Синдром Маркезани (Marchesani) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 21.01.2026
К основным общим симптомам относятся изменения костной системы (брахицефалия, короткие и широкие пальцы рук и ног, низкий рост), к симптомам поражения органа зрения — микросферофакия, хрусталиковая миопия, дислокация хрусталика (чаще книзу).
Диагноз и рекомендуемые клинические исследования
Дифференциальный диагноз
Дифференциальный диагноз проводят с аниридией, эктопией зрачка и хрусталика, дефицитом сульфитоксидазы и ксантиноксидазы.
Общие принципы лечения
Выбор терапии зависит от типа течения глаукомы.
При открытоугольной форме используют как медикаментозное (препараты, снижающие продукцию внутриглазной жидкости), так и хирургическое лечение (трабекулэктомия):
Тимолол (пролонгированного действия), 0,1% гель или 0,5% р-р, в конъюнктивальный мешок по 1 капле 1 р/сут или
ЛС обычного действия, 0,25—0,5% р-р, в конъюнктивальный мешок по 1 капле 2 р/сут, длительно
±
Бринзоламид, 1% р-р, в конъюнктивальный мешок по 1 капле 2 р/сут, длительно или
Дорзоламид, 2% р-р, в конъюнктивальный мешок по 1 капле 3 р/сут, длительно.
При закрытоугольной глаукоме показано удаление хрусталика.
Оценка эффективности лечения
Осложнения и побочные эффекты лечения
При использовании указанных препаратов возможно развитие аллергических реакций и побочных эффектов, свойственных данным ЛС.
Ошибки и необоснованные назначения
Несвоевременные диагностика и хирургическое вмешательство могут привести к стойкой потере зрения, помутнению роговицы.
Прогноз
При адекватной терапии и снижении уровня ВГД зрительные функции удается сохранить.
МАРКЕЗАНИ СИНДРОМ
МАРКЕЗАНИ СИНДРОМ (О. Marchesani, нем. офтальмолог, 1900— 1952; син.: синдром Вейлля — Маркезани, синдром микросферофакии-брахиморфии, врожденная гиперпластическая мезодермальная дисморфодистрофия) — наследственный порок развития, характеризующийся низким ростом, подвывихом хрусталика и брахидактилией.
Впервые описан в 1932 г. Вейллем (J. Weill), а в 1939 г. Маркезани выделил его как самостоятельный синдром. По мнению Фейнберга (S. В. Feinberg, 1960), М. с. наследуется как доминантный признак с низкой пенетрантностью гена либо как рецессивный с частичной экспрессивностью у гетерозигот; Кариотип нормален; встречается при родственных браках. В основе М. с. лежит системное наследственное поражение мезенхимальной ткани, к-рая под влиянием неизвестных факторов может развиться в гиперпластическом направлении с исходом в М. с. или в гипопластической направлении, обусловливая появление синдрома Марфана (см. Марфана синдром), для к-рого, как и для М. с., характерны глазные и дисморфологические аномалии.
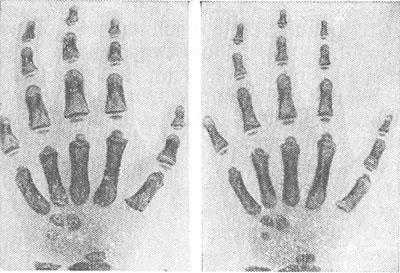
Рентгенограммы кистей: слева — укорочение и утолщение фаланг пальцев кисти при синдроме Маркезани; справа — нормальная кисть (дана для сравнения).
Морфологические изменения со стороны кожи отсутствуют, отмечается фиброз соединительной ткани яичка, в скелетных мышцах резко выражена поперечная исчерченности волокон. Для больных с М.С. характерны низкий рост, врожденные дефекты формы головы (брахицефалия), излишне развитые мышцы и подкожная клетчатка; верхние и нижние конечности настолько коротки, что больные не могут обхватить собственную голову руками. Пальцы уменьшены в размерах (брахидактилия), толстые. Рентгенологически выявляется асимметричный сдавленный череп с гипопластичными верхней челюстью и глазницами, отмечается неправильное формирование и расположение зубов (часто с отсутствием корней), укорочение пястных костей, укорочение и утолщение фаланг пальцев кистей (рис.) и стоп. Офтальмологически выявляются резкая миопии (см. Близорукость), глаукома (см.), уменьшение размеров и аномалии формы хрусталика — микросферофакия и подвывих (см. Хрусталик), нередко нистагм (см.) и нарушение конвергенции глаз (см.). Психическое развитие нормальное.
Лечение симптоматическое, направлено на снижение повышенного внутриглазного давления.
Прогноз для жизни благоприятный, но возможно развитие глаукомы.
Библиография Маккьюсик В. А. Наследственные признаки человека, пер. с англ., с. 514, М., 1976; Feinberg S. В. Congenital mesodermal dysmorpho-dystro-phy (brachymorphic type), Radiology, v. 74, p. 218, 1960; McGavicJ.S. Weill-Marche-sani syndrome, Amer. J. Ophthal., v. 62, p. 820, 1966; McKusick V. A. Heritable disorders of connective tissue, St Louis, 1972; Marchesani O. Brachydaktylie und angeborene Kugellinse als Systemer-krankung, Klin. Mbl. Augenheilk., Bd 103, S. 392, 1939; Rennert О. M. The Marchesani syndrome, Amer. J. Dis. Child., v. 117, p. 703, 1969.
Синдром Маркезани
Синдром Маркезани - наследственный порок развития, характеризующийся триадой:
- низкий рост,
- подвывих хрусталика глаза,
- брахидактилия.
Синонимы: синдром Вейлля-Маркезани, синдром микросферофакии-брахиморфии, врождённая гиперпластическая мезодермальная дисморфодистрофия.
Впервые описан в 1932 году J. Weill, а в 1939 году немецкий офтальмолог Маркезани выделил его как самостоятельный синдром.
Синдром Маркезани наиболее часто встречается при родственных браках.
Этиология и патогенез
По мнению S. B. Feinberg, представленному в работах 1960 года синдром Маркезани наследуется как доминантный признак с низкой пенетрантностью гена, либо как рецессивный с частичной экспрессивностью у гетерозигот. Кариотип нормален.
В основе синдрома Маркезани лежит системное наследственное поражение мезенхимальной ткани, которая под влиянием неизвестных факторов может развиться в гиперпластическом направлении с исходом в синдром Маркезани или в гипопластическом направлении, обусловливая появление синдрома Марфана , для которого, как и для синдрома Маркезани, характерны глазные и дисморфологические аномалии.
Патологическая анатомия
Морфологические изменения со стороны кожи отсутствуют, отмечается фиброз соединительной ткани яичка, в скелетных мышцах резко выражена поперечная исчерченность волокон.
Клинические проявления
Для больных с синдромом Маркезани характерны низкий рост, врождённые дефекты формы головы (брахицефалия), излишне развитые мышцы и подкожная клетчатка. Верхние и нижние конечности настолько коротки, что больные не могут обхватить собственную голову руками. Пальцы уменьшены в размерах (брахидактилия), толстые. На рентгенограммах выявляется:
- асимметричный сдавленный череп с гипопластичными верхней челюстью и глазницами,
- неправильное формирование и расположение зубов (часто с отсутствием корней),
- укорочение пястных костей,
- укорочение и утолщение фаланг пальцев кистей и стоп.
При офтальмологическом обследовании выявляются:
- резкая миопия ,
- глаукома ,
- уменьшение размеров и аномалии формы хрусталика - микросферофакия и подвывих ,
- нередко - нистагм и нарушение конвергенции глаз .
Психическое развитие больных нормальное.
Лечение
Лечение синдрома Маркезани симптоматическое, должно быть направлено на снижение повышенного внутриглазного давления.
Прогноз
Большая медицинская энциклопедия 1979 г.
Реклама, размещённая на сайте «Ваш дерматолог», является одним из источников его финансирования.
Наличие рекламы медицинских центров, лекарств, методов лечения, нельзя расценивать как рекомендацию владельца сайта к их посещению, приобретению или применению.
Последнее обновление страницы: 17.11.2014 Обратная связь Карта сайта
© NAU. При цитировании и копировании материалов убедительная просьба делать активную ссылку на сайт «Ваш дерматолог»
Представленная на сайте информация не должна использоваться для самостоятельной диагностики и лечения
и не может служить заменой очной консультации врача - дерматолога.
Детский врач
Медицинский сайт для студентов, интернов и практикующих врачей педиатров из России, Украины! Шпаргалки, статьи, лекции по педиатрии, конспекты, книги по медицине!
- Вы студент медик? Интерн? Детский врач? Добавьте наш сайт в социальные сети!

Синдром, описанный в 1939 г. Маркезани, представляет постоянное сочетание некоторых глазных аномалий (сферо- и микрофакия) и дисморфии (нанизм; брахиморфия; брахицефалия и т.д.).
Множество синонимов отражают одну и ту же клиническую картину данного синдрома Маркезани: «врожденная гиперпластическая мезодермальная дисплазия», «брахиморфия и сферофакия», «гиперпластическая мезодермальная дистрофия», «врожденная мезодермальная дисморфодистрофия» и «врожденная эктопия с брахиморфией».
Этиопатогенез синдрома Маркезани.
Этиология и патогенез неизвестны. Видимо, синдром является аномалией мезодермы, которая, под влиянием неизвестных факторов, может развиться в гиперпластическом направлении, доходя до появления синдрома Маркезани или, в гипопластическом направлении обусловливает появление синдрома Марфана (на который синдром Маркезани похож своими глазными и дисморфическими аномалиями).
Синдром Маркезани имеет семейный характер и чаще появляется в единокровных семьях. Передается наследственно, доминирующим образом, с повышенным проникновением в отношении брахидактилии и перемещения хрусталика, или рецессивным — в отношении микросферофакии.
Наличие случаев со стертыми признаками, при которых больной имеет только один симптом микросферофакии или только брахидактилию, позволяет предположить, что легкая брахидактилия представляет гетерозиготную форму, в то время как сферофакия, сопровождающаяся или нет костными аномалиями — гомозиготную рецессивную форму.
Синдром очень редкий и число случаев с поставленным диагнозом и опубликованных — небольшое.
Симптоматология синдрома Маркезани
—микросферофакия (хрусталик — малых размеров и очень выпуклый). Патологическая форма хрусталика всегда ведет к ранней эволютивной близорукости. Часто эта аномалия обусловливает постоянные головные боли; вывих хрусталика; двустороннюю глаукому. Глазная гипертония, всегда вторичная перемещению хрусталика, обусловливает, таким образом, увеличение глазного яблока (вторичная гидрофтальмия), катаракту; косоглазие.
- Нанизм проявляется с возрастом; ребенок принимает «коренастый» вид. Конечности короткие, подкожный жировой слой и мышцы хорошо развиты, а грудная клетка широкая;
- брахидактилия: ладони и ступни короткие и широкие, пальцы короткие.
- рахицефалия: широкий череп, широкий и выпуклый лоб.
Непостоянно сочетающиеся проявления:
- запоздалое психомоторное развитие;
- сердечнососудистые аномалии;
- стеноз клапанов или сосудов;
патологический вид папиллярных гребешков — нехарактерный.
Течение и прогноз синдрома Маркезани — весьма тяжелые. Среди глазных аномалий, глаукома является самым страшным осложнением. Она ухудшает прогноз, так как из-за ее двустороннего расположения, ее нельзя оперировать.
Лечение синдрома Маркезани.
Этиопатогенетического лечения не существует. В качестве симптоматического лечения для борьбы с глазной гипертензией, рекомендуется:
—инстилляции в глаза 1—2% раствором пилокарпина, 3—4 раза в день (пилокарпин понижает внутриглазное давление, способствуя выделению камерной жидкости в канал Шлема). Это лечение следует продолжать всю жизнь. Попытка хирургического лечения, состоящая в капсулотомии — бесполезна, так как внутриглазное повышенное давление — двустороннее.
Похожие медицинские статьи
- Синдром МориакаПод названием «синдром Мориака», «вторичный сахарный гликогеноз» или «детский сахарный […]Синдром Вернера. Генитально-склеродермическая дегенерацияВ 1904 г. Вернер описал синдром, характеризующийся постоянным сочетанием склеродермии, […]Синдром Бернара-ГорнераСиндром, описанный в 1852 г. CI. Bernard (Кл. Бернардом) и дополненный F. J. Horner […]
- уже при рождении отмечается брахицефалия с гипоплазией верхней челюсти, узким нёбом, мелкими глазницами и сложной врождённой патологией глаз;
- рефракция чаще миопическая (высокой степени), нередко обнаруживают микросферофакию, эктопию хрусталика (чаще кзади), а иногда - смещение хрусталика кпереди с развитием зрачкового блока и глаукомы. В результате зрачкового блока иногда формируются периферические синехии.
- выявляют и разнообразную патологию радужки (иридодонез в связи с дислокацией хрусталика, гипоплазию радужки, частичную аниридию, аномалии гониодисгенеза); иногда - отслойку, дегенеративные изменения сетчатки, атрофию зрительного нерва, обычно сопровождающиеся врождённой слепотой, развитием глаукомы, появлением нистагма и косоглазия;
- отмечают также скелетные деформации (карликовый рост с широкой грудной клеткой, короткими конечностями, брахидактилией, иногда - с тугоподвижными суставами);
- у этих детей нередко выявляют различные расстройства деятельности сердечно-сосудистой системы.
- Выделение признаков вегето-сосудистой дистонии. Виды лечения нейроциркуляторной дистонии
- Эффективность скрининговых тестов на злоупотребление алкоголем.
- Генетически модифицированные мыши в изучении сердечно-сосудистых заболеваний. Трансгенные мыши
- УЗИ при хроническом холецистите
- Причины нарушений прикуса. Этиология нарушений развития челюстей
Запись "Синдром Маркезани" опубликована в рубрике Офтальмология, СИНДРОМЫ в Пятница, Декабрь 20th, 2013 в 12:45 дп. К записи добавлены такие Метки: брахидактилия, глаза, катаракта, нанизм, синдром
5 коммент. к статье “Синдром Маркезани”
Отдельные симптомы синдрома Маркезани по частоте проявления, следуют в таком нисходящем порядке:
ожирение,
атипичный пигментный ретинит,
умственная отсталость,
гипогенитализм,
полидактилия.
Степень выраженности симптомов различная.
А может ли быть симптомом синдрома Маркезани аномальная форма хрусталика?
Возможны любые глазные аномалии при синдроме Маркезани.
В помощь тем, кто пишет рефераты по данному синдрому
=================
Синдром Маркеза́ни — синдром микросферофакии-брахиморфии, врождённая гиперпластическая мезодермальная дисморфодистрофия. Данная патология, характеризующаяся тремя основными признаками: низкий рост, подвывих хрусталика и брахидактилия. Также наблюдаются нарушение аккомодации, глаукома, брахицефалия и недоразвитие конечностей, ограничение подвижности в суставах, укорочение туловища и шеи.
=================
Синдром Маркезани - симптомы (клиническая картина).
Клиническая картина. Туловище, шея и конечности у таких больных короткие. Мускулатура и подкожный жировой слой хорошо развиты. Череп брахицефальный. Отмечаются ограничения в подвижности суставов, различные расстройства сердечно-сосудистой системы. Глазные симптомы: подвывих хрусталика книзу вследствие сферофакии или микрофакии, миопия высоких степеней (иногда до 40-50 дптр), отслойка сетчатки и вторичная глаукома. Хрусталик заметно уменьшен и имеет форму, близкую к сферической; его сагиттальный диаметр 3-5 мм, экваториальный 6-7 мм. При расширенном зрачке видны край хрусталика и недоразвитая циннова связка. Нередко наблюдаются подвывих хрусталика, приводящий к вторичной глаукоме. Зрение снижено из-за высокой миопии, причем миопия прогрессирует.
====================
Синдром Маркезани — синдром микросферофакии-брахиморфии, врождённая гиперпластическая мезодермальная дисморфодистрофия. Данная патология, характеризующаяся тремя основными признаками: низкий рост… Маркезани синдром (О. Магchesani, нем. офтальмолог, 1900— 1952; синонимы: синдром Вейлля — Маркезани, синдром микросферофакии-брахиморфии…Синдром Маркезани имеет семейный характер и чаще появляется в единокровных семьях. Передается наследственно, доминирующим образом…В науке есть своя версия о происхождении этой мутации. Фиолетовые глаза могут быть у людей, страдающих синдромом Маркезани.Фиолетовые глаза бывают в природе! Маркезани синдром (О. Marchesani, нем. офтальмолог, 1900—1952) — наследственная болезнь…
Вейля-Маркезани синдром
Вейля-Маркезани синдром (врождённая мезодермальная дисморфодистрофия, синдром сферофакии-брахиморфии) - является системным наследственным заболеванием соединительной ткани и характеризуется наличием у больных сферофакии, эктопии хрусталика, брахиморфизма. Тип наследования - аутосомно-рецессивный.
Микроскопическое исследование позволяет выявить дегенерацию фибрилл аппарата ресничного пояска, приводящую к накоплению на поверхности цилиарного эпителия ШИК-положительного вещества. Картина во многом идентична той, что обнаруживается при гомоцистинурии.
Клиническая симптоматика:
Прогноз: обычно эти больные живут не более 40-50 лет. Прогноз в отношении зрения зависит от выраженности врождённой патологии глаз и от развившихся осложнений.
Читайте также: