Синдром Моркио (Morquio) - синонимы, авторы, клиника
Добавил пользователь Евгений Кузнецов Обновлено: 09.01.2026
Мукополисахаридоз IV типа (MPS IV A) или синдром Моркио A (Syndrome Morquio A; MIM 253000 ) впервые описал Morquio в 1929 году ( Morquio L.,1929 ). При данном заболевании отмечаются значительные деформации скелета, особенно грудной клетки. В отличие от других типов мукополисахаридозов IV тип характеризуется отсутствием снижения интеллекта, помутнения роговицы, гепатоспленомегалии и гротескных черт лица ( Beck M.,1986 ; Guiney W.B.,1982 ; Hechit J.T.,1984 ; Holzgreve W.,1981 ; Nelson J.,1988 ; RaasRothschild A.,1999 ; Bargal B.,2001 ).
Мукополисахаридоз IV A типа подразделяется на тяжелую или классическую ( Morquio L.,1929 ), промежуточную ( Glossl J.,1981 ) и легкую ( Orii T.,1981 ; Hechit J.T.,1984 ; Beck M.,1986 ) формы.
Содержание
Наследование
Заболевание наследуется по аутосомно-рецессивному типу.
Локализация гена.
Ген N-ацетилгалактозамин-6-сульфат-сульфатазы (GALNS) локализуется на 16 хромосоме человека в позиции q24.3 ( Baker E.,1993 ; Masuno M.,1993).
Ген N-ацетилгалактозамин-6-сульфат-сульфатазы (GALNS)
Gene: [16q243/GALNS] N-acetylgalactosamine-6-sulfate sulfatase
В 1991 году Tomatsu S. клонировал и секвенировл полноразменую кДНК N-ацетилгалактозамин-6-сульфат-сульфатазы (GALNS) человека ( Tomatsu S.,1991 ). Кодирующая последовательность из 1566 нуклеотидов кодирует полипептид из 522 аминокислот, состоящий из 26 аминокислот сигнального пептида и 496 аминокислот зрелого пептида. Пропептид с молекулярным весом 60 kDa в результате протеолитического расщепления формирует 2 пептида с молекулярными весами 40 kDa 15 kDa.
Nakashima Y. и Morris C.P. анализировали структуру данного гена ( Nakashima Y.,1994 ; Morris C.P.,1994 ). По протяженности ген занимает около 50 kb и разделяется 13 интронами на 14 экзонов. Размер экзонов колеблется от 75 до 790 нуклеотидов, размер интронов варьирует от 380 до 14000 нуклеотидов. Экзон-интронные сочленения гена представлены на рис. MPS IV A int ( Рис. MPS IV A int ).
5'фланкирующая область гена утрачивает канонические "TATA-box" и "CCAAT-box" последовательности. Вместо данных участков промоторная область несет четыре инвертированных (antisense) "GC-box" (CCGCCC), 5 (sense) "GC-box" (GGGCGG) последовательностей, сайты связывания транскрипционных факторов Sp1. Сигнал полиаденилирования AATAAA локализуется в 14 экзоне через 33 нуклеотида от поли-A сайта. В 5 и 6 интроне локализуются Alu повторы и VNTR-подобные последовательности. В клетках выявляется единичная мРНК - 2,3 kb.
Нуклеотидная и аминокислотная последовательности гена N-ацетилгалактозамин-6-сульфат-сульфатазы рис. MPS IV A seq.
Строение гена
Биохимия
Биохимия N-ацетил-6-сульфат-сульфатазы (GLANS)
Мукополисахаридоз IV A типа обусловлен мутационными поражениями гена N-ацетилгалактозамин-6-сульфат-сульфатазы (GALNS). Лизосомный фермент - N-ацетилгалактозамин-6-сульфат-сульфатаза (N-acetylgalactosamine-6-sulfatase; GALNS; EC 3.1.6.4 ) осуществляет гидролиз сульфатно-фосфорной группы N-ацетилгалактозамин-6-сульфата у хондроитин-6-сульфата и кератансульфата. Дефицит N-ацетилгалактозамин- 6-сульфат-сульфатазы сопровождается внутрилизосомным накоплением и уринальной экскрецией недградированных или частично деградированных гликозамингликанов - хондроитин-6-сульфата и кератансульфата ( DiFerrante N.M.,1978 ; Fujimoto A.,1983 ; Gibson G.J.,1987 ; Linker A.,1970 ; Matalon R.,1974a ; Matalon R.,1974b ; Pedrini V.,1962 ; Robins M.M.,1963 ; Singh J.,1976 ; Horwitz A.L.,1978 ; Maroteaux P.,1967 ; Yuen M.,1985 ). N-ацетилгалактозамин-6-сульфат-сульфатаза выделена в чистом виде из печени и плаценты человека ( Bielicki J.,1991 ; Masue M.,1991 ).
Мутации гена
Первая мутация в гене N-ацетилгалактозамин-6-сульфат-сульфатазы (GALNS) была выявлена в 1992 году Fukuda S. ( Fukuda S.,1992 ). Полный спектр идентифицированных мутаций в гене N-ацетилгалактозамин- 6-сульфат-сульфатазы представлен в таблице MPS IV A m ( Таблица MPS IV A m ).
Мукополисахаридоз IV типа у детей
Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящее к поражению органов и тканей. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул.
Мукополисахаридоз IV типа (Синдром Моркио) - (синонимы: болезнь Моркио, спондило-эпифизарная дисплазия, хондроостеодистрофия, деформирующая остеохондродистрофия, Моркио - Брайлсфорда синдром, Моркио - Ульриха синдром, К - мукополисахаридоз, эксцентрохондроплазия, Дугве - Мелхиора - Клаузена синдром) - наследственная болезнь накопления, обусловленная дефицитом лизосомных гидролаз: галактозамин-6-сульфат-сульфатазы (МПС IVА) или β-галактозидазы (МПС IVВ), обусловлена отложением в соединительной ткани кератансульфата и характеризуется значительной деформацией скелета и отставанием в росте. Все вышеперечисленные признаки приводят к инвалидизации, а при тяжелом течении болезни - к летальному исходу 3.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
• Мукополисахаридоз IVА типа. Стеноз шейного и верхнегрудного отдела позвоночного канала. Миелопатия шейного отдела спинного мозга. Вальгусная деформация нижних конечностей, состояние после оперативного лечения. Правосторонний экссудативный отит. Аденоиды I степени. Правосторонняя кондуктивная тугоухость II степени. Двусторонний экссудативный отит. