Синдром Лаверье (Laverie) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 01.02.2026
К сожалению, до сих пор есть заболевания, которые за считанные часы доводят, казалось бы, совершенно здорового человека до критического состояния и имеют крайне неблагоприятный прогноз.Одним из таких заболеваний является синдром Лайелла (L51.2 по МКБ-10).
В СМИ широко обсуждался скандальный случай смерти от данной патологии — гибель в июле 2003 года журналиста, депутата Государственной Думы третьего созыва Юрия Щекочихина.
Свидетели отмечали необъяснимое резкое ухудшение его состояния: «за две недели превратился в глубокого старика, волосы выпадали клоками, с тела сошла кожа, практически вся, один за другим отказывали внутренние органы» («Новая газета», выпуск № 20 от 24 марта 2008 г.).
По факту смерти депутата было заведено уголовное дело по статье 105 УК РФ ч. 1 — «Убийство». Родственники и подхватившая ажиотаж пресса твердили об отравлении, в частности таллием. Семь месяцев проводились соответствующие экспертизы, эксгумировали тело депутата. Лишь к марту 2004 года были установлены обстоятельства стремительного ухудшения состояния Щекочихина. Диагноз и причина смерти — синдром Лайелла, острая сердечно-сосудистая недостаточность.
Из истории синдрома
Лайелл заменил принятое ранее обозначение «острый пемфигус» на «токсический эпидермальный некролиз» (ТЭН).Дело в том, что у всех пациентов отмечалась сыпь, «имеющая сходство с ошпариванием кожи объективно и субъективно». Сыпь и послужила основным диагностическим признаком для выделения новой нозологической единицы. Термин «токсический» Лайелл ввел, полагая, что заболевание вызывается токсемией — циркулирующим в организме определенным токсином. «Некролиз» — медицинский неологизм, придуманный самим Лайеллом, скомбинировавшим в нем основной клинический признак — «эпидермолиз» и гистопатологический признак — «некроз».
Также Лайелл отметил тяжесть поражения слизистых оболочек и необычную слабость воспалительных процессов в дерме — «дермальное безмолвие».
В научном мире заболевание получило имя своего первооткрывателя еще при жизни Лайелла — в 60‑е годы. Однако сам дерматолог всегда использовал обозначение ТЭН.
В 1967 году, проведя целевой опрос коллег по всей Великобритании, Лайелл представил самое обширное на сегодняшний день обобщение по ТЭН — 128 случаев заболевания. В том же году он описал 4 формы ТЭН, соответствующие определенной этиологии: стафилококковая, лекарственная, смешанная и идиопатическая.
«Некролиз» — медицинский неологизм, придуманный самим Лайеллом, скомбинировавшим в нем основной клинический признак — «эпидермолиз» и гистопатологический признак — «некроз».
В настоящее время стафилококковая форма синдрома Лайелла выделена в отдельную нозологическую единицу: стафилококковый синдром ошпаренной кожи (staphylococcal scalded skin syndrome, SSSS, L00 по МКБ-10).
Эпидемиология
С 70‑х годов в медицине ведется ежегодный учет распространенности синдрома Лайелла. По данным разных авторов, показатели колеблются в пределах 0,4-1,3 случая на 1 млн населения в год. Показатели смертности составляют 20-60 % от числа заболевших, в зависимости от состояния системы здравоохранения региона. Таким образом число смертей от данной патологии по всему миру за год исчисляется тысячами. Среди аллергических реакций немедленного типа по показателю смертности синдром Лайелла уступает лишь анафилактическому шоку. В общей структуре лекарственных аллергий доля синдрома Лайелла составляет 0,3 %. Чаще болеют женщины: соотношение с пациентами-мужчинами 1,5:1. Прямой зависимости риска заболевания от возраста не выявлено. Группой риска считаются пациенты, ранее перенесшие синдром Лайелла, особенно в детском возрасте.
Самая распространенная форма синдрома — лекарственная, до 80 % всех случаев.После лекарственной, второй по частоте причиной выступают злокачественные новообразования (особенно лимфомы). Частота идиопатических случаев синдрома составляет 5-10 %.
У детей Лайелла синдром чаще — до 60 % случаев — имеет инфекционную этиологию. Особая группа риска проявления болезни среди детей — это перенесшие в раннем возрасте ОРВИ и прошедшие в связи с этим курс лечения НПВП.
Этиология
Дискуссии об этиологии и патогенезе синдрома Лайелла продолжаются. Наиболее изучена лекарственная форма заболевания, которая развивается из‑за нарушения способности организма к обезвреживанию реактивных промежуточных лекарственных метаболитов. Данные метаболиты взаимодействуют с тканями, в результате формируется антигенный комплекс, иммунный ответ на который и инициирует развитие болезни.
Фактически доказана и генетическая предрасположенность к данной патологии, в частности у лиц с рядом антигенов комплекса гистологической совместимости HLA: A2, А29, В12, В27, DR7. Наличие в организме хронических очагов инфекции (синусит, тонзиллит, холецистит и т. п.), приводящих к снижению иммунитета, — увеличивает риск заболевания. Особую группу риска составляют ВИЧ-инфицированные пациенты: у них риск развития синдрома Лайелла в 1000 раз выше, чем в общей популяции.
Диагностика
Для дифференциальной и лабораторной диагностики провокационные пробы не проводятся, поскольку высок риск неконтролируемых осложнений. Наиболее распространены внеорганизменные диагностические пробы, основанные на реакциях клеток крови пациента на сенсибилизировавшее организм вещество. К ним относятся: тест дегрануляции базофилов по Шелли, реакция агломерации лейкоцитов по Флеку, реакция бластной трансформации лимфоцитов, гемолитические тесты.
У ВИЧ-инфицированных пациентов риск развития синдрома Лайелла в 1000 раз выше, чем в общей популяции.
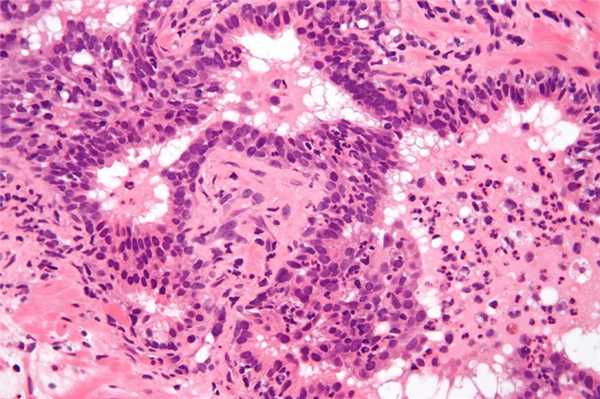
Апоптоз кератиноцитов — один из первых тканевых морфологических признаков синдрома Лайелла. Всё большее распространение приобретает пункционная биопсия с использованием замороженных срезов кожи. При этом выявляются отсутствие типичных акантолитических клеток, тотальный эпидермальный некролиз, под- и внутриэпидермальные пузыри.
Течение
По характеру течения выделяют три варианта клиники синдрома Лайелла:
- Молниеносная форма — до 10 % всех случаев. Развивается в течение нескольких часов. Этиология: идиопатическая или лекарственная. Кожные поражения за сутки охватывают до 90 % поверхности тела. Нарушение сознания вплоть до коматозного. Острая почечная недостаточность — анурия. Ввиду того, что эти пациенты в большинстве своем не доживают до госпитализации или поступают уже в терминальном состоянии, летальный исход составляет 95% в течение 2-3 суток. На вскрытии внутренние органы обычно интактны.
- Острая форма — 50-60 % случаев. Поражение кожи и слизистых проходит весь спектр созревания: от высыпаний до некролиза. Площадь некролизированных поверхностей может достигать 70 % поверхности тела. Заболевание длится от 7 до 20 суток. Начиная с 3-4 суток появляются симптомы почечной, печеночной, сердечно-сосудистой, легочной недостаточности, инфекционные осложнения — прежде всего пульмональные и инфекции мочеполовой системы, а при прогрессировании заболевания — сепсис. Летальность достигает 60 %.
- Благоприятное течение (сглаженная форма) — частота до 30 % случаев. Поражение кожных покровов и слизистых оболочек не превышает 50 % поверхности тела. Клинические проявления достигают пика на 5-6 сутки заболевания. Затем, в течение 3-6 недель, наблюдается улучшение состояния пациента до полного восстановления здоровья.
У подавляющего числа пациентов — более 90 % — присутствуют эрозивные изменения слизистых оболочек. Типичны жалобы на болезненность по ходу уретры при мочеиспускании и светобоязнь.
