическими методиками. Специфического лечения заболевания не существует (за исключением трансплантации печени), терапия сводится к увеличению разрушения и элиминации билирубина из организма (гемосорбция, фототерапия, плазмаферез, прием барбитуратов).
Общие сведения
Синдром Криглера-Найяра - тяжелое генетическое заболевание, характеризующееся нарушением связывания билирубина с глюкуроновой кислотой, что является ключевым этапом его обезвреживания и выведения из организма. Впервые это заболевание было описано в 1952 году двумя американскими педиатрами - Джоном Криглером и Виктором Найяром. Дальнейшее изучение синдрома Криглера-Найяра показало, что это состояние имеет генетическую природу и аутосомно-рецессивный характер наследования, кроме того, удалось выявить две клинические разновидности данной патологии. Заболевание достаточно редкое, поэтому точные цифры встречаемости не определены - большинство исследователей полагает, что она находится на уровне 1:1 000 000. Половое распределение больных синдромом Криглера-Найяра не имеет особенностей, заболевание с одинаковой частотой поражает как мальчиков, так и девочек. В лечении этого состояния крайне важна ранняя (в идеале - пренатальная) диагностика, так как от своевременности начатой терапии очень сильно зависят прогноз заболевания и качество жизни больного.
Причины
Синдром Криглера-Найяра относят к классу ферментопатий (по другой классификации - к группе неконъюгированных гипербилирубинемий), причина этого заболевания кроется в недостаточности уридиндифосфатглюкуронидазы 1, функцией которой является связывание билирубина с двумя молекулами глюкуроновой кислоты. В итоге этого биохимического процесса билирубин становится способным растворяться в воде, выводиться в составе желчи и, главное, значительно падает его токсичность. При синдроме Криглера-Найяра этот процесс резко замедлен или не происходит совсем, вследствие чего возникает задержка элиминации билирубина из организма и его накопление.
Билирубин обладает выраженной нейротоксичностью, при повышении концентрации в крови это вещество начинает откладываться в тканях кожных покровов и слизистых оболочек, приводя к развитию желтухи. Когда концентрация билирубина превышает определенный порог, соединение начинает проникать через гематоэнцефалический барьер в головной мозг, приводя к тяжелой энцефалопатии (особенно повреждаются базальные ядра). При отсутствии лечения больные синдромом Криглера-Найяра погибают от многочисленных неврологических расстройств и нарастающей печеночной комы.
Причиной низкой активности уридиндифосфатглюкуронидазы являются мутации гена UGT1A1, который располагается на 2-й хромосоме, отвечает за аминокислотную последовательность и выделение этого фермента. Помимо синдрома Криглера-Найяра дефекты этого гена могут приводить к ряду других нарушений билирубинового обмена наследственного характера - синдрому Жильбера, транзиторной неонатальной билирубинемии семейного типа. Механизм наследования мутаций гена UGT1A1 при синдроме Криглера-Найяра аутосомно-рецессивный. При этом описано несколько вариантов возможного повреждения этого гена, которые приводят к разному течению данного заболевания.
Классификация и симптомы синдрома Криглера-Найяра
В настоящее время описаны две основные клинические формы синдрома Криглера-Найяра, в основном различающиеся между собой тяжестью проявлений и прогнозом заболевания. Это обусловлено типом генетического дефекта в UGT1A1. Первый тип заболевания (СКН-1) вызывается миссенс-мутациями, приводящими к появлению неполноценного фермента, имеющего сигнальную последовательность аминокислот, характерную для подвергающихся внутриклеточной утилизации белков. Таким образом, при этой форме дефект гена поражает кодирующие участки (экзоны), что вызывает развитие патологии у гомозигот. Вскоре после своего образования уридиндифосфатглюкуронидаза 1 разрушается и конъюгации билирубина не происходит совсем.
Синдром Криглера-Найяра 1-го типа характеризуется тяжелым и стремительным течением - первые признаки гипербилирубинемии в виде желтухи обнаруживаются уже через несколько часов после рождения. Со временем к ним присоединяются неврологические нарушения - нистагм, судорожные приступы, иногда возникает опистотонус. Желтуха сохраняется на протяжении всей жизни ребенка, его умственное развитие резко отстает от такового у сверстников, симптомы заболевания неуклонно нарастают даже при интенсивном лечении. Обычно больные синдромом Криглера-Найяра 1-го типа умирают на протяжении первого года жизни из-за интоксикации билирубином и поражения базальных подкорковых ядер (ядерная энцефалопатия).
Причиной синдрома Криглера-Найяра 2-го типа также являются миссенс-мутации гена UGT1A1, однако они могут возникать как в кодирующей последовательности (экзонах), так и в промоторе - участке, отвечающем за экспрессию данного гена. У большинства больных СКН-2 наблюдается наличие на одной хромосоме дефекта экзона, на другой - промотора, то есть, такие лица являются компаунд-гетерозиготами. Результатом нарушения является продукция дефектной формы фермента уридиндифосфатглюкуронидазы, которая не разрушается, но имеет пониженную (на уровне 20-25% от нормы) функциональную активность. Поэтому синдром Криглера-Найяра 2-го типа характеризуется менее тяжелой клинической картиной и более благоприятным прогнозом.
В первые месяцы и даже годы жизни больных синдром Криглера-Найяра этого типа нередко проявляется только незначительной желтухой, при отсутствии лечения к подростковому периоду могут развиваться неврологические отклонения. В ряде случаев, особенно при правильно назначенных терапевтических мероприятиях, никаких нарушений со стороны центральной нервной системы не возникает вовсе. Проявления желтухи различной степени выраженности у больных синдромом Криглера-Найяра 2-го типа могут сохраняться на протяжении всей жизни и нередко расцениваются как индикатор осложнений и ухудшения состояния пациента. С возрастом иногда появляется нистагм, могут регистрироваться судорожные припадки, однако течение и выраженность симптомов заболевания всецело зависят от качества лечения и выполнения рекомендаций специалистов.
Диагностика
Диагностика синдрома Криглера-Найяра производится на основании данных общего осмотра ребенка, биохимических исследований крови, желчи и мочи, молекулярно-генетических анализов. При осмотре выявляется желтуха, возникшая в первые часы (при СКН-1) или месяцы (СКН-2) жизни, признаки неврологических нарушений (опистотонус, нистагм, длительное сохранение транзиторных рефлексов). У больных 2-м типом синдрома Криглера-Найяра неврологические расстройства могут регистрироваться во взрослом возрасте, тогда как у детей наблюдается только желтуха. Также с возрастом могут присоединяться такие проявления, как нейросенсорная глухота или хореоатетоз.
При биохимическом исследовании крови выявляется выраженная непрямая гипербилирубинемия (вплоть 200-350 мкмоль/л), отсутствие (при синдроме Криглера-Найяра 1-го типа) или резкое снижение концентрации прямого билирубина. Конъюгированная фракция этого соединения отсутствует в желчи при СКН-1 и присутствует в незначительных количествах при СКН-2. Фенобарбиталовая проба при синдроме Криглера-Найяра положительна только в случае наличия уридиндифосфатглюкуронидазы, то есть при СКН-2. Изучение концентрации неконъюгированного билирубина в моче показывает его увеличение. Молекулярно-генетическая диагностика синдрома Криглера-Найяра производится врачом-генетиком - он совершает прямое секвенирование последовательности гена UGT1A1 с целью выявления мутаций. При отягощенной по этому заболеванию наследственности у родителей может осуществляться пренатальная диагностика патологии. Дифференциальный диагноз следует проводить с обычной транзиторной желтухой новорожденных и синдромом Жильбера.
Лечение синдрома Криглера-Найяра
Специфического или этиотропного лечения синдрома Криглера-Найяра на сегодняшний день не существует, все терапевтические мероприятия назначаются для ускорения распада билирубина, его выведения из организма и защиты ЦНС. Особых отличий в терапии 1-го или 2-го типа заболевания нет (за исключением активизации микросомального окисления барбитуратами, которая не производится при 1-м типе), однако при СКН-1 лечение лишь незначительно оттягивает наступление летального исхода. Самым радикальным методом лечения синдрома Криглера-Найяра в настоящее время является операция по аллотрансплантации печени от родственника или генетически сходного донора - в этом органе происходит образование уридиндифосфатглюкуронидазы.
Синдром Криглера-Найяра 2-го типа лечат назначением умеренных доз барбитуратов для активации окисления билирубина и увеличения образования нужного фермента. Кроме того, показаны плазмаферез, гемосорбция, заместительное переливание крови - все эти процедуры направлены на удаление неконъюгированного билирубина из организма. Неплохие результаты у больных синдромом Криглера-Найяра дает фототерапия - облучение кожных покровов приводит к частичному разрушению билирубина и освобождению рецепторов тканей для новых порций этого токсина, что снижает его концентрацию в крови. Правильный питьевой режим и повышенное потребление жидкости ускоряет выведение токсина из организма, поэтому следует избегать обезвоживания. Необходим постоянный мониторинг уровня этого вещества в плазме крови, особенно опасным считается его количество свыше 300-340 мкмоль/л - при такой концентрации билирубин становится способным проникать через гематоэнцефалический барьер.
Прогноз и профилактика
Прогноз синдрома Криглера-Найяра 1-го типа исключительно плохой - из-за полного отсутствия активности фермента уридиндифосфатглюкуронидазы 1 больные умирают на протяжении первого года жизни из-за осложнений ядерной энцефалопатии. Течение СКН-2 зависит от таких факторов, как выраженность проявлений, своевременность диагностики и начала лечения, соблюдения рекомендаций специалистов, наличия или отсутствия сопутствующих заболеваний. В большинстве случаев прогноз относительно благоприятный - больные синдромом Криглера-Найяра 2-го типа могут прожить до преклонного возраста, из характерных проявлений патологии их может беспокоить только желтуха. Профилактика этого состояния возможна только в рамках консультации генетика для родителей, имеющих отягощенную наследственность по этому заболеванию, а также при помощи пренатальной диагностики.
Синдром Кирле (Kyrle) - синонимы, авторы, клиника
Красноярский государственный медицинский университет им. проф. В.Ф. Войно-Ясенецкого
ГОУ ВПО Красноярский государственный медицинский университет им. проф. В.Ф. Войно-Ясенецкого
КГБУЗ Красноярский краевой кожно-венерологический диспансер №1
Медицинский колледж Университета министерства обороны, г. Аддис-Абеба;
Красноярский государственный медицинский университет им. проф. В.Ф. Войно-Ясенецкого
Журнал: Клиническая дерматология и венерология. 2013;11(3): 48‑50
Прохоренков В.И., Карачева Ю.В., Гузей Н.М., Гузей Т.Н. Болезнь Кирле. Клиническая дерматология и венерология. 2013;11(3):48‑50.
Prokhorenkov VI, Karacheva IuV, Guzeĭ NM, Guzeĭ TN. Kyrle's disease. Klinicheskaya Dermatologiya i Venerologiya. 2013;11(3):48‑50. (In Russ.).
Представлен редкий случай генодерматоза - болезни Кирле у ребенка.
Кератоз фолликулярный и парафолликулярный Кирле (синоним — Кирле болезнь) — редкая форма фолликулярного перфорирующего кератоза, обусловленного преждевременной кератинизацией и проникновением ороговевших клеток в дерму [1]. Заболевание описано Дж. Кирле в 1916 г.
Первичный дефект неизвестен. Возможное значение имеют обменные нарушения, гиповитаминоз А. Описано развитие заболевания в процессе лечения купренилом. Наследование предположительно аутосомно-рецессивное [2]. Придают патогенетическое значение преждевременному ороговению клеток нижних слоев эпидермиса с проникновением в дерму под влиянием механической травмы [3].
Заболевание развивается обычно после 30 лет, редко у детей, в том числе новорожденных. Описаны семейные случаи дерматоза [3]. Клиническая картина характеризуется фолликулярными, реже — парафолликулярными плотноватыми узелками с ороговением в центре в виде роговой пробки с локализацией преимущественно на коже голеней, предплечий, ягодиц, иногда на лице, туловище, волосистой части головы, значительно реже — на ладонях, подошвах и слизистых оболочках [2, 3]. Сначала величина узелков соответствует булавочной головке, но затем они постепенно увеличиваются, достигая 1 см и более. Высыпания вокруг ороговевающего центра сероватого, коричневато-красноватого цвета, без воспалительных изменений по периферии элементов. Роговые массы могут придавать поверхности папул бородавчатый вид. Сыпь склонна к периферическому росту и слиянию с образованием сухих бородавчатых бляшек, глубоко проникающих в дерму с воспалительно-гранулематозной перифокальной реакцией [4]. После удаления роговой пробки обнаруживают слегка влажное или кровоточащее кратерообразное углубление. На месте регресса элементов остаются пигментированные рубчики. Может быть положительный феномен Кебнера. Заболевание следует дифференцировать от бородавчатой формы красного плоского лишая, болезни Дарье, порокератоза Мибелли, красного волосяного лишая Девержи, эластоза серпигинозного перфорирующего Мишера—Лютца, фолликулярного псориаза, множественной кератоакантомы. Многие авторы [1] идентифицируют заболевание с лентикулярным стойким кератозом Флегеля на основе общего характера происхождения и клинической картины. Течение заболевания хроническое, длительное, возможны рецидивы, прогноз благоприятный, однако полное выздоровление сомнительно. Описаны сочетания данного заболевания с сахарным диабетом, функциональными нарушениями печени, почек, удвоением почечной лоханки и мочеточника [5]. Для лечения рекомендуются препараты витамина А, витамин С и группы В, при упорном течении — ароматические ретиноиды (тигазон, этретинат, ацитретин) по 0,5 мг на 1 кг массы тела в сутки, наружно — серно-салициловые мази, мази с мочевиной, на основе ретиноидов, ретин-А, айрол, атредерм, после удаления роговых наслоений — облучение ультрафиолетовыми лучами. Отдельные очаги можно удалять электрокоагуляцией [4, 6].
Таким образом, данный генодерматоз является редким, поэтому описание клинических случаев представляет практический интерес для дерматологов.
Приводим наше наблюдение.
Девочка П. родилась 08.02.2008 г. Родители здоровы, у бабушки по линии матери — рак яичника, у дедушки по линии отца — псориаз. Ребенок от 1-й беременности. Беременность сопровождалась токсикозом в I и II половине (изжога, рвота). Роды в срок без осложнений. Масса тела при рождении — 3 кг, рост — 48 см. На грудном вскармливании находилась до 1,5 года, прикорм введен с 6 мес. Ребенок развивался соответственно возрасту. При рождении мать заметила высыпания на коже ребенка в области ягодицы, лобка и левого бедра в виде «черных точек», которые в дальнейшем как бы проваливались в кожу (образовывались углубления).
При осмотре ребенка в возрасте 1 года 7 мес на коже в области левой боковой поверхности туловища, лобка, левой ягодицы и наружнобоковой поверхности левого бедра и голени выявлены линейные папулы с ороговением на поверхности черного цвета, расположенные в углублениях (рис.1). Рисунок 1. Больная П. в возрасте 1 года 7 мес. Проникающий фолликулит и парафолликулит (болезнь Кирле). Высыпания на коже в области ягодицы, левого бедра (a) и лобка (б). На основании анамнеза и клинической картины диагностирован фолликулярный и парафолликулярный кератоз (болезнь Кирле). Рекомендовано для уточнения диагноза произвести биопсию. С целью лечения рекомендованы курсы β-каротина 2 раза в год, наружно — гель дифферин, крем эмолиум с 3% мочевины. Биопсия проведена через 1 год. Гистологическая картина: резко выраженный гиперкератоз с массивными роговыми пробками в углублениях эпидермиса, в подлежащей дерме вокруг углублений — инфильтрат, представленный лимфоцитами и полиморфноядерными лейкоцитами. Заключение: патогистологическая картина может быть при болезни Кирле (рис. 2). Рисунок 2. Гистологическая картина: резко выраженный гиперкератоз, в углублениях эпидермиса — массивные роговые пробки. В течение этого периода высыпания постепенно прогрессировали, в местах поражения появлялись инфильтраты с гнойным расплавлением, некоторые из них были вскрыты хирургическим путем. Процесс сопровождался лихорадкой. В течение 2 лет девочка перенесла ветряную оспу и 3 раза острую респираторную вирусную инфекцию. При осмотре в 4 года на коже в области боковой поверхности туловища, левой ягодицы, лобка, бедра и голени видны множественные атрофические глубокие рубцы (рис. 3). Рисунок 3. Больная П. в возрасте 4 лет. Множественные атрофические рубцы на коже в области боковой поверхности туловища (a), ягодицы и бедра (б), голени (в).
Развернутый анализ крови от 08.02.2011: лейкоциты — 10 10 /л, эритроциты — 4,54×10 12 /л, гемоглобин — 122 г/л, гематокрит — 35,2%, тромбоциты — 2,79×10 12 /л, ретикулоциты — 0,2%; средний объем эритроцита — 77, MCH — 26,8; MCHC — 346, RDW — 12,8, MPV — 6,9, лимфоциты — 54,3%, моноциты — 6,5%, гранулоциты — 39,2%, скорость оседания эритроцитов — 5 мм/ч. Общий анализ мочи от 08.02.11: цвет — светло-желтый, глюкоза — отрицательно, белок — отрицательно, кетоновые тела — отрицательно, удельный вес — 1010, билирубин — отрицательно, рН 5,5, уробилиноген — 3,2, нитраты — отрицательно, лейкоциты — 70/мл. Уровень глюкозы в крови — 4,5 ммоль/л. Гликозилированный гемоглобин от 16.05.11 — 4,5% (норма 4,8—5,9%). При обследовании органов брюшной полости патологии не выявлено.
Таким образом, особенностями данного клинического случая являются его раннее развитие (с рождения), генерализация процесса, воспалительный характер высыпаний с образованием множественных атрофических рубцов, что может вызвать затруднения при диагностике данного дерматоза.
Болезнь Кирле
Болезнь Кирле - дерматоз неясной этиологии, характеризующийся рецидивирующим течением с образованием в дерме околофолликулярных очагов гиперкератоза. Клинически проявляется высыпанием на разгибательных поверхностях рук и ног мелких папул грязно-красного цвета, покрытых роговыми чешуйками, способных к диссеминации. Снятие роговых чешуек открывает мокнущие поверхности с последующим исходом в неглубокий пигментированный рубчик. Диагноз ставится на основании анамнеза, клинической картины, наличия сопутствующей патологии, клинико-лабораторного обследования. Морфология неспецифична. Лечение направлено на радикальное удаление очагов гиперкератоза.
Болезнь Кирле (проникающий гиперкератоз) - редкое дерматологическое заболевание, относящееся к группе хронических гиперкератозов, в основе которых лежит нарушение нормального механизма ороговения клеток эпидермиса с последующим проникновением очагов кератоза в дерму. Впервые описал клинические проявления заболевания австрийский дерматолог Йозеф Кирле в 1916 году. Заслуга автора, имя которого с тех пор носит гиперкератоз, состоит в том, что он выделил данную патологию в качестве самостоятельной нозологической единицы, тогда как до него проникающий гиперкератоз рассматривали только как симптом многих кожных заболеваний. Интересно, что поспособствовал этому характер его работы. Й. Кирле занимался гистопатологией в венской кожной клинике, основным трудом его жизни стал двухтомник по гистобиологии кожи. На основании сравнительных морфологических характеристик многих кератозов учёному удалось доказать уникальность этой патологии (проникающий характер гиперкератоза). Актуальность заболевания определяется наличием системных нарушений, способных привести к летальному исходу.
Причины болезни Кирле
Этиология заболевания неизвестна. Болезнь Кирле может возникнуть спонтанно, как результат обменных нарушений (синтеза витамина А, липидов), иметь генетическую составляющую (семейная история заболевания). В подавляющем большинстве случаев болезнь Кирле связывают с системными нарушениями (хроническая почечная и сердечно-сосудистая недостаточность, сахарный диабет, цирроз печени).
Единого мнения о патогенезе заболевания в дерматологии не существует. Одни исследователи считают заболевание кожными проявлениями почечной недостаточности, другие - папулёзной крапивницы. Приоритет принадлежит теории патологического ороговения. В норме клетки эпидермиса синтезируют белок кератин, регулирующий физиологический процесс ороговения. При болезни Кирле клетки эпидермиса начинают активно размножаться, продуцируя избыток кератина и ороговевая раньше срока. Граница между обычными и ороговевшими клетками под тяжестью «лишнего» кератина сползает в базальный слой эпидермиса. Возникает воспаление, нарушается целостность базальной мембраны, роговые массы проникают в дерму, замещая собой часть её клеток, которые вместе с кератином «всплывают» наружу, создавая гиперкератоз на поверхности кожи.
Симптомы болезни Кирле
Первичным элементом является бурая папула (фолликулярный узелок) величиной с просо, желтовато-серебристого оттенка в центре (кератин). Локализуется сыпь чаще на конечностях, в локтевых и подколенных сгибах. Папулы трансформируются в бляшки, которые, сливаясь между собой, образуют эффлоресценции (локальные очаги высыпаний) с очагами некроза либо гиперкератоза в центре. Дерматоз обладает тенденцией к генерализации процесса (за исключением ладоней и подошв). При удалении роговых наслоений обнажается эрозия в форме неглубокой воронки, мокнущая, незначительно кровоточащая, оставляющая после себя пигментированный поверхностный рубчик. Исход самостоятельной инволюции элементов - атрофическое рубцевание.
Субъективные ощущения, связанные с кожными проявлениями, отсутствуют. В редких случаях пациента беспокоит зуд. Объективное состояние зависит от наличия сопутствующей патологии. Некоторые практикующие врачи-дерматологи считают, что возникновение проникающего кератоза - основной симптом грядущего системного заболевания. Дерматоз имеет хроническое течение, не исключены рецидивы. Возрастная и гендерная составляющие отсутствуют.
Диагностика болезни Кирле
Из-за отсутствия специфической морфологии диагностика болезни Кирле проводится методом сравнения данной патологии с другими заболеваниями, симптомом которых является гиперкератоз. Клинически к болезни Кирле близка болезнь Дарье, но в её симптоматике отсутствуют поверхностные «воронки» на коже после удаления кератиновых наслоений, а в биоптате присутствует дискератоз, отсутствующий в гистологии болезни Кирле.
От бородавчатой формы красного плоского лишая болезнь Кирле отличается отсутствием зуда и первичными элементами: папулы с кератиновым «беретом» в центре (в отличие от папул с центральным вдавлением при красном плоском лишае). Красный волосяной лишай отличается от болезни Кирле наличием конусов Бенье - чёрных точечных папул - на тыле пальцев рук, бессимптомным снятием роговых корочек. Парапсориаз Муха‐Габермана различается морфологически (воспалительные изменения в эндотелии сосудистой стенки), а аллергический ангиит - симптомом пальпируемой пурпуры (геморрагическая, а не папулёзная сыпь).
Основное внимание при диагностике дерматоза уделяют наличию сопутствующей системной патологии. Проводят консилиум врачей в составе дерматолога, эндокринолога, нефролога, невролога, гастроэнтеролога, кардиолога; полное клинико-лабораторное обследование (ОАК, ОАМ, биохимия, специальные нагрузочные пробы с целью установления сохранности функций органа).
Лечение болезни Кирле
Солитарные, ограниченные очаги высыпаний удаляют радикально: мелкие папулы - лазером, радиоволновой терапией, крупные - хирургически. Показана консультация дерматокосметолога с целью проведения электрокоагуляции или криодеструкции очагов локального кератоза, фототерапии. Консервативная терапия включает коррекцию углеводного, липидного обмена, патологии почек, заболеваний сердечно-сосудистой системы, другой сопутствующей патологии. Фоновая терапия предполагает проведение длительных курсов витамина А, Е в капсулах, при резистентности процесса показано назначение глюкокортикоидов (бетаметазон). Наружно применяют кератолитические мази, УФО, ванны с отрубями, грязелечение. Прогноз с точки зрения дерматологической патологии благоприятен. Соматическая патология требует к себе постоянного внимания.
Болезнь Кирле
МКБ-10 коды
Описание
Болезнь Кирле. Дерматоз неясной этиологии, характеризующийся рецидивирующим течением с образованием в дерме околофолликулярных очагов гиперкератоза. Клинически проявляется высыпанием на разгибательных поверхностях рук и ног мелких папул грязно-красного цвета, покрытых роговыми чешуйками, способных к диссеминации. Снятие роговых чешуек открывает мокнущие поверхности с последующим исходом в неглубокий пигментированный рубчик. Диагноз ставится на основании анамнеза, клинической картины, наличия сопутствующей патологии, клинико-лабораторного обследования. Морфология неспецифична. Лечение направлено на радикальное удаление очагов гиперкератоза.
Дополнительные факты
Этиология заболевания неизвестна. Болезнь Кирле может возникнуть спонтанно, как результат обменных нарушений (синтеза витамина А, липидов), иметь генетическую составляющую (семейная история заболевания). В подавляющем большинстве случаев болезнь Кирле связывают с системными нарушениями (хроническая почечная и сердечно-сосудистая недостаточность, сахарный диабет, цирроз печени).
Единого мнения о патогенезе заболевания в дерматологии не существует. Одни исследователи считают заболевание кожными проявлениями почечной недостаточности, другие - папулёзной крапивницы. Приоритет принадлежит теории патологического ороговения. В норме клетки эпидермиса синтезируют белок кератин, регулирующий физиологический процесс ороговения. При болезни Кирле клетки эпидермиса начинают активно размножаться, продуцируя избыток кератина и ороговевая раньше срока. Граница между обычными и ороговевшими клетками под тяжестью «лишнего» кератина сползает в базальный слой эпидермиса. Возникает воспаление, нарушается целостность базальной мембраны, роговые массы проникают в дерму, замещая собой часть её клеток, которые вместе с кератином «всплывают» наружу, создавая гиперкератоз на поверхности кожи.
Клиническая картина
Первичным элементом является бурая папула (фолликулярный узелок) величиной с просо, желтовато-серебристого оттенка в центре (кератин). Локализуется сыпь чаще на конечностях, в локтевых и подколенных сгибах. Папулы трансформируются в бляшки, которые, сливаясь между собой, образуют эффлоресценции (локальные очаги высыпаний) с очагами некроза либо гиперкератоза в центре. Дерматоз обладает тенденцией к генерализации процесса (за исключением ладоней и подошв). При удалении роговых наслоений обнажается эрозия в форме неглубокой воронки, мокнущая, незначительно кровоточащая, оставляющая после себя пигментированный поверхностный рубчик. Исход самостоятельной инволюции элементов - атрофическое рубцевание.
Субъективные ощущения, связанные с кожными проявлениями, отсутствуют. В редких случаях пациента беспокоит зуд. Объективное состояние зависит от наличия сопутствующей патологии. Некоторые практикующие врачи-дерматологи считают, что возникновение проникающего кератоза - основной симптом грядущего системного заболевания. Дерматоз имеет хроническое течение, не исключены рецидивы. Возрастная и гендерная составляющие отсутствуют.
Из-за отсутствия специфической морфологии диагностика болезни Кирле проводится методом сравнения данной патологии с другими заболеваниями, симптомом которых является гиперкератоз. Клинически к болезни Кирле близка болезнь Дарье, но в её симптоматике отсутствуют поверхностные «воронки» на коже после удаления кератиновых наслоений, а в биоптате присутствует дискератоз, отсутствующий в гистологии болезни Кирле.
От бородавчатой формы красного плоского лишая болезнь Кирле отличается отсутствием зуда и первичными элементами: папулы с кератиновым «беретом» в центре (в отличие от папул с центральным вдавлением при красном плоском лишае). Красный волосяной лишай отличается от болезни Кирле наличием конусов Бенье - чёрных точечных папул - на тыле пальцев рук, бессимптомным снятием роговых корочек. Парапсориаз Муха‐Габермана различается морфологически (воспалительные изменения в эндотелии сосудистой стенки), а аллергический ангиит - симптомом пальпируемой пурпуры (геморрагическая, а не папулёзная сыпь).
Основное внимание при диагностике дерматоза уделяют наличию сопутствующей системной патологии. Проводят консилиум врачей в составе дерматолога, эндокринолога, нефролога, невролога, гастроэнтеролога, кардиолога; полное клинико-лабораторное обследование (ОАК, ОАМ, биохимия, специальные нагрузочные пробы с целью установления сохранности функций органа).
Лечение
Читайте также: