Синдром Мартена-Олбрайта (Martin-Albright) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 21.01.2026
В статье представлены результаты клинического наблюдения порока развития костной ткани - фиброзной дисплазии. В настоящее время нет единого подхода к лечению этой нозологии. Большинством авторов отдается предпочтение хирургическому лечению, предлагаемые тактика и объем хирургического вмешательства различны. В приведенном наблюдении у пациентки диагностирован достаточно редкий синдром - синдром Олбрайта. Выполнена гайморотомия доступом через переднюю стенку, что привело к декомпрессии ветвей тройничного нерва. После оперативного вмешательства купировались головные боли, ощущение онемения левой половины лица. Сделан вывод о том, что хирургическое лечение возможно при функциональных нарушениях и выраженном косметическом дефекте и не должно быть радикальным.
В статье представлены результаты клинического наблюдения порока развития костной ткани - фиброзной дисплазии.
Для цитирования. Добротин В.Е. Синдром Олбрайта как форма фиброзной дисплазии // РМЖ. 2015. No 23. С. 1422-1424.
Резюме: в статье представлены результаты клинического наблюдения порока развития костной ткани - фиброзной дисплазии. В настоящее время нет единого подхода к лечению этой нозологии. Большинством авторов отдается предпочтение хирургическому лечению, предлагаемые тактика и объем хирургического вмешательства различны. В приведенном наблюдении у пациентки диагностирован достаточно редкий синдром - синдром Олбрайта. Выполнена гайморотомия доступом через переднюю стенку, что привело к декомпрессии ветвей тройничного нерва. После оперативного вмешательства купировались головные боли, ощущение онемения левой половины лица. Сделан вывод о том, что хирургическое лечение возможно при функциональных нарушениях и выраженном космети- ческом дефекте и не должно быть радикальным.
Ключевые слова: фиброзная дисплазия, болезнь Лихтенштейна, синдром Олбрайта, патология основания черепа, гайморотомия.
Фиброзную дисплазию, или болезнь Брайцева - Лихтенштейна, относят к порокам развития костной ткани вследствие неправильного развития остеогенной мезенхимы. По своей морфологии она близка к истинным опухолям, ее относят к опухолеподобным процессам. Этиология ее связана с мутацией гена, кодирующего α субъединицу G белка в хромосоме 20q13.2-13.3 3. В 1927 г. В.Р. Брайцев впервые описал эту патологию, назвав ее фиброзной остеодистрофией [4]. А уже в 1937 г. L. Lichtenstein предложил термин «фиброзная дисплазия» [5]. Это доброкачественное заболевание, однако опухоль обладает способностью к быстрому росту, сдавлению и нарушению функции близлежащих органов. Чаще всего развивается у лиц молодого возраста, преимущественно женщин [5, 6]. Составляет около 3% всех опухолей костей, а среди первичных опухолей и опухолеподобных образований челюстей - 7,5% [7, 8]. В исследовании М. Cai проведен ретроспективный анализ 36 случаев поражения костей черепа. У большинства больных поражались лобные кости (52,78%), височные кости были поражены в 30,56% случаев, клиновидные - в 25%, теменные - в 19,44% и орбитальные - в 13,89% [9]. Однако, по другим данным (L.R. Lustig et al.), чаще всего подвергались поражению костные структуры решетчатого лабиринта (71%). Клиновидная кость была поражена в 43% случаев, лобная - в 33%, верхнечелюстная - в 29%, височная - в 24%, теменная - в 14%, затылочная - в 5% [10].
Выделяют моно- и полиоссальную формы фиброзной дисплазии, а также синдром Олбрайта; по характеру поражения - очаговую и диффузную. А.А. Колесов дифференцирует эту патологию на 3 гистологические формы. При первой - основной - форме отмечается разрастание клеточно-волокнистой ткани, на фоне которой разбросаны костные образования в виде немногочисленных балочек. При второй - пролиферирующей - форме вместе с зонами костных балочек на отдельных участках клетки расположены плотно. При третьей - остеокластической - форме обнаруживается большое число остеокластов, собранных в узелки [11].
Монооссальная форма - самая распространенная, встречается в 70% случаев. Чаще всего поражаются ребра и черепно-лицевая область, тогда как остальные отделы скелета сохраняют нормальное строение [12]. Поражение костей может протекать бессимптомно. Полиоссальная форма встречается в 30% случаев.
Наиболее тяжелой формой является синдром Олбрайта, который характеризуется триадой признаков: полиоссальной формой фиброзной дисплазии, преждевременным половым развитием (менархе в возрасте 3 лет) и пигментацией кожи (пятна светло-кофейного цвета, не возвышающиеся над уровнем кожи).
Предполагают, что этиология синдрома Олбрайта связана с неврогенными влияниями и эндокринными сдвигами вследствие врожденных изменений в гипоталамической области. Для него характерна преждевременная продукция гонадотропного гормона передней доли гипофиза на фоне склероза и гиперостоза основания черепа, преимущественно в области турецкого седла [13].
Клиническая картина фиброзной дисплазии разнообразна и зависит от локализации и вовлечения различных структур в патологический процесс. Сроки первичных проявлений фиброзной дисплазии зависят от морфологической структуры диспластической ткани [14]. Трудности в диагностировании фиброзной дисплазии связаны с тем, что заболевание долгое время протекает латентно. Одним из ранних и частых симптомов является головная боль. В дальнейшем, при прогрессировании процесса, появляются жалобы на косметический дефект лицевого черепа, затруднение носового дыхания, экзофтальм, а также онемение лица, амавроз вследствие компрессии лицевого и зрительного нервов, снижение слуха за счет сужения наружного слухового прохода.
Следует отметить разнообразие рентгенологической картины фиброзной дисплазии. Характерны участки разрежения и уплотнения кости, так называемая картина «матового стекла», увеличение кости в объеме, истончение кортикального слоя, а также мелкосетчатый, пятнистый или «ватный» рисунок. При очаговых формах в кости отмечаются очаги просветления овальной формы с характерным ячеистым рисунком [15].
Окончательный диагноз фиброзной дисплазии ставится после биопсии ткани на основании данных гистологического исследования.
В настоящее время не существует единого подхода к лечению этой патологии. По данным литературы, при полиоссальной форме применяют препараты, предотвращающие резорбцию костной ткани, - бисфосфонаты. В ряде исследований был использован памидронат, что привело к улучшению клинической картины, повышению минеральной плотности костей 16. Однако в большинстве случаев выполняется хирургическое вмешательство, объем которого зависит от локализации и распространения поражения, степени выраженности функциональных нарушений, темпов прогрессирования процесса [10, 21-23].
В нашей практике отмечен следующий случай фиброзной дисплазии. Пациентка С. (возраст - 51 год) при поступлении 17.06.2013 г. в УКБ № 1 Первого МГМУ им. И.М. Сеченова предъявляла жалобы на приступы боли в левой лобно-теменно-затылочной области, затруднение дыхания через нос, ухудшение зрения, беспокоящие ее в течение 10 лет. Два года назад присоединились жалобы на ощущение онемения левой половины лица. Лечилась самостоятельно (НПВС, антибиотики) с временным удовлетворительным эффектом. В анамнезе: преждевременное половое развитие (маточные кровотечения в возрасте 3 и 5 лет, менархе в 11 лет).
Объективно: пигментные пятна светло-кофейного цвета в области шеи и верхнего плечевого пояса. Форма наружного носа не изменена. Носовое дыхание умеренно затруднено, больше слева. При риноскопии: перегородка умеренно искривлена влево, слизистая оболочка полости носа розовая, нижние носовые раковины умеренно отечные, после анемизации хорошо сокращаются, отделяемого в носовых ходах нет. Со стороны других ЛОР-органов - без патологии. На КТ околоносовых пазух (ОНП): негомогенное заполнение тканью левой верхнечелюстной, лобной, клиновидной пазух, частично клеток решетчатого лабиринта слева (рис. 1А). Ткань из клиновидной пазухи распространяется в среднюю черепную ямку, с тенденцией к сдавлению вещества мозга и деструкции основания черепа. Соответствует костным структурам различной степени плотности (рис. 1Б). Естественное соустье левой верхнечелюстной пазухи проходимо. Помимо КТ ОНП с целью определения границ распространения патологической ткани в костях черепа больной была проведена остеосцинтиграфия - метод радионуклидной диагностики, основанный на введении, накоплении и регистрации в организме радиофармацевтического препарата, тропного к костной ткани. На остеосцинтиграмме определялась патологическая гиперфиксация индикатора в костях черепа - области носовой кости с переходом на нижнюю стенку левой орбиты, левую височную кость, верхнюю стенку орбиты и верхнюю челюсть, левую часть затылочной кости, а также в ребрах, верхних и нижних конечностях слева, грудном отделе позвоночника (рис. 2).
Больная обследована терапевтом. Патологии со стороны внутренних органов не выявлено. Проведено дуплексное сканирование брахиоцефальных сосудов, при котором атеросклеротических изменений экстракраниальных отделов БЦА не наблюдалось. Выявлен вариант развития левой позвоночной артерии (гипоплазия, вхождение в канал на уровне С5). Больная консультирована неврологом. Отмечена гипостезия левой половины лица. Заключение: болезнь Брайцева - Лихтенштейна (фиброзная дисплазия околоносовых пазух). Заключение окулиста: пресбиопия, заднекапсулярная катаракта.
24.06.2013 г. была выполнена операция - гайморотомия. Доступ через переднюю стенку. Верхние и заднелатеральные отделы пазухи были заполнены губчатой костной тканью, которая по возможности была удалена. В средних и медиальных отделах пазухи ткань удалена фрагментами. В альвеолярном отростке определялась полость в виде пещеры с гладкими стенками без какого-либо патологического содержимого. Через нижний носовой ход было сформировано соустье с верхнечелюстной пазухой диаметром 7 мм. После оперативного вмешательства купировались головные боли, ощущение онемения левой половины лица, что, очевидно, связано с декомпрессией ветвей тройничного нерва. Диагноз «фиброзная дисплазия» подтвержден гистологическим исследованием.
Жалобы на приступы боли в левой лобно-теменно-затылочной области, затруднение дыхания через нос, ухудшение зрения, ощущение онемения левой половины лица; анамнез: преждевременное половое развитие; объективные данные: пигментные пятна светло-кофейного цвета в области шеи и верхнего плечевого пояса; результаты КТ: негомогенное затемнение околоносовых пазух слева, распространяющееся в среднюю черепную ямку, с тенденцией к сдавлению вещества мозга и деструкции основания черепа; данные остеосцинтиграфии: патологическая гиперфиксация индикатора в костях черепа, а также в ребрах, верхних и нижних конечностях слева, грудном отделе позвоночника позволили поставить больной С. диагноз: «фиброзная дисплазия, синдром Олбрайта».
С учетом относительно доброкачественной природы заболевания и данных литературы о наблюдении больных в послеоперационном периоде хирургическое лечение не должно быть радикальным, т. к. это провоцирует активный рост патологической ткани. Операция должна быть направлена на сохранение существующих функций. Оперативное вмешательство возможно при функциональных нарушениях и выраженном косметическом дефекте. Фиброзная дисплазия является редким заболеванием, однако современные методы визуализации упрощают диагностику.
Список литературы Свернуть Развернуть
1. Weinstein L.S., Shenker A., Gejman P.V., Merino M.J., Friedman E., Spiegel A.M. Activating mutations of the stimulating G protein in the McCune-Albright syndrome // N Engl J Med. 1991. Vol. 325. Р. 1688-1695.
2. Weinstein L.S., Chen M., Liu J. Gs(alpha) mutations and imprinting defects in human disease // Ann NY Acad Sci. 2002. Vol. 968. Р. 173-197.
3. DiCaprio M.R., Enneking W.F. Fibrous Dysplasia // Pathophysiology, Evaluation and Treatment J Bone Joint Surg Am. 2005. Vol. 87. Р. 1848-1864.
4. Брайцев В.Р. Osteodystrophia fibrosa localisata: Мат-лы 19-го съезда российских хирургов. Л., 1927. С. 301-315.
5. Lichtenstein L. et al. Fibrous dysplasia of bone // Arch Patol. 1942. Vol. 33. Р. 777-816.
6. Воловик Н.Е. Полиоссальная фиброзная дисплазия костей черепа // Вестник оториноларингологии. 1968. № 2. С. 111-113.
7. Coley B.L. Neoplasms of bone and related conditions; etiology, pathogenesis, diagnosis, and treatment. 2nd ed. New York: Hoeber, 1960.
8. Campanacci M. Bone and soft tissue tumors: clinical features, imaging, pathology and treatment. 2nd ed. New York: Springer, 1999.
9. Cai M., Ma L., Xu G., Gruen P., Li J., Yang M., Pan L., Guan H., Chen G., Gong J., Hu J., Qin S. Clinical and radiological observation in a surgical series of 36 cases of fibrous dysplasia of the skull // Clin Neurol Neurosurg. 2012 Apr. Vol. 114 (3). Р. 254-259.
10. Lustig L.R., Holliday M.J., McCarthy E.F., Nager G.T. Fibrous Dysplasia Involving the Skull Base and Temporal Bone // Arch Otolaryngol Head Neck Surg. 2001. Vol. 127. Р. 1239-1247.
11. Колесов А.А. Новообразования лицевого скелета. М., 1969.
12. Jaffe H.L. Tumors and tumorous conditions of the bones and joints. London, 1958.
13. Виноградова Т.П. // Вопросы патологии костной системы. М., 1957. С. 159-174.
14. Котова Е.Н. Фиброзная дисплазия ЛОР-органов у детей: Дис. … канд. мед. наук. М., 2006.
15. Терехова Т.Н., Кушнер А.Н., Кармалькова Е.А. Хирургическая стоматология детского возраста: учебно-методическое пособие / Минск.: БГМУ, 2009.
16. Zacharin M., O’Sullivan M. Intravenous pamidronate treatment of polyostotic fibrous dysplasia associated with the McCune Albright syndrome // J Pediatr. 2000. Vol. 137. Р. 403-409.
17. Pfeilschifter J., Ziegler R. Effect of pamidronate on clinical symptoms and bone metabolism in fibrous dysplasia and McCune-Albright syndrome // Med Klin (Munich). 1998. Vol. 93. Р. 352-359.
18. Weinstein R.S. Long-term aminobisphosphonate treatment of fibrous dysplasia: spectacular increase in bone density // J Bone Miner Res. 1997. Vol. 12. Р. 1314-1315.
19. Parisi M.S., Oliveri B., Mautalen C.A. Effect of intravenous pamidronate on bone markers and local bone mineral density in fibrous dysplasia // Bone. 2003. Vol. 33. Р. 582-588.
20. O’Sullivan M., Zacharin M. Intramedullary rodding and bisphosphonate treatment of polyostotic fibrous dysplasia associated with the McCune-Albright syndrome // J Pediatr Orthop. 2002. Vol. 22. Р. 255-260.
21. Волков М.В., Самойлова Л.И. Фиброзная остеодисплазия. М., 1973.
22.Zimmerman D.C. et al. Fibrous dysplasia of the maxilla and mandible. Oral Surg 1958; 11: 1: 55-68.
23.Espinosa J.M. et al. Fibrous dysplasia of the maxilla. Ann Otol Rhinol Laryngol 1998; 107: 175-177
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Детский врач
Медицинский сайт для студентов, интернов и практикующих врачей педиатров из России, Украины! Шпаргалки, статьи, лекции по педиатрии, конспекты, книги по медицине!
Синдром Мартина - Олбрайта (ALBRIGHT - MARTIN)
- Вы студент медик? Интерн? Детский врач? Добавьте наш сайт в социальные сети!

Лекция для интернов педиатрического профиля на базе «ОХМАТДЕТ»
В 1942 г., F. Albright (Олбрайт) и Е. Martin (Мартин) описывают клинический и биологический синдром — сходный с синдромом гипопаратиреоза — вследствие расстройств рассасывания кальция на уровне почечных канальцев.
В зависимости от клинических и биологических проявлений, наблюдающихся при этой метаболической аномалии, «синдром Albright - Martin» (Олбрайта - Мартина) появляется в медицинской литературе и под другими названиями:
- «идиопатическая гиперкальциурия»,
- «псевдогипопаратиреоз»,
- «гипопаратиреоидный кретинизм»,
- «конституциональная хроническая гипокальцемия» ,
- «наследтственная остеодистрофия Albright (Олбрайта)»,
- «синдром Albright-Bantam (Олбрайт - Бантам)»,
- «синдром Sea bright-Bantam».
Этиопатогенез. Этиология синдрома не известна. Патогенетический механизм синдрома состоит в расстройстве функции почечных канальцев, вызванном, по всей вероятности, понижением тормозящего действия паратгормона на ретрорассасывание фосфатов из почечных канальцев, а не отсутствием этого гормона.
Симптоматология
Дистрофические аномалии:
- нанизм, появляющийся у ребенка старше 3—4 лет, который с возрастом становится все более выраженным;
- ранее ожирение;
- короткая шея и круглое лицо;
- короткие конечности, вследствие замедления роста длинных костей в длину;
- дистрофия зубов, с изменением их формы и появлением множественных кариесов.
Дистрофические аномалии костей и хрящей:
- короткие пястные кости и/или кости плюсневые;
- деформация фаланг;
- экзостозы разной локализации;
- cubitus valgus;
- coxa vara;
- выпуклый лоб;
- короткие, широкие и хрупкие ногти.
Нейропсихические аномалии:
- тетанические проявления — конвульсии и даже самопроизвольные обмороки — в детском возрасте и даже у взрослых;
- иногда отмечаются инфраклинические формы, выявляемые только электромиограммой;
- изредка слабоумие.
Мочекаменная болезнь проявляется клинически только в редких случаях, в виде почечно — уретральной или мочепузырной колики и в большинстве случаев выявляется только при анализе осадка мочи.
Биохимические исследования выявляют значительные изменения, как например, гипокальциемия, гипофосфатурия, гипокальциурия, и только в некоторых формах, гиперкальцируя.
В настоящее время описываются две клинические формы синдрома Albright-Martin.
- Форма псевдогипопаратиреоза описана в 1942 г. этими двумя авторами.
- Форма ложного псевдогипопаратиреоза (описана в 1952 г. Albright), которая отличается от первой отсутствием проявлений тетании и гипокальцемии.
Во многих из 269 случаев, опубликованных до 1970 г., можно было установить семейный, а в большинстве и наследственный характер синдрома; генетические исследования не выявили наличие характерных изменении кариотипа.
Течение и прогноз почти всегда благоприятные; все клинические симптомы стабилизируются в короткое время от их появления; кроме того, они переносятся легко большинством больных.
Лечение. Глубокие изменения почечной функции делают неэффективными любые терапевтические попытки.
Похожие медицинские статьи
- Синдром Бартера (BARTTER)В 1962 г. Fr. Barttor (Бартер) описал клинический синдром, редко встречающийся у […]Синдром Эллис-Ван КревельдаСиндром, описанный в 1940 г. Эллисом и Ван Кревельдом, представляет полиморфное […]СИНДРОМ АРТРОГРИППОЗА«Синдром артрогриппоза» известен и под названием «врожденный множественный […]
- 7777 к записи Скарлатина у детей
- Аленка к записи Как перераспределиться на интернатуре?
- Detvrach - Детский врач к записи Синдром Криста-Сименса-Турена. Ангидротическая эктодермальная дисплазия
- Ylia к записи Синдром Криста-Сименса-Турена. Ангидротическая эктодермальная дисплазия
- Detvrach - Детский врач к записи Синдром Прадера — Вилли
- скрытое заболевание - без каких-либо признаков костной или эндокринной патологии, а также без симптоматики необычной пигментации;
- неполные формы синдрома, например, в процесс патогенеза вовлечения костная ткань, возможны эндокринные изменения и псевдогипопаратиреоз, но нет кожных проявлений;
- псевдогипопаратиреозили наследственная остеодистрофия Олбрайта — заболевание, которое также поражает костные ткани, но механизм и этиология отличаются — патология вызвана нарушением резистентности к паратгормону, метаболизма кальция и фосфора; псевдогипопаратиреоз также проявляется в детском возрасте и выражается в виде низкого роста, круглого лица, умственной отсталости, ожирения, гипотиреоза, гипогонадизма, сахарного диабета и пр.
- преждевременного гонадотропиннезависимого полового развития, развивающегося позже и медленнее нежели при других формах ранней половозрелости;
- пятнистой пигментации кожи - лентиго, пятна с четкими границами светло-коричневого цвета обычно есть от рождения на груди, спине пояснице, бедрах и в местах костных деформаций;
- эндокринных расстройствах, вызывающих тахикардию, необъяснимую утрату веса, глазные симптомы гипертиреоидизма;
- полиоссальной фиброзной остеодисплазии, мышечной слабости и остеопороза, вовлекающей в процесс чаще длинные кости, нарушения проявляются в виде изуродованных черт лица, более вытянутом телосложении, искривленных и асимметричных, укороченных запястьях, что затрудняет движения во время ходьбы, вызывает хромоту, сильные боли и повышает вероятность патологических переломов.
- рентгенологические исследования, которые выявляют псевдокисты, сегментарное поражение любых отделов скелета - чаще нижних конечностей, в более редких случаях — верхнего плечевого пояса и черепа;
- УЗИ брюшной полости позволяет обнаружить кисты яичников;
- лабораторные исследования для выявления повышенного уровня щелочной фосфатазы в кровотоке, сниженного количества гонадотропинов - в анализах мочи и повышенной экскреции лютеинизирующего гормона, 17-гидрокси и 17-оксикортикостероидов.
- бисфосфонаты;
- ингибиторы антагонистов остеокластов.
- минимизации психических отклонений (прием психостимуляторов, антидепрессантов)
- коррекции изменений при атаксическом синдроме (прием ноотропов, препаратов на основе лития, которые улучшают вербальную память, повышают способность к социальной адаптации)
- регуляции гормонов при первичной недостаточности яичников;
- при судорожных приступах применение противоэпилептических средств
- Клинический опрос, осмотр. В беседе с ребенком на первый план выходит снижение интеллекта, гиперактивность и расторможенность поведения, нарушение коммуникативных навыков. Уровень психического развития не соответствует возрасту, методики исследования интеллекта выявляют олигофрению (IQ - 40-79 баллов). Внешне наблюдаются характерные фенотипические признаки, при неврологическом осмотре выявляется мышечный гипотонус, усиленные сухожильные рефлексы, паракинезы.
- Генеалогический анализ. В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники с данным заболеванием, чаще - мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
- Генетическое исследование. В лабораторных условиях исследуется строение ДНК: определяется количество ЦГГ-повторов и статус метилирования. Применяется ПЦР и цитогенетический метод. Диагноз подтверждается, если количество триплетных повторов составляет более 200. При результате 60-199 возможны легкие фенотипические проявления болезни, риск развития патологии в следующем поколении (если показатель диагностирован у женщины).
Запись "Синдром Мартина - Олбрайта (ALBRIGHT - MARTIN)" опубликована в рубрике СИНДРОМЫ, Эндокринология в Вторник, Май 8th, 2012 в 1:17 пп. К записи добавлены такие Метки: гипопаратиреоз, дистрофия, почечные канальцы, синдром
Оставьте комментарий
Для того, чтобы оставить комментарий, Вы должны зарегистрироваться ЗДЕСЬ
Детского врача не бойся,
Не волнуйся, успокойся,
И, конечно же, не плачь,
Это добрый детский врач!
Информация о диагностике и лечении болезней для детских врачей, интернов, студентов и заботливых мам!
Отделения больницы
Поиск по признаку
«5 — минутка»
История болезни
Вопрос для врача
Когда выписываем?
| Пн | Вт | Ср | Чт | Пт | Сб | Вс |
|---|---|---|---|---|---|---|
| « Окт | ||||||
| 1 | 2 | |||||
| 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| 10 | 11 | 12 | 13 | 14 | 15 | 16 |
| 17 | 18 | 19 | 20 | 21 | 22 | 23 |
| 24 | 25 | 26 | 27 | 28 | 29 | 30 |
| 31 | ||||||
Всего медицинских статей - 488 Copyright © 2022 Детский врач - Медицинский сайт для студентов, интернов и практикующих врачей из Украины, России! Шпаргалки, статьи, лекции по педиатрии, конспекты, книги по медицине! All rights reserved. Wordpress
Детский врач ™ Детврач.ком ® © 2011-2012-2013-2014-2015 Медицинский сайт для врачей педиатров, студентов, интернов и практикующих детских врачей из Украины, России!
Этот сайт будет полезен в помощь студентам медикам и интернам. На сайте Детский врач Вы сможете найти шпаргалки, медицинские статьи, лекции по педиатрии. Кроме этого мы размещаем конспекты занятий из медицинских ВУЗов, справочники болезней и синдромов, энциклопедии и книги по медицине. Пройдясь по разделам сайта Детский врач, Вы прочтете истории детских болезней, современные методы и приемы, применяемые при лечении детских болезней, описания синдромов заболеваний. Студенты медики обнаружат тесты на КРОК, которые сдают в медицинских университетах.
Много полезного материала могут прочесть и молодые мамы, которые задают очень много вопросов в комментариях к нашим медицинским статьям!
Сайт "Детский врач" - это сайт для студентов-медиков, интернов, педиатров, врачей и молодых мам! Заходите, читайте, спрашивайте!
Синдром Олбрайта

Синдром Мак-Кьюна-Олбрайта является гетерогенным заболеванием, которое независимо друг от друга описали американский педиатр D. J. McCune и эндокринолог F. Albright в 1936-1937 гг. Заболевание редкое, врожденное, но не наследственное, сопровождается метаболическими нарушениями, признаками преждевременного полового развития и деформаций скелета. Первые клинические проявления отмечаются в раннем детстве или подростковом возрасте в виде нарушений развития, полиоссальной фиброзной дисплазии костей, кожной пигментации по типу «кофе с молоком» и эндокринных аномалий, включая тиреотоксикоз, синдром Кушинга, акромегалию.
Приблизительно в 10 раз чаще диагностируют синдром Олбрайта Мак-Кьюна у девочек, чем у мальчиков. Википедия отмечает, что частота встречаемости в человеческой популяции — 1 на 100 тыс. или на миллион особ.
Патогенез
В результате постзиготной мутации гена GNAS1, участвующего в клеточном сигнализировании белков GS-α , обеспечивающих сопряжение рецепторов лютеинизирующих и фоликолостимулирующих гормонов с молекулами аденилатциклазы в цитологических структурах яичников. Постоянное влияние мутантного белка активизирует аденилатциклазу и повышение внутриклеточного цАМФ, а также стимулирует усиление секреции стероидных женских половых гормонов фолликулярными эстрогенпродуцирующими кистами, вызывающими преждевременное половое развитие даже при отсутствии гонадотропинов. Такая же картина наблюдается и «автономное производство» наблюдается с гормонами щитовидной железы, кортизолом и гормонами роста. Прогрессирование патологии вызвано образованием клеток, уже содержащих мутантные белки.
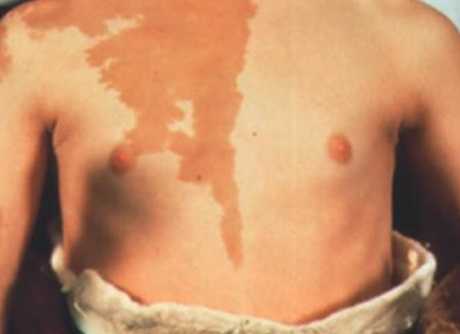
Пигментация при синдроме Олбрайта
Кроме того, рост уровня цАМФ приводит к увеличению продукции меланина и образованию лентигиозных пятен, очаговой гиперпигментации на фоне нормальных значений АКТГ и меланотропина.
Фиброзно-кистозная дисплазия в результате усиленной кальцинации под действием эстрогенов осуществляется путем замещения нормальных костных структур и образованных кальцинатов фиброзными массами, при этом нарушается равновесие процессов резорбции и регенерации костей, пролиферации мезенхимальных стромальных клеток. Наблюдается формирование очагов деструкций и псевдоопухолей.
Классификация
Болезнь Олбрайта бывает разной, в зависимости от тяжести течения различают:
Причины
Синдром Олбрайта Маккьюна возникает в результате генетических нарушений на ранних этапах эмбриогенеза. До сих пор не установлен тип наследования, все описанные случаи были спорадические.
Симптомы
Клиническая картина заболевания проявляется в виде целого симпатокомплекса, состоящего из:

Первые признаки преждевременной половой зрелости начинаются с маточных кровотечений, не имеющих цикличного характера и вызваны кратковременными повышениями концентраций эстрогенов и гонадотропных гормонов в кровяном русле. Такие колебания провоцируют образования кист в яичниках. У детей в возрасте 5-10 лет наблюдается ранний рост груди и гинекомастия. Её нежная этиология не приводит к формированию вторичных половых признаков — Синдром Мак-Кьюна-Олбрайта-Брайцева не вызывает раннего полового оволосения или дифференцировки фигуры. Истинное половое созревание возможно после окончания пубертатного периода.
Анализы и диагностика
При подозрении на синдром Мак Кьюна Олбрайт проводят:
Лечение
Лечение назначают симптоматическое, оно имеет свои сложности в зависимости от индивидуальных особенностей и клинической картины. Аналоги гонадолиберина не оказывают терапевтического эффекта, так как процесс секреции эстрогенов спровоцирован не избытком гонадотропинов. Для купирования процесса гонадального стероидогенеза у особ мужского пола эффективным может оказываться кетоконазол и андрокур. При лечении медроксипрогестерона ацетатом возможно развитие гипокальциемии, поэтому, если процесс развития ранней половозрелости сопровождается тяжёлыми множественными поражениями костных тканей, то терапию необходимо назначать с осторожностью.
Для угнетения гиперэстрогенизиции и удлинения скелета могут быть назначены препараты тестостерона. Чтобы противодействовать дисплазии костей следует вводить:
Больным необходимо регулярно, каждые 3 мес. посещать эндокринолога для коррекции проблем с щитовидной и другими железами.
Синдром Мартина Белла - симптомы и лечение
Синдром Мартина-Белла - относительное новое генетическое заболевание, которое чаще поражает мальчиков. Впервые болезнь была описана в середине прошлого века врачами Мартином и Беллом. В честь этих докторов болезнь и была названа. Иногда можно встретить и другое название болезни - «болезнь ломкой хромосомы». Дело в том, что синдром Мартина-Белла связан с истончением хромосомы в определенном месте, то есть Х хромосома становится уязвима всегда в одном и том же участке. Эту ломкость (хрупкость) обнаружил врач Lubs в конце шестидесятых 20 века, а в начале девяностых был выявлен ген FMR1, отвечающий за появление данной «хрупкости» и влияющий на прекращение выработки определенного белка, который необходим для правильного созревания нервной системы.
Благодаря быстрому научному прогрессу в 20 веке и интересу многих врачей к этому синдрому, в последнее время появилось много публикаций, которые подробно рассказывают о данной проблеме. Почему врачи и ученые всего мира так заинтересовались этой болезнью? Дело в том, что болезнь Мартина-Белла дает высокий процент инвалидности, а по частоте распространения она занимает второе место после синдрома Дауна среди болезней, сопровождающихся умственной отсталостью. На 2000 новорожденных в мире рождается один с синдромом Мартина-Белла, причем эта патология не зависит от места жительства, от экологических факторов, от материального достатка родителей. Известно только, что болезнь поражает чаще мальчиков и прогрессирует с самого раннего возраста. Что же выявили ученые?
Причины рождения детей с синдромом Мартина-Белла
Синдром Мартина-Белла ученые относят к группе генетических заболеваний, сцепленных с полом, а именно с Х-хромосомой. Как всем известно, женский хромосомный набор половых клеток состоит их двух Х хромосом (ХХ), а у мужской - из ХУ. В этой генетической информации, передающейся ребенку, заложены все сведения о будущем организме.
При болезни Мартина - Белла концы Х хромосомы истончаются. Это сужение происходит из-за повторяющихся определенных единичных тринуклеотидов цитозин-гуанин-гуанин (ЦГГ). В норме таких повторов может быть от 29 до 31, а при болезни Мартина -Белла их количество может увеличиваться до 4000 раз! Чрезмерный повтор ЦГГ называется генетическим «заиканием». В результате полной мутации нарушается работа специфического гена — FMR1, который ответственен за нормальное развитие нервной системы, за связь нервных клеток друг с другом.
Так как у мужчин в половой клетке всего одна Х хромосома, то синдром выражен у них более ярко и проходит тяжелее. У женщин синдром Мартина-Белла развивается, если встречаются две пораженные Х хромосомы. Но такое встречается крайне редко. Поэтому женщины обычно переносят болезнь, причем они их передают как мальчикам, так и девочкам в одинаковых пропорциях. Мужчина же может передать патологическую Х хромосому только девочке, мальчики получают Y хромосому. Из рода в род происходит накопление повторов ЦГГ (парадокс Шермана). Мужчины с полной мутацией, как правило, бесплодны.
Пока неизвестно, что именно провоцирует премутацию гена, но благодаря научным исследованиям в этой области на сегодняшний день появилась возможность не только выявить эту болезнь, но и лечить ее. По крайней мере, уже синтезированы лекарственные средства, которые способны улучшить состояние больного.
Симптомы и признаки синдрома Мартина-Белла
Симптомы и признаки синдрома Мартина-Белла зависят от количества повторений (генетического заикания) остатков аминокислот (ЦГГ).
Если повторов ЦГГ от 40 до 60, то клинических изменений может не быть. Врачи называют такое состояние - промежуточным или аллеей серой зоны. Но по наследству эта патология будет передаваться, повторы ЦГГ накапливаться, и через несколько поколений признаки болезни будут налицо.
Если повторы доходят до 200, то видимых поражений центральной нервной системы не будет. Такое состояние врачи-генетики называют премутацией. Если у пациента премутация, то в пожилом возрасте (у каждого третьего больного) может развиваться атаксический синдром (двигательные и координационные расстройства). Для женщин характерен ранний климакс - в возрасте около 40 лет. Вероятность передачи по наследству в случае премутации высокая.
Если повторов более 200, то это уже полная мутация. То есть ген FMR1 в Х - хромосоме полностью отключается, а значит и прекращается выработка специфического белка, который отвечает за развитие мозга.
Эта болезнь очень напоминает аутизм, поэтому распознать синдром Мартина-Белла клинически сложно. Судя по симптомам, болезнь затрагивает не только нервную систему, но и соединительную ткань, так как поражается и кожа, и связочный аппарат, и костная система.
Клинические проявления синдрома Мартина-Белла у детей
Ребенок, как правило, рождается массой от 3,5 кг до 4. Родители больного ребенка предъявляют жалобы на заторможенность, робость, боязнь толпы. С возрастом становится все более заметной задержка в психо-моторном развитии. Но у девочек интеллектуальные способности страдают меньше. Ребенок значительно позже своих сверстников начинает сидеть, ходить, разговаривать. В некоторых случаях речь может отсутствовать. У него большая голова с широким лбом, иногда очень светлая радужка и светлые волосы, могут отмечаться глазодвигательные нарушения, очень гибкие пальцы.
В дальнейшем ребенок может быть агрессивен, гиперактивен, двигательно расторможен. Возможны стереотипные гримасы (зажмуривание глаз, нахмуривание), он размахивает руками, хлопает в ладоши, что-то бормочет про себя, монотонно хнычет, его могут беспокоить навязчивые идеи. Он чувствителен к свету, к громким звукам, не переносит, когда смотрят в его глаза - отводит взгляд. И вообще, ребенок тревожный и пугливый, есть склонность к истерикам. Эти проявления сходные с клинической картиной детской шизофрении, диагноз которой иногда ошибочно ставят. Для мальчиков и для девочек характерна склонность к ожирению.
В школе могут быть проблемы по некоторым предметам, например, математикой из-за рассеянного внимания. Ребенку сложно общаться со сверстниками, у него ограничены социальные навыки.
Высокоспецифическим симптомом болезни Мартина-Белла будет увеличение в размере яичек у мальчиков в пубертатном периоде. Также будет специфичным раннее половое созревание у девочек, при этом и у мальчиков, и у девочек эндокринные функции сохранены.
Особенности проявления синдрома Мартина-Белла у взрослых
Прежде всего, у больных с синдромом Мартина-Белла обращает на себя внимание их внешность: крупная голова, низко расположенные оттопыренные уши, клювовидный, но округлый кончик носа с широким основанием, тяжелый внушительный лоб, узкое лицо с выступающим подбородком.
Со стороны конечностей - большие кисти и стопы, плоскостопие, суставы очень гибкие. Кожа сверхэластичная, легко растяжимая.
Речь больных воспринимается с трудом, так как она своеобразная и очень быстрая. Для нее характерно частое повторение целых фраз, предложений, могут непроизвольно повторяться слова говорящего или окончания этих слов. Речь ускоряется, слова опережают друг друга. Нарушения речи могут быть и в виде заикания.
Больной невнимателен, ему трудно находиться на одном месте. Типичны вычурные позы, он может совершать бессмысленные движения, похлопывания, постукивания, возможно появление аффективной агрессии, Психическое состояние больных с синдромом Мартина-Белла характеризуется недоразвитием интеллекта (IQ = 30—75).
Неврологические расстройства не очень выражены (гиперкинезы в виде зажмуривания глаз, нахмуривание бровей, тремор), но в тяжелых случаях возможны эпилептические припадки (у 10% больных).
У женщин появляется преждевременная менопауза, для них особенно характерны симптомы вегето-сосудистой дистонии.
В пожилом возрасте, если пациент является носителем премутации, может возникнуть тремор. Пациент начинает жаловаться на забывчивость (особенно стираются из памяти недавние события - нарушение кратковременной памяти). Больной перестает понимать речь и не может читать. Постепенно развивается слабоумие.
Диагностика синдрома Мартина-Белла
Для того, чтобы был поставлен диагноз «синдром Мартина-Белла» необходимо обратиться к генетику. Только врач-генетик может подтвердить или опровергнуть эту болезнь, так как применяет специальные методики для диагностики. Конечно, определенную роль играет осмотр пациента и выявление таких специфических признаков, как оттопыренные низкопосаженные уши, клювовидный нос, большая голова, низкий интеллект, своеобразная речь. Но все эти явления не дают основания поставить диагноз «ломкой хромосомы».
Для подтверждения диагноза врачи применяют цитогенетический метод. Точность современных диагностических технологий в Германии достигает более 95%.
У пациента берут клеточный материал, в который добавляют фолиевую кислоту, которая будет провоцировать изменения в хромосомах. Если появляется участок хромосомы с истончением, то можно предполагать синдром Мартина-Белла (перед исследованием надо убедиться, что накануне не было приема поливитаминов, которые содержат фолиевую кислоту).
Другим высокоспецифичным методом является исследование повторений ЦГГ с помощью полимеразной цепной реакции(ПЦР). На ЭЭГ также отмечаются характерные для болезни признаки. Все технологии базируются на применении самой современной аппаратуры, которой оснащены все клиники Германии.
К счастью, эту болезнь можно выявить и с помощью пренатальной диагностики, которая находится в Германии на самом высоком уровне. Взятие хориона и амниотической жидкости на проведение генетического теста позволяет обнаружить премутацию и мутацию хромосом.
Если во время исследования обнаружены признаки генной мутации, паре предлагаются дальнейшие шаги по предупреждению или лечению данной патологии.
Лечение синдрома Мартина-Белла
Лечение детей с синдромом Мартина-Белла эффективнее у детей, чем у взрослых. Специфической терапии пока не существует. Если вовремя предложена адекватная схема лечения и правильная программа обучения, то большинство детей с синдромом Мартина-Белла можно научить ходить, разговаривать, читать, писать.
Лечение синдрома Мартина-Белла основывается на современных технологиях и новых достижений мировой науки и медицины. Она сводится к симптоматической терапии, которая способствует:
Эффективность лечения больного с синдромом Мартина-Белла является не очень высокой и результативной, но правильно назначенная терапия может тормозить развитие болезни.
В Германии с каждым больным работает психиатр и психоневролог, а занятия с логопедом помогают избавиться от дефектов речевого построения. Благодаря активному взаимодействию высококвалифицированных врачей разных специальностей, применению новейших достижений фармакологической промышленности, инновационным технологиям лечение синдрома Мартина-Белла в клиниках Германии помогает ослабить симптомы заболевания.
Прогноз больных с синдромом Мартина-Белла
Средняя продолжительность жизни у лиц, страдающих синдромом Мартина-Белла, такая же, как и у других людей. Дети чаще болеют инфекционными заболеваниями, более повержены травмам из-за повышенной подвижности.
Профилактика синдрома Мартина-Белла
Главный и единственный способ профилактики - это обследование беременных. В Германии проводится скрининг на ранних стадиях беременности, когда еще есть возможность прервать ее. Если пара только планирует беременность, то врачи могут предложить экстракорпоральное оплодотворение, которое поможет будущему ребёнку унаследовать «здоровую» Х-хромосому.
Синдром Мартина-Белл ( Синдром ломкой X-хромосомы )
Синдром Мартина-Белл - это наследственная болезнь, которая характеризуется стойким интеллектуальным снижением, расстройствами аутистического спектра и специфическими фенотипическими особенностями. Ключевой симптом - недостаточность познавательных функций. Отмечается гиперактивность, дефицит коммуникативных способностей, замкнутость. Лицо удлиненное, ушные раковины большие, лоб выступающий, кончик носа загнутый. Диагностика основывается на клинико-анамнестических данных и результатах биогенетического анализа. Лечение симптоматическое, включает использование медикаментов и психолого-педагогическую коррекцию.
МКБ-10
Общие сведения
Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в X-хромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая - резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана.
При секвенировании FMR1-гена было выявлено, что основой симптоматики и цитогенетически определяемой ломкости хромосомы X является многократное увеличение количества единичных тринуклеотидов ЦГГ. Это приводит к подавлению транскрипции и последующему недостаточному производству белка FMR1, ответственного за развитие центральной нервной системы, а именно - за формирование аксонов и синапсов, появление и усложнение нейронных связей, успешность процессов обучения и запоминания.
Участок хромосом, подверженный структурным изменениям при наследственном синдроме Мартина-Белл, может находиться в четырех состояниях, характеризующихся различным удлинением повторяющихся последовательностей тринуклеотидов. При отсутствии болезни и носительства определяется нормальное количество повторов - от 6 до 39. В промежуточном состоянии диагностируется 40-60 повторов, в состоянии премутации - 55-200. В обоих случаях заболевание отсутствует. Поскольку экспансия тринуклеотидов возможна лишь в период гаметогенеза, премутация способна превратиться в полную мутацию. Это происходит при передаче измененного материнского гена, аллель «утяжеляется» во время овогенеза. При полной мутации выявляется больше 200 повторов ЦГГ, чаще всего - от 230 до 4 000.
Дети рождаются с увеличенной массой тела, в среднем - 3,5-4 кг. Первыми обращают на себя внимание фенотипические особенности младенцев. Характерен макроорхизм - увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Фенотипические признаки могут быть выражены по-разному, от одного-двух едва определяемых до полного комплекса.
Ключевое клиническое проявление заболевания - умственная отсталость. Стойкое интеллектуальное снижение проявляется слабым развитием сложных форм мышления и памяти. Пациентам недоступно понимание абстрактно-логических высказываний и явлений, использование категорий, установление аналогий. Сравнение, анализ и обобщение могут осуществляться на простом уровне, например, в конкретных бытовых ситуациях. Словарный запас обеднен. У многих мальчиков IQ равен 40-50 баллам, реже достигает 70-79. Относительно сохранна номинативная речь и зрительное восприятие. У девочек когнитивное снижение менее выраженное, соответствует легкой степени олигофрении или пограничному уровню интеллектуального развития.
Другой типичный симптом заболевания - своеобразие речи. Она ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети часто проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм - полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самопровреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации.
Неврологические симптомы неспецифичны. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. Недостаточное развитие мелкой моторики затрудняет освоение письма, некоторых игровых и бытовых навыков (сборки конструктора, рисования, шитья и др.). У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы, например, зажмуривание глаз, нахмуривание бровей, гримасничанье. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников.
Диагностика
При выраженных фенотипических изменениях заболевание может быть обнаружено с первых месяцев жизни ребенка - неонатологи и врачи-педиатры обращают внимание на увеличенные размеры яичек и характерные особенности лица. В иных случаях подозрение на умственную отсталость возникает в возрасте от полугода до 2-3 лет. В этот период прослеживается отставание умственного развития, поведенческие и речевые нарушения. Дифференциальная диагностика нацелена на исключение РАС, в частности раннего детского аутизма, а также умственной отсталости другого происхождения (не связанной с ломкостью хромосомы Х). Обследование проводится психиатрами, неврологами и врачами-генетиками, включает:
Лечение синдрома Мартина-Белл
Методы специфической терапии синдрома в настоящее время отсутствуют. Проводится симптоматическое медикаментозное лечение и психолого-педагогическая коррекция. Усилия врачей и специальных психологов направлены на минимизацию эмоционально-поведенческих отклонений, овладение навыками ходьбы, речи и общения, чтения и письма. Медикаментозная терапия включает прием психостимуляторов, антидепрессантов, ноотропов, противоэпилептических средств и гормональных препаратов (при первичной недостаточности яичников). Обучение пациентов проводится по специальным коррекционно-развивающим программам. Для улучшения социальных навыков используются методы когнитивно-поведенческой терапии, групповые тренинги.
Прогноз и профилактика
Синдром Мартина-Белла не имеет осложнений и не сокращает продолжительность жизни больных, поэтому при своевременной и адекватной медико-психолого-педагогической помощи прогноз достаточно благоприятный: пациенты осваивают навыки общения и самообслуживания, обучаются в специальных школах, иногда овладевают рабочими профессиями. Профилактика основана на медико-генетическом консультировании пар из групп риска и пренатальной диагностике синдрома. Эти меры необходимы женщинам с синдромом преждевременного истощения яичников, семьям, в которых диагностированы премутационные состояния FMR1 или выявлены случаи интеллектуальной недостаточности у мальчиков и мужчин.
Читайте также: