Синдром Ландау Клеффнера: причины, симптомы и лечение
Добавил пользователь Cypher Обновлено: 08.01.2026
Что такое Приобретенная афазия с эпилепсией (синдром Ландау - Клеффнера) -
Представляет собой регресс речевых навыков после периода нормального речевого развития. Основные черты - сенсомоторная афазия, эпилептические изменения ЭЭГ в виде мультифокальных спайков и комплексов спайк-волна (заинтересованы височные отделы мозга, чаще билатерально) и эпилептические припадки.
Что провоцирует / Причины Приобретенной афазии с эпилепсией (синдрома Ландау - Клеффнера):
В большинстве случаев неизвестна. Предположительно наличие энцефалитического процесса. У 12% детей с синдромом Ландау - Клеффнера в семейном анамнезе выявляются случаи эпилепсии. Инструментальные методы исследования (пневмоэнцефалография, КТ, артериография) не выявляют морфологических нарушений. Биопсия мозга и серологические исследования дают неоднозначные результаты и не позволяют подтвердить наличие специфической энцефалопатии.
Симптомы Приобретенной афазии с эпилепсией (синдрома Ландау - Клеффнера):
Типично начало в возрасте 3-7 лет, но может возникать раньше и позже. В начале заболевания наблюдается относительно медленно прогрессирующее нарушение понимания речи. С нейропсихологической точки зрения, развивается слуховая вербальная агнозия. В дальнейшем к нарушению понимания речи присоединяются расстройства экспрессивной речи. Спонтанная речь исчезает в течение нескольких недель или месяцев. Часто наблюдается полная утрата речи. Операциональная сторона мышления остается сохранной. У 50% детей с синдромом Ландау - Клеффнера выявляются расстройства поведения, в первую очередь гипердинамический синдром. Клинически эпилептические приступы проявляются только в 70% случаев. В 1/3 случаев отмечается единичный приступ или эпилептический статус в начале заболевания. После достижения 10-летнего возраста приступы наблюдаются только у 20% больных, а после 15 лет прекращаются. На ЭЭГ регистрируются множественные, билатеральные высокоамплитудные спайки и комплексы спайк-волна, наиболее выраженные в височных областях. С возрастом эпилептические проявления в ЭЭГ становятся менее заметными и к 15-16 годам исчезают у всех больных. В подростковом возрасте отмечается незначительное улучшение речи. Однако при сенсомоторной афазии речь полностью не восстанавливается. Прогноз восстановления речи зависит от возраста манифестации и времени начала противоэпилептической терапии и восстановительных логопедических занятий.
Большинство детей с данным расстройством чаще попадает в поле зрения клиницистов даже не по поводу припадков и, тем более, не речевых расстройств, а по поводу поведенческих нарушений - «расторможенность», гиперкинезы. Нейрофизиологи указывают, что ЭЭГ - это единственный патогномоничный критерий выявления синдрома на тех стадиях, когда правильное лечение еще может спасти речь, хотя прогноз неблагоприятный.
Лечение Приобретенной афазии с эпилепсией (синдрома Ландау - Клеффнера):
Предполагается положительный эффект от приема кортикостероидов в начале заболевания. В течение всего заболевания рекомендуется прием антиконвульсантов. Препаратами первого выбора являются карбамазепины (финлепсин), второго выбора - ламиктал (ламотриджин). Речевая терапия и семейная терапия рекомендованы в течение всего заболевания.
К каким докторам следует обращаться если у Вас Приобретенная афазия с эпилепсией (синдром Ландау - Клеффнера):
Вас что-то беспокоит? Вы хотите узнать более детальную информацию о Приобретенной афазии с эпилепсией (синдрома Ландау - Клеффнера), ее причинах, симптомах, методах лечения и профилактики, ходе течения болезни и соблюдении диеты после нее? Или же Вам необходим осмотр? Вы можете записаться на прием к доктору .
Синдром Клайнфельтера
Синдром Клайнфельтера - хромосомная патология, обусловленная наличием в мужском кариотипе одной или нескольких дополнительных женских половых хромосом. Синдром Клайнфельтера характеризуется первичным гипогонадизмом, маленькими размерами тестикул, бесплодием, гинекомастией, неглубоким снижением интеллекта. Решающая роль в диагностике синдрома Клайнфельтера принадлежит кариотипированию; также проводится анализ фенотипических признаков, определение полового хроматина, экскреции фолликулостимулирующего гормона с мочой, спермограмма и пр. Лечение при синдроме Клайнфельтера включает гормональную терапию, возможно - оперативную коррекцию гинекомастии, однако полное излечение синдрома невозможно.
Общие сведения
Синдром Клайнфельтера - дисомия или полисомия по женской половой хромосоме, при которой у лиц мужского пола имеется не менее двух Х-хромосом и одна Y-хромосома. Синдром Клайнфельтера встречается с частотой 1 случай на 850-1000 новорожденных мальчиков. Среди детей, страдающих олигофренией, распространенность синдрома Клайнфельтера составляет 1-2%. Синдром получил название по фамилии американского врача Гарри Клайнфельтера, впервые описавшего его в 1942 г. Кариотип таких больных с дополнительной Х-хромосомой был определен в 1959 г. Поскольку ведущим клиническим проявлением синдрома Клайнфельтера является первичный гипогонадизм, ведением таких пациентов занимаются специалисты в области эндокринологии и андрологии.
Причины синдрома Клайнфельтера
Как и в случае синдрома Дауна, хромосомная аберрация при синдроме Клайнфельтера связана с нерасхождением хромосом (в последнем случае - половых) в процессе мейоза либо нарушением деления зиготы. При этом значительно чаще (в 60%) мальчики с синдром Клайнфельтера получают лишнюю материнскую Х-хромосому, чем отцовскую.
Среди возможных причин подобного рода хромосомных аномалий называются вирусные инфекции, поздняя беременность, неполноценность регуляторных механизмов материнской и отцовской иммунной системы.
При наличии лишней X-хромосомы развивается аплазия эпителия яичек, их последующая гиалинизация и атрофия, что во взрослом возрасте сопровождается азооспермией и эндокринным бесплодием. Среди причин мужского бесплодия синдром Клайнфельтера составляет 10%, о чем всегда должны помнить специалисты в области репродуктивной медицины.
Наиболее частым цитогенетическим типом является полный вариант синдрома Клайнфельтера с кариотипом 47,ХХY. Реже встречается мозаицизм (46XY/47XXY; 46XX/47XXY), еще реже - полисомия 48,XXXY; 48,XXYY; 49,XXXXY и т. д. При мозаичном варианте (около 10% случаев) часть клеток имеет нормальный кариотип, поэтому мужчины с синдром Клайнфельтера могут иметь нормально развитые и функционирующие половые железы и сохранные репродуктивные способности.
Симптомы синдрома Клайнфельтера
Ребенок с синдромом Клайнфельтера рождается с нормальными росто-весовыми показателями, правильной дифференцировкой наружных гениталий, обычными размерами тестикул. В раннем возрасте у мальчиков с синдромом Клайнфельтера может отмечаться частая заболеваемость ОРВИ, бронхитом, пневмониями. Такие дети обычно отстают в моторном развитии (позднее начинают держать головку, сидеть, стоять, ходить), имеют задержку речевого развития. Уже в возрасте 5-8 лет мальчики с синдромом Клайнфельтера отличаются высоким ростом, диспропорциональным телосложением (длинными конечностями, высокой талией). В допубертатном возрасте может обнаруживаться одно или двусторонний крипторхизм.
Умственная отсталость умеренной степени, трудности установления контакта со сверстниками, нарушения поведения отмечаются у половины больных синдромом Клайнфельтера.
Отчетливые внешние признаки, свидетельствующие о наличии у ребенка синдрома Клайнфельтера, проявляются в препубертатном и пубертатном периодах развития. К ним относятся евнухоидный тип телосложения, позднее появление вторичных половых признаков, гипоплазия яичек, малый половой член, гинекомастия. В постпубертатном периоде онтогенеза наблюдается инволюция тестикул, сопровождающаяся потерей фертильности. При осмотре подростка с синдромом Клайнфельтера выявляется отсутствие или скудный рост волос на лице и в подмышечных впадинах, оволосение на лобке по женскому типу. У большинства больных присутствуют редкие поллюции, эрекция, сохранно половое влечение, однако из-за выраженного андрогенного дефицита в среднем к 30 годам происходит снижение либидо и развивается импотенция.
Синдрому Клайнфельтера часто сопутствуют аномалии скелета (деформации грудной клетки, остеопороз), нарушения прикуса, врожденные пороки сердца и др. Характерно преобладание ваготонических реакций: брадикардии, акроцианоза, потливости ладоней и стоп. Со стороны органа зрения нередко отмечается нистагм, астигматизм, птоз века.
Больные с синдромом Клайнфельтера предрасположены к развитию сопутствующих заболеваний: эпилепсии, рака молочной железы, сахарного диабета, ХОБЛ, желчнокаменной болезни, варикозного расширения вен, ожирения, гипертонической болезни, ИБС, ревматоидного артрита, острого миелоидного лейкоза. Могут отмечаться психические заболевания - маниакально-депрессивный психоз, шизофрения и др. Есть данные, подтверждающие склонность больных с синдромом Клайнфельтера к алкоголизму, наркомании и гомосексуализму.
Диагностика синдрома Клайнфельтера
Как и другие хромосомные аномалии, синдром Клайнфельтера у плода может быть обнаружен еще на этапе беременности при проведении инвазивной пренатальной диагностики (амниоцетеза, биопсии хориона или кордоцентеза с последующим анализом кариотипа или КФ-ПЦР).
Постнатальная диагностика синдрома Клайнфельтера проводится эндокринологами, андрологами и генетиками. При исследовании полового хроматина в клетках слизистой оболочки полости рта присутствуют тельца Бара, что является маркером синдрома Клайнфельтера. Другими характерными признаками служат особые изменения кожного рисунка на пальцах. Тем не менее, окончательный диагноз хромосомной аномалии может быть установлен только после исследования кариотипа.
УЗИ мошонки выявляет уменьшение объема яичек. При исследовании андрогенного профиля уровень тестостерона в крови больных синдромом Клайнфельтера понижен, однако при этом отмечается повышение уровня фолликулостимулирующего и лютеинизирующего гормонов. При анализе спермограммы выявляется олиго- или азооспермия. Морфологическое исследование материала, полученного путем биопсии яичек, выявляет гиалиноз семенных канальцев, гиперплазию клеток Лейдига, уменьшение числа клеток Сертоли, отсутствие сперматогенеза.
В течение жизни мужчины с синдромом Клайнфельтера могут обращаться к андрологу, сексологу, эндокринологу с проблемами бесплодия, импотенции, гинекомастии, остеопороза и др., однако нередко основное заболевание так и остается нераспознанным.
Лечение синдрома Клайнфельтера
Полностью излечиться от синдрома Клайнфельтера не представляется возможным. Тем не мене, все больные нуждаются в проведении симптоматической и патогенетической терапии. В детском возрасте необходима профилактика инфекционных заболеваний, закаливание, занятия ЛФК, коррекция нарушений речи с помощью логопеда.
С подросткового возраста пациентам с синдромом Клайнфельтера назначается пожизненная заместительная терапия половыми гормонами (внутримышечные инъекции тестостерон-пропионата, сустанона-250; сублингвальный прием метилтестостерона и др.). Ранняя и адекватная гормонотерапия препятствует атрофии яичек, способствует повышению полового влечения, развитию вторичных половых признаков. При резко выраженном увеличении молочных желез проводится операция по коррекции гинекомастии.
С целью повышения трудоспособности и социальной адаптации, предупреждения психопатизации личности и ее асоциальной направленности показана психотерапия.
Прогноз и профилактика синдрома Клайнфельтера
Пациенты с синдромом Клайнфельтера имеют нормальную продолжительность жизни, однако склонность к развитию хронических заболеваний может стать риск-фактором ранней смертности. Большинство больных с синдромом Клайнфельтера бесплодны; единственно возможным вариантом рождения детей в семьях, где партнер болен, является использование донорской спермы. Тем не менее, при мозаичной форме синдрома Клайнфельтера мужчины могут стать отцами самостоятельно или воспользовавшись вспомогательными репродуктивными технологиями (ЭКО).
Для оценки вероятности рождения ребенка с синдромом Клайнфельтера в процессе ведения беременности женщинам предлагается прохождение пренатального скрининга. Однако даже в случае получения положительных данных за наличие синдрома Клайнфельтера у плода настаивание на прерывании беременности со стороны акушера-гинеколога является недопустимым. Решение вопроса о целесообразности пролонгирования беременности должно приниматься родителями. При нормальном кариотипе родителей риск повторного появления ребенка с такой же хромосомной аномалией составляет не более 1%.
Диспансерное наблюдение больных с синдромом Клайнфельтера осуществляется эндокринологом.
Синдром Ландау-Клеффнера
Синдром Ландау-Клеффнера — сочетание прогрессирующей утраты речевого навыка с эпилептиформными изменениями электроэнцефалограммы, клинически проявляющимися в виде эпиприступов лишь у 70% пациентов. Диагностируется синдром Ландау-Клеффнера на основании клинических данных и обнаружения длительной эпилептиформной активности на ЭЭГ при условии исключения органической патологии мозга. В комплексную терапию включают противоэпилептические фармпрепараты, глюкокортикостероидные гормоны, логопедические занятия и нейропсихологическую коррекцию. Прогноз по эпилепсии благоприятный, по речевой дисфункции — серьезный.
Синдром Ландау-Клеффнера представляет собой симптомокомплекс, включающий постепенный регресс развитой в соответствии с возрастом речи и наличие эпилептиформной активности на ЭЭГ. Описан в 1957 году. Изменения ЭЭГ не всегда имеют клиническую выраженность: эпилептические пароксизмы отмечаются только у 70% больных. Однако наличие пароксизмального ЭЭГ-паттерна у 100% заболевших позволяет предположить, что именно эпилептическая активность головного мозга лежит в основе возникающих нарушений речи. В связи с этим синдром Ландау-Клеффнера носит синонимичное название «приобретенная эпилептическая афазия».
Заболеваемость наблюдается среди детей в возрастном периоде от 1,5 до 13 лет, наиболее часто в возрасте от 4 до 7 лет. Поскольку синдром Ландау-Клеффнера является редким заболеванием, точные данные о его распространенности отсутствуют. Исследование японских ученых, проведенное в конце ХХ века, показало, что у детей 5-14 лет синдром встречается с частотой 1 случай на 1 млн. чел. Среди заболевших наблюдается небольшое преобладание мальчиков (гендерное соотношение около 1,7:1). В половине случаев синдром Ландау-Клеффнера сопровождается нарушениями поведения, которые у некоторых пациентов выходят на первый план, что вынуждает их родных обращаться, прежде всего, к психологу или психиатру.
Причины
Этиология остается неизвестной. У 12% пациентов в семейном анамнезе выявляются случаи эпилепсии у родственников, однако синдром Ландау-Клеффнера не имеет прослеживаемый наследственный характер. Предполагается, что патология представляет собой дисфункцию речевой зоны коры мозга, обусловленную эпилептогенной активностью. В пользу этой гипотезы свидетельствует необычайная продолжительность эпилептиформной активности, регистрируемой на ЭЭГ, у многих пациентов носящей почти непрерывный характер всю фазу медленного сна. В редких случаях синдром является симптоматическим. В литературе описано его возникновение при астроцитоме головного мозга, локализующейся в височной доле, церебральном цистицеркозе, черепно-мозговой травме.
Симптомы синдрома Ландау-Клеффнера
До манифестации синдрома психомоторное и речевое развитие ребенка идет соответственно возрасту. Как правило, дебют заболевания происходит с нарушений восприятия обращенной к ребенку речи (сенсорной афазии). Родители замечают неадекватную реакцию ребенка на их слова. У 50% заболевших детей возникают эмоциональная лабильность, повышенная возбудимость, поведенческие расстройства (гиперактивность, агрессивность, тревожность, негативизм, замкнутость).
Прогрессирование речевых нарушений происходит в период от нескольких недель до месяцев, в отдельных случаях — за несколько дней. Сенсорная афазия достигает такой степени, что у ребенка отсутствует реакция на внешние звуки, хотя слух не нарушен. Со временем к сенсорной присоединяется моторная афазия — расстройство экспрессивной речи. В разговоре ребенок начинает употреблять только простые фразы, затем отдельные слова и, наконец, вообще утрачивает способность говорить — развивается мутизм.
Сопровождающие синдром Ландау-Клеффнера эпилептические пароксизмы представлены атипичными абсансами и парциальными моторными приступами (наиболее часто гемифасциальными или оральными). Могут отмечаться миоклонические и атонические пароксизмы, вторичная генерализация приступов. У большинства больных эпиприступы возникают в период отхода ко сну или просыпания. Наблюдаются достаточно редко. У 30% пациентов они отсутствуют, возможен единичный эпиприступ в анамнезе. Отмечается тенденция к исчезновению эпилептических пароксизмов по мере взросления ребенка. Так, в 10-летнем возрасте они наблюдаются только у 20% заболевших, а в 15-летнем отсутствуют практически у 100% пациентов.
Диагностика
Диагностика осуществляется совместными усилиями специалистов в области неврологии, логопедии, эпилептологии, психиатрии и педиатрии. Зачастую, особенно при отсутствии эпиприступов, диагностировать синдром Ландау-Клеффнера бывает достаточно трудно. При неврологическом и нейропсихологическом обследовании выявляется сенсомоторная афазия, изменения поведения. Для исключения кохлеарного неврита и других нарушений слуха, пациентов направляют на консультацию сурдолога, аудиометрию и исследование слуховых ВП.
При проведении ЭЭГ у всех больных выявляют эпилептиформные изменения. Патогномоничны высокие пик-волны, регистрируемые в височных областях и наиболее выраженные в фазу медленного сна. Последнее диктует необходимость проведения ЭЭГ сна, которая зачастую регистрирует электрический эпилептический статус медленного сна — постоянную диффузную эпилептическую активность в период медленноволнового сна. При отсутствии изменении на ЭЭГ сна рекомендуется длительное ЭЭГ-мониторирование.
МРТ и КТ головного мозга выявляют патологические изменения его морфологии только при симптоматическом характере синдрома. Данные ПЭТ головного мозга обычно указывают на одно- или двусторонние изменения метаболизма в височных областях мозга. Дифференцировать синдром Ландау-Клеффнера необходимо от аутизма, других видов эпилепсии у детей (синдрома Леннокса-Гасто), психических расстройств (шизофрении, психоза). Следует также исключить органическую церебральную патологию: опухоли головного мозга, цереброваскулярные нарушения, энцефалит, демиелинизирующие заболевания.
Лечение и прогноз синдрома Ландау-Клеффнера
Противоэпилептическая терапия является обязательной для всех пациентов вне зависимости от наличия эпиприступов. Она может быть назначена неврологом или эпилептологом. У пациентов без клинических проявлений эпилепсии препаратами выбора являются сукцинимиды или бензодиазепины (клобазам). При наличии эпилептических пароксизмов рекомендованы вальпроаты, топирамат, леветирацетам или их комбинации. В политерапии возможно сочетание вальпроатов с клобазамом. Использование в лечении карбамазепина противопоказано, поскольку клинические наблюдения показали, что в ряде случаев он способствовал усилению речевой дисфункции и учащению эпиприступов.
Параллельно с противоэпилептической терапией может проводиться лечение глюкокортикостероидами. Оно показано при отсутствии должного эффекта от использования антиконвульсантов (урежения приступов, купирования электрического эпистатуса на ЭЭГ сна). Фармпрепаратами выбора выступают дексаметазон, тетракозактид, преднизолон. Лечение начинают с внутримышечного введения и наращивания дозы. Затем постепенно снижают дозировку и переходят на пероральный прием поддерживающих доз. Наряду с медикаментозным лечением пациентам рекомендованы занятия с логопедом и нейропсихологическая коррекция, проводится психологическое консультирование родителей.
Синдром Ландау-Клеффнера не имеет однозначного прогноза. Относительно эпиприступов он благоприятный: у всех пациентов отмечается исчезновение пароксизмов к возрасту 15-16 лет. Речевые и поведенческие нарушения могут сохраняться на протяжении всей жизни пациента. Возможно некоторое восстановление речевых навыков, но у большинства пациентов взрослого возраста наблюдается выраженная речевая дисфункция. Наиболее часто грубые расстройства речи отмечаются при неадекватной начальной терапии синдрома вследствие трудностей с распознаванием его эпилептического генеза. Некоторые авторы указывают на неблагоприятный речевой прогноз при сохранении электрического эпистатуса медленного сна более 3-х лет.
Детские афазии
Детские афазии - это гетерогенная группа патологий ЦНС, которые проявляются частичной или полной потерей ранее присутствовавшей речи. Кроме того, они могут сопровождаться нарушениями чтения, письма, восприятия, счета, эмоциональными и поведенческими реакциями. Основа диагностики детских афазий - непосредственная оценка речевых и неречевых функций ЦНС, а также определение структурных и функциональных нарушений коры головного мозга при помощи КТ, МРТ и ЭЭГ. Лечение включает в себя прохождение курса специального восстановительного обучения и ликвидацию этиологического фактора (по возможности). При синдроме Ландау-Клеффнера также проводится противосудорожная терапия.
Детские афазии - это группа полиэтиологических нарушений центральной нервной системы, которые характеризуются частичной или тотальной потерей речевых функций у детей с ранее сформировавшейся речью. В педиатрии такие состояния встречаются редко - данные патологии больше распространены у взрослых. Суммарная встречаемость среди детей - менее 1%. Детские афазии чаще наблюдаются у мальчиков. В детском возрасте афазии проявляются менее разнообразно, чем у взрослых, поскольку речь у детей не столь развита. Чем младше ребенок - тем менее разнообразны клинические симптомы детской афазии. Также для пациентов детского возраста характерен быстрый регресс возникших симптомов - спустя несколько месяцев речевые функции могут полностью восстановиться.
Причины детских афазий
Детская афазия - это гетерогенное состояние. Он развивается в результате поражения речевых систем ЦНС в период сформированной речи. В большинстве случаев подобные состояния диагностируются на фоне черепно-мозговых травм и патологии сосудов, кровоснабжающих головной мозг - внутренней сонной или средней мозговой артерии. Среди ЧМТ ведущую роль играют открытые повреждения, сопровождающиеся потерей мозгового вещества. Закрытые травмы головного мозга провоцируют детские афазии значительно реже.
Также в роли этиологических факторов выступают опухоли, аневризмы, гематомы, абсцессы головного мозга, энцефалит. При синдроме Ландау-Клеффнера потеря речи возникает совместно с эпилептическими припадками. Точная этиология этой формы детской афазии не установлена. По мнению многих авторов, она может быть вызвана генетической или приобретенной структурной склонностью к эпилептиформной активности. Развитие приобретенной эпилептической афазии предположительно может провоцировать ранее перенесенный энцефалит.
Классификация детских афазий
Согласно Международной классификации болезней (МКБ-10) детские афазии можно разделить на две группы:
1. Детские афазии, возникшие в результате органического или структурного изменения коры головного мозга. Сюда относятся речевые нарушения вследствие опухолей, травм, патологии сосудов и др. В зависимости от места поражения и патогенетических механизмов эта группа разделяется на подгруппы, которые будут рассмотрены далее.
2. Синдром Ландау-Клеффнера или приобретенная эпилептическая детская афазия. В данном случае речевые нарушения возникают без органических патологий головного мозга, основой их развития является эпилептиформная активность.
Отдельно стоит выделить сочетание этих синдромов. Данное состояние развивается в тех ситуациях, когда на фоне новообразования, гематомы или других структурных изменений головного мозга появляются судорожные приступы, существенно утяжеляющие клиническую картину и стимулирующие прогрессирование детской афазии.
Симптомы детских афазий
Характерный возраст для детских афазий - 3-7 лет. Однако во многих случаях время начала заболевания зависит от того, в какой момент подействовал этиологический фактор - возникла гематома или произошла травма. В зависимости от речевых и неречевых симптомов, а также локализации поражения в педиатрии и логопедии выделяют следующие формы структурных детских афазий: акустико-гностическая или сенсорная, акустико-мнестическая, афферентная и эфферентная моторные, динамическая. Присутствующей у взрослых семантической формы в детском возрасте не наблюдается, поскольку в этом периоде еще не сформирована система символически-знакового обобщения сигналов.
Акустико-гностическая или сенсорная форма. Зона поражения - задняя 1/3 верхней темпоральной извилины левой половины головного мозга. Эта форма детской афазии возникает из-за нарушения акустического анализа и обработки звуков речи, что характеризуется поражением фонематического слуха. Клинически проявляется нарушением всех форм устной и письменной речи, чтения и устного счета, ритмическим воспроизведением. Также у таких детей наблюдается чрезмерная тревога и возбудимость, эмоциональная нестабильность.
Акустико-мнестическая афазия. Локализация поражения - средние и задние участки темпоральной области. Суть этой детской афазии - повышение тормозимости слуховых следов, приводящее к нарушению слуховой и речевой памяти. Также присутствует дефект зрительных и предметных образов-представлений. Такие дети не понимают подтекста, аллегорий, не могут называть предметы. Отмечается умеренное нарушение устной речи и ее восприятия. Может возникать повышенная активность и эмоциональная нестабильность, тревога.
Афферентная моторная афазия. Место поражения - нижние париетальные участки доминирующего полушария. Патогенетически основывается на нарушениях кинестетического восприятия. Основной признак - аномалии мелких артикуляционных движений губ и языка. Такие дети или неспособны к экспрессивной речи или имеют большое количество литеральных парафазий. Непроизвольная и автоматизированная (песни, стихи) речь, письмо и чтение сохранены.
Эфферентная моторная форма детской афазии. При этой форме поражаются задние лобные участки. Страдает инертность сформировавшихся стереотипов, что проявляется персеверациями. Способность к устным высказываниям минимальная или полностью отсутствует. Могут сохраняться отдельные звуки, автоматизированная речь. Наблюдается нарушение чтения, письма, апраксия.
Динамическая афазия. Крайне редкая форма в педиатрии, может наблюдаться у детей старших возрастных групп. Локализация патологического очага - задние лобные отделы. Патогенетически данная разновидность заболевания обусловлена дефектами внутренней речи, нарушением сукцессивной организации высказывания. Проявляется расстройством продуктивной речи, неспособностью активного общения - нормальные предложения заменяются стереотипами или шаблонами, глаголы полностью отсутствуют. Больные с данной формой детской афазии почти никогда ничего не спрашивают и не вступают в диалоги, но охотно отвечают на поставленные вопросы. Чтение и письмо могут быть сохранены.
Синдром Ландау-Клеффнера. Локализация пароксизмальной активности может быть разной, наиболее часто поражаются височные области. Потеря речи может происходить как резко (чаще всего), так и постепенно, на протяжении нескольких месяцев. Также теряется способность к восприятию речи, возможны нарушения поведения и эмоциональной сферы - гипервозбудимость, эмоциональная лабильность. Характерная черта данной формы детской афазии - судорожные припадки, которые, однако, наблюдаются не у всех больных.
Диагностика детских афазий
Диагностика детских афазий включает в себя сбор анамнестических данных, объективный осмотр и общение с ребенком, лабораторные и инструментальные методы исследования. При выяснении анамнеза у родителей устанавливают этиологические факторы (травмы, сопутствующие заболевания), а также динамику симптомов от возникновения до момента обследования. При объективном осмотре ребенка обращают внимание на возможные неврологические расстройства, которые могут указывать на характер поражения головного мозга. При общении с ребенком педиатр или детский психиатр оценивают способность пациента к устной речи, письму, чтению и счету, другие речевые и неречевые функции, что позволяет определить форму детской афазии.
Лабораторные анализы, как правило, малоинформативны. В некоторых случаях они могут указывать на возможную этиологию (лейкоцитоз со сдвигом формулы влево при абсцессе и т. д.). Среди инструментальных методов применяются ЭЭГ, рентгенография черепа, КТ и МРТ. ЭЭГ используется для оценки активности того или иного участка коры головного мозга, а при синдроме Ландау-Клеффнера - для выявления эпилептиформных припадков. Рентгенография и КТ черепа показаны при травмах головы, поскольку позволяют определить состояние костей черепа, диагностировать их переломы. МРТ головного мозга - наиболее информативный метод оценки структуры ЦНС. Она почти всегда дает возможность установить этиологический фактор детской афазии и распространенность патологического процесса и определить дальнейшею терапевтическую тактику.
Лечение детских афазий
Лечение детских афазий подразумевает специальное восстановительное обучение под контролем логопеда. Его суть заключается в активации компенсаторных механизмов головного мозга при помощи прямых и непрямых способов. Прямые методы показаны на ранних сроках, основаны на использовании активации резервных способностей клеток. Непрямые или обходные методы компенсируют утраченные функции за счет функциональных перестроек. В зависимости от ситуации и формы детской афазии в качестве материалов для обучения применяются тексты, карточки, картинки, компьютерные программы, различные предметы, однако ведущую роль играют упражнения с логопедом.
Эффективность восстановительного обучения зависит от целого ряда факторов: формы и длительности заболевания, тяжести поражения ЦНС, этиологического фактора, возраста ребенка и момента начала терапии. Детский мозг очень пластичен, поэтому при легких формах зачастую наблюдается быстрый регресс симптомов. При структурных детских афазиях легкой степени тяжести способность к коммуникации возвращается на протяжении 3-5 недель, при средней - через 1-6 месяцев. При синдроме Ландау-Клеффнера помимо обучения может использоваться соответствующее медикаментозное противосудорожное лечение. Однако даже на фоне позитивной динамики возрастной нормы достичь удается достаточно редко.
Прогноз и профилактика детских афазий
Прогноз при детской афазии зачастую благоприятный. При ранней диагностике и своевременно начатом восстановительном обучении на протяжении первых нескольких недель или месяцев удается добиться быстрого регресса патологии. При тяжелых формах поражения ЦНС или синдроме Ландау-Клеффнера прогноз сомнительный. Прогностически неблагоприятным признаком считается отсутствие позитивной динамики на протяжении первых нескольких недель. Специфической профилактики детских афазий не существует. Неспецифические меры подразумевают исключение всех возможных этиологических факторов: раннюю диагностику и лечение фоновых заболеваний ЦНС и сосудов, которые могут вызвать ишемию коры головного мозга, минимизацию риска травм головы.
Синдром Ландау — Клеффнера - симптомы и лечение
Что такое синдром Ландау — Клеффнера? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гладун Ксении Викторовны, невролога со стажем в 9 лет.
Над статьей доктора Гладун Ксении Викторовны работали литературный редактор Вера Васина , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Ландау — Клеффнера (ЛКС) — редкое заболевание, которое проявляется приступами эпилепсии с постепенной или внезапной регрессией речевого развития. Нарушение выражается в афазии — неспособности понять и использовать речь. Точные причины ЛКС неизвестны. До описания синдрома Уильямом Ландау и Фрэнком Клеффнером в 1957 году снижение и/или утрата речи на фоне судорог не выделялось в отдельное заболевание. Патологию относили к проявлению эпилепсии и называли "приобретённой эпилептической афазией".
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Ландау — Клеффнера
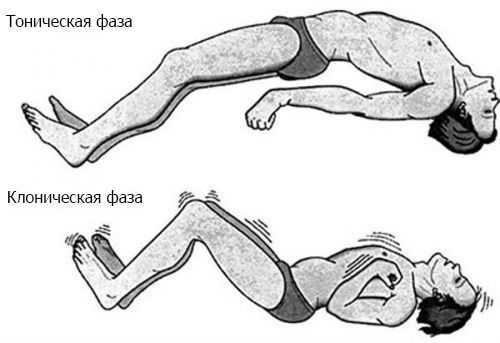
Как правило, дети с ЛКС развиваются нормально, но затем без видимой причины теряют речевые навыки. Многие пациенты страдают от судорог, но у некоторых болезнь протекает без них. Наиболее распространённым типом судорог, наблюдаемых при синдроме Ландау — Клеффнера, является фокальный моторный приступ. Он проявляется сокращением одной мышцы или группы мышц, например в шее или руке. Очаговые приступы могут прогрессировать, затрагивая оба полушария мозга, и/или распространяясь на соседние области. Когда это происходит, клиническая картина включает тонико-клонические приступы — резкие сокращения мышц в начале приступа с последующими их ритмическими сокращениями [1] .
ЛКС сложен для диагностики. При постановке диагноза опираются на следующие симптомы:
- нормальное развитие до начала заболевания;
- ночные вздрагивания, судороги, приступы (могут отсутствовать или быть однократными);
- отсроченный и прогрессирующий дефицит высших психических функций, соответствующих возрасту ребёнка, с акцентом на речевую функцию.
Если электроэнцефалограмма показывает аномалии в лобных отделах головного мозга, то пациент имеет поведенческие проблемы, такие как синдром дефицита внимания и гиперактивности (СДВГ). Нарушения проявляются в снижении концентрации, гиперактивности, агрессии или импульсивности. В старшем возрасте в поведении могут преобладать аутистические черты [19] .
Патогенез синдрома Ландау — Клеффнера
Патогенез Синдрома Ландау — Клеффнера полностью не изучен. Фундаментальные научные исследования находятся в стадии разработки, многие авторы связывают развитие заболевания с нейровоспалением. Причины воспаления в нервной ткани при ЛКС не выявлены, но есть предположение о воздействии аутоиммунных факторов [15] . Исследования показали, что у детей с ЛКС наблюдается повышенный уровень аутоантител, направленных против нейротрофического фактора мозга (белка, стимулирующего и поддерживающего развитие нейронов) [16] . Потенциальное вовлечение иммунной системы и/или воспалительные каскады реакций при ЛКС объясняют эффективность гормональной терапии [17] .
Предполагается, что причиной ЛКС являются врождённые пороки развития головного мозга, генетические нарушения или метаболические состояния. Точная ассоциация ЛКС с генетическим фактором неизвестна, но за последнее время обнаружены новые генетические мутации, характерные для данного заболевания. В основном доказана связь с геном GRIN2A [11] . Ген кодирует белок GluN2A (ранее известный как NR2A). GluN2A содержится в нервных клетках головного и спинного мозга, в том числе в областях мозга, участвующих в формировании речи, и является одним из компонентов NMDA-рецепторов. Эти рецепторы регулируют нейрональную возбудимость и синаптическую пластичность [13] . В подтверждение гипотезы исследователи выявили троих неродственных пациентов с различными делециями шестнадцатой хромосомы (утраты её участка), включая ген GRIN2A.
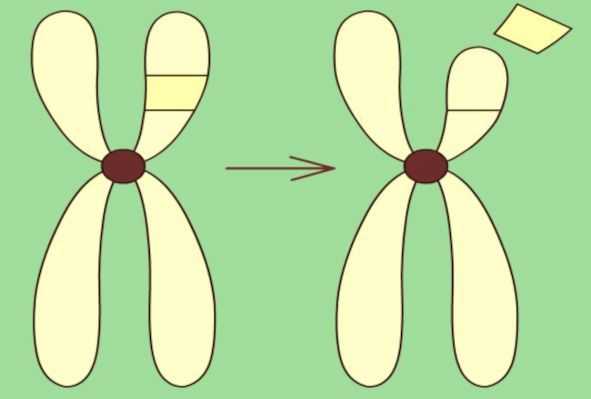
Больные страдали ранней фокальной эпилепсией, тяжёлой умственной нетрудоспособностью, отсутствием речи или задержкой речевого развития [11] . Помимо гена GRIN2A, изучается ряд дополнительных генов-кандидатов: RELN, BSN, EPHB2 и NID2 предположительно ассоциированных с ЛКС [20] [12] .
Классификация и стадии развития синдрома Ландау — Клеффнера
Диагноз ЛКС в соответствии с МКБ-10 относят в отдел "Специфические расстройства развития речи и языка", рубрику (F80). Диагноз звучит так: "Приобретённая афазия с эпилепсией [Ландау — Клефнера]" (F80.3).
Специальная группа по классификации Международной лиги против эпилепсии предложила группу синдромов, называемых эпилептическими энцефалопатиями, которые включают эпилепсию с непрерывным всплеском во время медленного сна и синдром Ландау — Клеффнера.
Согласно данным, размещённым Международной противоэпилептической лигой от 2017 года, эпилепсии подразделяют на следующие виды [28] :
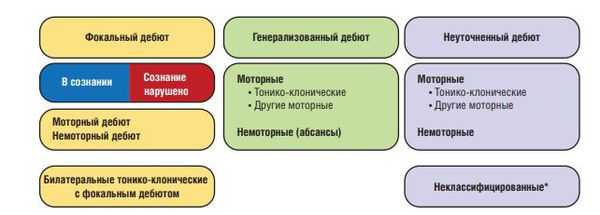
Фокальные приступы возникают в сетевых структурах, ограниченных одним полушарием. Они могут быть локализованы, либо же распространяться на соседние зоны или другое полушарие мозга. Первично-генерализованные приступы возникают одномоментно, с быстрым вовлечением сетевых структур обоих полушарий. Немоторные приступы могут проявляться вегетативными симптомами, заторможенностью поведенческих реакций, когнитивными, эмоциональными и сенсорными нарушениями.
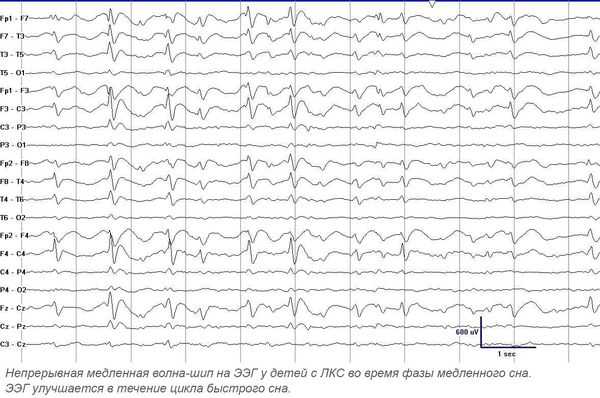
Утверждённой классификации по данным ЭЭГ нет. Изменения на ЭЭГ при ЛКС имеют чёткую зависимость от возраста: возникают в 3-9 лет и, как правило, постепенно исчезают к 13-15 годам [1] [7] [8] . Продолжительность максимально выраженных диффузных ЭЭГ-аномалий составляет в среднем от 1 до 5 лет. Обычно максимальная выраженность диффузной эпилептиформной активности на ЭЭГ у больных ЛКС наблюдается в возрасте 7-8 лет [7] .
Осложнения синдрома Ландау — Клеффнера
Синдром Ландау — Клеффнера может привести к развитию энцефалопатии. Энцефалопатия — это заболевание, проявляющееся дистрофией нервной ткани в верхних слоях головного мозга или в коре больших полушарий с постепенным снижением функции коры. Злокачественное течение энцефалопатии обусловлено тем, в каком возрасте началась эпилепсия — ранний дебют заболевания имеет более серьёзные последствия. Эпилепсия младенчества с мигрирующими очаговыми припадками является крайне тяжёлым синдромом развития.
Важно отметить, что клинические проявления (афазия) не зависят от тяжести и частоты судорог. У некоторых детей наблюдается тяжёлая афазия с полной потерей речи и её понимания, несмотря на отсутствие судорог. Таким образом, эпилептическая активность, а не судороги, вероятно, коррелирует с нарушением языка и его степенью [5] .
ЛКС может вызывать изменение настроения, беспокойство и депрессию, нарушения сна, ухудшение рабочей памяти (но не долговременной), гиперчувствительность к звукам [21] [22] .
Следует учитывать дозозависимый эффект и взаимодействие противоэпилептических препаратов с другими медикаментами для предотвращения побочного действия лекарственных средств. Эффективность противоэпилептических препаратов измеряется, прежде всего, по его влиянию на частоту приступов, а не по воздействию на картину нарушения на ЭЭГ.
Диагностика синдрома Ландау — Клеффнера
Синдром Ландау — Клеффнера требует привлечения различных специалистов: педиатра, нейропсихолога, детского психиатра, психолога, логопеда, эрготерапевта (специалиста по восстановлению социальных, бытовых, рабочих, функциональных и двигательных навыков), отоларинголога.
Основной метод диагностики — электроэнцефалография (ЭЭГ). Повышенная эпилептиформная активность может быть подтверждена длительным ЭЭГ видео-мониторингом (суточным или ночным). Эпилептиформная активность представляет собой электрические колебания головного мозга в виде острых волн и пиков. Она значительно (более чем на 50 %) отличается от фоновой активности и, как правило, обнаруживается на ЭЭГ у лиц, страдающих эпилепсией. Ночной мониторинг выполняется в виде непрерывной регистрации электроэнцефалограммы в течение ночи. При ЛКС в период сна происходит увеличение патологической активности — появление непрерывного всплеска и пик-волновой активности во время медленного сна, в основном с частотой 1,5-2,5 Гц. В 70-80 % случаев ЛКС сопровождают моторные эпилептические припадки.
Другим методом диагностики является магниторезонансная томография (МРТ). Описаны случаи уменьшения объёма областей мозга, ответственных за развитие речи. Эти данные, однако, не несут диагностической пользы, но нужны для исключения структурных поражений, таких как опухоли головного мозга [4] .
Дифференциальный диагноз при ЛКС включает в себя следующие заболевания:
- Эпилептический синдром с усилением эпилептиформной активности во время сна — по сравнению с ЛКС отсутствуют речевые нарушения.
- Доброкачественная затылочная эпилепсия детского возраста с ранним дебютом (синдром Панайотопулоса) — также отсутствуют речевые нарушения.
- Доброкачественная эпилепсия с центро-темпоральными спайками (роландическая эпилепсия) — отсутствуют речевые нарушения.
- Детская затылочная эпилепсия с дебютом в старшем возрасте (тип Гасто).
- Синдромом Леннокса-Гасто — крайне редки моторные приступы.
- Синдром Ретта — более раннее начало. .
- Дефекты слуха.
Диагностика степени тяжести энцефалопатии проводится при помощи ЭЭГ и проявляется различными аномалиями электроэнцефалограммы в виде медленноволновой активности чаще всего вокруг височно-теменных областей головного мозга.
Лечение синдрома Ландау — Клеффнера
В большинстве случаев для лечения используется противоэпилептическая (ПЭП) или стероидная терапия. Эффект после фармакотерапии является переменным и непредсказуемым. В исследовании Marescaux et al. (1990) сообщается, что использование противосудорожных средств обычно не приводит к улучшению языковых способностей. Применение высоких доз гормональной терапии кортикостероидов может улучшить общее состояние детей, но речевые и интеллектуальные нарушения при этом сохраняются [3] .
Есть исследования о более длительном эффекте от терапии кортикостероидов, в сравнении с терапией ПЭП [6] . Кортикостероиды (например, преднизон перорально 1 мг / кг / день в течение 6 месяцев или преднизолон перорально 2 мг / кг / день в течение не менее трёх месяцев перед постепенным снижением) могут быть полезны для улучшения и/или стабилизации речевой функции, когнитивных и поведенческих навыков [23] . Их применение в сочетании с бензодиазепинами (БДЗ) рекомендуется при эпилептической активности и языковых нарушениях, сохраняющихся более 10 месяцев, несмотря на терапию [24] .
При тяжёлом течении применяют хирургическое лечение:
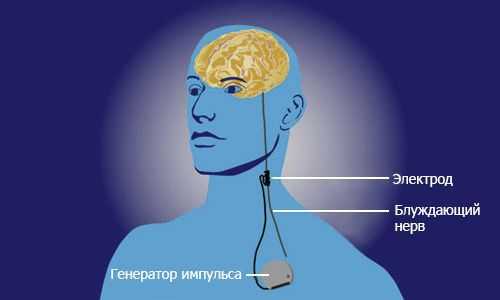
- Стимуляция блуждающего нерва. Операция заключается в том, что нерв, по которому импульсы поступают в головной мозг, окутывается электродом. Через него в головной мозг из стимулятора, вшиваемого под кожу, посылаются противоэпилептические стимулы. Это предупреждает развитие судорожного припадка. Методика применяется с 1990 года.
- Метод множественного субпиального транскортикального рассечения. Данный метод предназначен для устранения способности кортикальной ткани генерировать патологическую активность нейронов или субклиническую эпилептиформную активность. При этом сохраняются корковые функции оперируемых областей. Предварительно проводится трепанация черепа в проекции оперируемой области под общим наркозом, эффект достигается благодаря рассечению и, следовательно, предотвращению распространения пароксизмальной активности на соседние области головного мозга.
После операции большое значение имеет речевая реабилитация, включающая как занятия с логопедом, так и специализированный массаж и артикуляционную гимнастику. Методы логопедии заметно улучшают речевые способности большинства людей с ЛКС, однако зачастую в полном объёме речь не восстанавливается. Результаты речевой реабилитации зависят от многих факторов: уровня манифестации заболевания, стратегии гормональной терапии, начала и интенсивности реабилитационных мероприятий.
Спорным методом лечения является кетогенная диета. Кетогенная диета (КД) используется с 1920-х годов для терапии резистентных к лечению видов эпилепсии [14] . При КД пациент потребляет большое количество жиров (90 %) и низкое содержание белков и углеводов. Данные некоторых исследователей показывают, что индивидуально подобранная КД является хорошей альтернативой нехирургическому и после хирургическому вмешательству фармакорезистентных пациентов с эпилепсией [10] . Однако другие исследователи отмечают нехватку клинических данных, чтобы можно было сделать такие выводы [14] .
При лечении заболевания требуется наблюдение клинического фармаколога. Специалист поощряет соблюдение правил приёма лекарств, проверяет взаимодействие медикаментов и предупреждает врача и пациента о препаратах, которые могут усугубить состояние. Специалисты по неврологии могут помочь в мониторинге лечения, а также в консультировании пациентов и/или родителей.
Кроме того, важную роли играет социальная служба. Социальный работник должен помочь пациенту обеспечить дома адекватные для восстановления условия и доступ к вспомогательным услугам.
Прогноз. Профилактика
Синдром редкий, поэтому точного прогноза нет. Патологию считают доброкачественной с точки зрения развития эпилептической энцефалопатии. Прогноз варьируется в основном в отношении афазии. У детей может наблюдаться постепенное снижение внимания, памяти, мышления и общей когнитивной функции через 1-2 года после начала судорог. Результаты послеоперационного лечения в раннем детском возрасте показывают, что улучшение функции речи чаще всего наблюдается через годы после операции. У некоторых детей речевые нарушения могут сохраниться и со временем приобрести более тяжёлое течение. У других пациентов возможно восстановление большей части речевых навыков, хотя реабилитация занимает длительное время. В некоторых случаях может произойти ремиссия с последующим рецидивом.
Прогноз улучшается, если расстройство наступило после шести лет, а также при ранней реабилитации и сопровождении логопеда. Судороги обычно исчезают в зрелом возрасте, к тому времени эпилептическая активность на ЭЭГ также снижается.
Поскольку заболевание связано с генетическими нарушениями, его профилактики на данный момент нет. При высоком генетическом риске во время планирования беременности обязательна консультация врача-генетика.
Читайте также:
- Электрическая ось и электрическая позиция сердца
- Предоперационная подготовка при туберкулезе. Комбинации препаратов при устойчивом туберкулезе
- Электрическое оборудование для лапароскопии. Методы окклюзии сосудов
- Прогноз хронического лимфолейкоза
- Влияние адреналина на сосуды. Применение тиреоидных гормонов в лечении атеросклероза