Характерен положительный симптом Никольского: отслойка эпидермиса на внешне неизмененной коже при скользящем надавливании и отслойка околопузырного эпидермиса при потягивании за обрывок пузырной покрышки. При особо тяжелых формах наблюдается тотальная отслойка эпидермиса при трении по всей поверхности тела пациента.
Синдром Стивена — Джонсона или синдром Лайелла?
Один из главных прогностических факторов при синдроме Лайелла — площадь некролизированной поверхности. Поэтому очень важна правильная оценка ее протяженности. Для этого используются те же правила, что и в комбустиологии: правило «девятки» — когда руки, грудь и живот считаются по 9 % поверхности тела, а спина и ноги — по 18 %; и правило «однопроцентной ладони». Для более точной оценки рекомендуется измерять площадь отделенного и отделяемого эпидермиса не только на участках с выраженными эритематозными изменениями, но и на отдаленных от «эпицентра» участках тела.В зависимости от площади пораженной некролизом поверхности выделяют клинические отличия синдрома Лайелла от синдрома Стивена — Джонсона.
На протяжении 30 лет — с конца 70‑х — кортикостероиды считались обязательным звеном в терапии синдрома Лайелла. В последние годы появились критические замечания по поводу их эффективности: в частности, указывается, что при их использовании возрастает риск септических осложнений.
В последние годы набирает популярность теория, что эти синдромы являются двумя вариантами тяжелого исхода эпидермолитической кожной реакции на медикаменты и отличаются лишь степенью отслойки кожного покрова.
Системная терапия
К сожалению, единых клинических рекомендаций при диагностированном синдроме Лайелла не существует. Это связано прежде всего с полиорганностью поражений, что предполагает в первую очередь разнонаправленную посимптомную терапию.Для лечения синдрома Лайелла правило «отмены последнего» назначенного медикамента чаще всего неэффективно, так как время проявления заболевания варьирует от нескольких часов до нескольких суток. Поэтому рекомендуется отмена всех ранее назначенных медикаментов и экстренная симптоматическая терапия. Подтверждено, что быстрая и тотальная отмена всех медикаментов при невозможности четкого выявления определенного аллергена — улучшает прогноз заболевания на 30 %. В рамках специфического лечения аллергического синдрома Лайелла большинством исследователей однозначно рекомендованы:
- экстренная, желательно в первые же часы заболевания, каскадная плазмофильтрация (плазмаферез), позволяющая удалить до 80 % плазмы пациента. При технической невозможности плазмафереза рекомендована гемосорбция;
- массивная системная кортикостероидная терапия. Рекомендуемые схемы кортикостероидов варьируют от 3-4 мг/кг массы тела в первые сутки заболевания до полного запрета при ухудшении состояния пациента;
- внутривенные иммуноглобулины. Ежесуточная доза начиная с первого дня заболевания варьируется от 0,2 до 0,75 г/кг массы тела пациента. Длительность цикла — от 4 до 12 дней. Эффективность иммуноглобулинов обусловлена содержанием в них естественных анти-Fas-антител, регулирующих апоптоз и пролиферацию в интактных и трансформированных лимфоидных клетках. Они снижают частоту бактериальных осложнений и предотвращают прогрессирование синдрома;
- гипербарическая оксигенация при отсутствии в легких пациента абсцессов, каверн и свободной проходимости дыхательных путей.
Лечение поражений кожного покрова и слизистых проводят по принципам терапии ожогов. Соблюдается режим парентерального питания. Коррекция инфузий осуществляется в зависимости от состояния пациента.
В период нахождения в стационаре обязательны регулярные — не реже одного раза каждые 72 часа — осмотры урологом, окулистом, стоматологом, отоларингологом, чтобы вовремя выявить и назначить соответствующую терапию при поражении слизистых оболочек.
Прогноз
С 2011 года на Западе распространена SCORTEN шкала оценки тяжести синдрома Лайелла. В ней учитываются следующие прогностические факторы:
- возраст пациента > 40 лет;
- ЧСС > 120 уд. в мин.;
- наличие сопутствующего злокачественного онкологического заболевания;
- площадь пораженной поверхности тела > 10 %;
- уровень мочевины крови > 10 ммоль/л;
- уровень бикарбонатов плазмы 14 ммоль/л.
Наличие каждого фактора увеличивает риск летального исхода. Так, примерный риск смерти составляет: при наличии 1 фактора — до 3,2 %; 2‑х факторов — 12,1 %; 3‑х факторов — 35,3 %; 4‑х факторов — 58,3 %; 5 и более факторов — 90 %.
Клинический случай
В 2009 году в Донецке я стал свидетелем редчайшего для Донбасса (всего 6 случаев в Донецкой области за период с 1991 по 2013 годы!) случая синдрома Лайелла. Пациентка: 37 лет. Аллергический и инфекционный анамнезы — не отягощены. За трое суток до поступления заболела простудным заболеванием. Лечилась «знакомыми» лекарствами: таблетки от кашля, глазные, носовые капли, витамины. С вечера второго дня появились зуд, высыпания. Самостоятельно принимала супрастин. После кратковременного облегчения — состояние ухудшилось. Вечером на третьи сутки, после обморока доставлена БСМП в городскую больницу, в течение нескольких часов переведена в областную клинику.
Поступила женщина с гиперемированной кожей на грудной клетке, плечах, внутренних поверхностях бедер. На третий день появились наполненные мутноватым содержимым пузыри. Объем иных достигал 100 мл. На протяжении всего периода сохранялась гиперемия век, склер, слизистых оболочек ротовой полости, перианальной области.
С третьих суток комбустиологом неоднократно предпринимались попытки закрыть раневую поверхность ксенокожей. Однако кратковременные периоды спокойствия сменялись у пациентки психомоторным возбуждением, что приводило к смещению повязок и ксенокожи. Ни один лоскут не прижился. На протяжении двух недель в реанимационном отделении пациентка более половины времени провела в вынужденном медикаментозном сне. А с шестых суток, когда состояние пациентки стало критическим, ее перевели на постоянную ИВЛ.
В данном случае синдром Лайелла диагностирован в течение двух суток после поступления пациентки в стационар; течение заболевания было не молниеносным, а скорее острым; проводилась посимптомная терапия, но спасти женщину не удалось.
laverie
laverie — [ lavri ] n. f. • 1776; « lavage » 1555; de laver 1 ♦ Techn. Lieu, usine où on lave le minerai, la houille. 2 ♦ Laverie (automatique) : établissement mettant à la disposition des clients des machines à laver en libre accès. ● laverie nom féminin… … Encyclopédie Universelle
LAVERIE — n. f. Endroit où on lave le minerai. Il se dit aussi de l’Endroit où on lave la vaisselle … Dictionnaire de l'Academie Francaise, 8eme edition (1935)
laverie — (la ve rie) s. f. Usine où s opère le lavage des minerais. Lieu où l on lave dans les usines à sucre … Dictionnaire de la Langue Française d'Émile Littré
laverie — nf. => Blanchisserie … Dictionnaire Français-Savoyard
Laverie automatique — Blanchisserie Devanture d une blanchisserie traditionnelle Une blanchisserie est un établissement, usine ou boutique où le linge, après avoir été blanchi, est repassé pour être livré aux clients[1] … Wikipédia en Français
Mines du Laurion — Carte du Laurion antique Les mines du Laurion sont d anciennes mines d argent, situées dans la pointe méridionale de l Attique, entre Thorikos et le cap Sounion, à une cinquantaine de kilomètres au sud d Athènes, en Grèce. De nombreux vestiges de … Wikipédia en Français
Amsterdam (navire de croisière) — MS Amsterdam Amsterdam Le Amsterdam Noms : Ms Amsterdam … Wikipédia en Français
MS Amsterdam — Amsterdam Le Amsterdam Autres noms Ms Amsterdam Type Navire de croisière Histoire … Wikipédia en Français
MS Rotterdam — Rotterdam Type Navire de croisière Histoire Quille posée 1997 Lancement … Wikipédia en Français
Charmion — For other and similar uses, see Charmian (disambiguation) Charmion on a vaudeville promotional button Laverie Vallee née Cooper (July 18, 1875 - February 6, 1949), best known by her stage name Charmion, was an American vaudeville trapeze artist… … Wikipedia
Bohéries — Vadencourt (Aisne) Vadencourt Église de Vadencourt Administration Pays France Région Picardie Département … Wikipédia en Français
Синдром Прадера-Вилли ( Синдром гипотонии-ожирения )
Синдром Прадера-Вилли - это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость. Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано. Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
МКБ-10

Общие сведения
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли. Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 - 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.
Причины
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е. обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% - наследованием обеих 15 хромосом от матери. Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патогенез
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Симптомы
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении. Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела. В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом - полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы. В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение. Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек - недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием. Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп). Типичны гипопигментация кожи, светлые волосы.
Осложнения
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения - ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза - показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации - основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития - синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Лечение синдрома Прадера-Вилли
Консервативная терапия
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция - увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии - бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия - прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина - карбетоцина. Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами. В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Прогноз и профилактика
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике - предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
1. Синдром Прадера-Вилли у детей: новое в этиологии, патогенезе и лечении/ Казанцева Л.З., Новиков П.В., Семячкина А.Н., Николаева Е.А., Курбатов М.Б., Добрыкина Э.В.// Российский вестник перинатологии и педиатрии - 1999 - №4.
2. Синдром Прадера-Вилли в Беларуси: генетическая структура и фенотипическая характеристика/ Хурс О.И., Политыко А.Д., Румянцева Н.В. и др.//Известия Национальной Академии наук Беларуси - 2010 - № 1.
3. Clinical report-health supervision for Children with Prader-Willi Syndrome, the Committee on Genetics/ McCandless S.E.// Pediatrics - 2011 - №1.
4. Prader-Willi syndrome: clinical and genetic fi ndings/ Butler M.G., Thompson T.// Endocrinologist - 2000 - №10.
Словари
Толковый словарь русского языка. Поиск по слову, типу, синониму, антониму и описанию. Словарь ударений.
сущ., кол-во синонимов: 1
ЛАВЁР а, м. laveur à gaz m. Лавёр, газопромыватель. Пищепром 350. ♦ Лавёрная вода. eau de lavage de gaz. Пищепром.
ЛАВЕРАК - ЛАВЕРА́К, то же, что английский сеттер (см. АНГЛИЙСКИЙ СЕТТЕР).
- Порода собак, английский сеттер.
- Французский врач, паразитолог, лауреат Нобелевской премии (1907 г.).
- Французский врач, паразитолог 19-20 вв., открывший возбудителя малярии (1880 г.).
ЛАВЕРАН (Laveran) Шарль Луи Альфонс (1845-1922) - французский врач, паразитолог. Открыл возбудителя малярии (1880). Труды по этиологии и профилактике протозойных болезней, военной гигиене. Нобелевская премия (1907).
Лавера́н Шарль Луи Альфонс (Laveran) (1845-1922), французский врач, паразитолог. Открыл возбудителя малярии (1880). Труды по этиологии и профилактике протозойных болезней, военной гигиене. Нобелевская премия (1907).
ЛАВЕРАН Шарль Луи Альфонс - ЛАВЕРА́Н (Laveran) Шарль Луи Альфонс (18 июня 1845 - 18 мая 1922), французский врач, паразитолог. Открыл возбудителя малярии (см. МАЛЯРИЯ) (1880). Труды по этиологии и профилактике протозойных болезней, военной гигиене. Нобелевская премия (1907).
ЛАВЕРАН Шарль Луи Альфонс (Laveran, Charles Louis Alphonse)
ЛАВЕРАН (Laveran) Шарль Луи Альфонс (1845 -1922), французский врач, паразитолог. Открыл возбудителя малярии (1880). Труды по этиологии и профилактике протозойных (вызываемых простейшими) болезней, военной гигиене. Нобелевская премия (1907).
Синдром Лайелла: симптомы и лечение

Синдром Лайелла, или токсический эпидермальный некролиз — это острое состояние, в основе развития которого лежит тяжелая аллергия. Такая патология сопровождается не только нарушением общего состояния больного человека, но и приводит к появлению буллезных элементов на кожных покровах и слизистых оболочках. Данное заболевание имеет высокую степень летальности при неоказании своевременной медицинской помощи.
Впервые синдром Лайелла был описан в тысяча девятьсот пятьдесят шестом году шотландским дерматологом, в честь которого в последующем он и был назван. Ранее считалось, что такая болезнь является крайней степенью токсидермии. Однако затем она была выделена в качестве самостоятельной нозологической единицы. Данная аллергия занимает второе место по тяжести и количеству летальных исходов после анафилактического шока. При этом наиболее часто такое состояние диагностируется у детей и у лиц молодого возраста. По различным данным уровень смертности при синдроме Лайелла составляет от тридцати до семидесяти процентов.
Причины и классификация синдрома Лайелла
В зависимости от того, каким фактором была спровоцирована аллергия, синдром Лайелла разделяется на четыре формы. Первая форма устанавливается в том случае, если аллергическая реакция развилась в ответ на проникновение в организм инфекционной флоры. При этом чаще всего такое состояние бывает обусловлено золотистым стафилококком.
Вторая форма этой болезни имеет место в том случае, если возникновение клинической картины спровоцировал прием лекарственных препаратов. Наиболее часто отмечается наличие повышенной чувствительности организма к средствам сульфаниламидной группы. Однако свою роль могут сыграть и другие препараты, например, антибиотики или нестероидные противовоспалительные средства. В последние годы все чаще встречается подобная реакция в ответ на витамины и биологически активные добавки.
Третья форма также называется идиопатической. Другими словами, причины развития патологического процесса остаются неизвестными. Четвертая форма подразумевает под собой совокупность и лекарственного, и инфекционного факторов. Как правило, формирование аллергии происходит на фоне приема лекарственных средств, предназначенных для лечения инфекционного заболевания.
Наиболее часто люди, столкнувшиеся с синдромом Лайелла, имеют и другие аллергические патологии. Они могут страдать от экземы, аллергического дерматита, бронхиальной астмы и так далее. Механизм развития данного состояния заключается в том, что антигены, в качестве которых выступают лекарственные вещества или продукты жизнедеятельности инфекционной флоры, прикрепляются к белку, входящему в состав эпидермального слоя. В результате этого иммунная система оказывает повреждающее воздействие не только на антигены, но и на клетки кожи. Помимо этого, наблюдается параллельное накопление в организме токсических веществ, приводящих к поражению практически всех внутренних органов.
Симптомы при синдроме Лайелла
Как мы уже говорили ранее, симптомы при данном заболевании нарастают внезапно и стремительно. В первую очередь больной человек обращает внимание на резкое и беспричинное повышение температуры тела до фебрильных значений. В течение нескольких часов на поверхности кожных покровов и слизистых оболочек формируются эритемы, имеющие различные размеры. Такие эритемы отечны и болезненны при пальпации. В некоторых случаях они сливаются между собой с образованием обширных очагов поражения.
По мере прогрессирования аллергии на коже появляются участки отслаивания эпидермиса. Они выглядят как пузыри, имеющие тонкую стенку и неправильную форму. Величина этих пузырей бывает совсем небольшой, а иногда даже превышает десять сантиметров в диаметре. После того, как пузырь вскрывается, на его месте остаются эрозированные участки. Отмечаются такие симптомы, как отечность и покраснение кожи вокруг эрозий, а также выделение из них большого количества жидкости, имеющей серозно-кровянистый характер.
Обязательным моментом является наличие симптома Никольского. Симптом Никольского характеризуется отслаиванием эпидермального слоя в ответ даже на небольшое механическое воздействие. За счет этого, на тех участках тела, которые подвергались давлению или трению, образуются эрозированные поверхности без предшествующих им пузырей.
Как правило, при синдроме Лайелла все тело больного человека быстро становится гиперемированным, а при дотрагивании до кожи отмечается выраженная болезненность. При осмотре пациента может создаться ощущение, что он подвергся ожогу. В некоторых случаях клиническая картина дополняется мелкими петехиями.
Как правило, параллельно местным симптомам отмечается стремительное ухудшение общего состояния больного человека. Он начинает жаловаться на повышенную жажду, низкое слюноотделение, значительную головную боль. За счет нарастающей интоксикации постепенно угнетается работа внутренних органов.
Диагностика болезни

Диагностика этой болезни в первую очередь основывается на сопутствующей клинической картине. В обязательном порядке проводят общий и биохимический анализы крови, а также коагулограмму. Перечисленные анализы позволяют выявить признаки, характерные именно для синдрома Лайелла. Для определения вещества, на которое развилась аллергическая реакция, назначают ряд иммунологических тестов. В сомнительных случаях показана биопсия кожи с последующим гистологическим исследованием.
Лечение аллергии и ее профилактика
Лечение такой аллергии проводится в условиях реанимационного отделения. Пациенту назначаются большие дозы глюкокортикостероидов. Параллельно этому проводят экстракорпоральную гемокоррекцию и инфузионную терапию. Местное лечение сводится к применению топических глюкокортикостероидов, антибактериальных и эпителизирующих средств.
Для профилактики такого состояния следует при обращении к врачу обязательно рассказывать ему об имеющихся аллергических реакциях на лекарственные препараты и избегать самолечения. Кроме этого, не рекомендуется одновременно принимать более пяти медикаментозных средств — только если это не предусмотрено схемой лечения.
Читайте также: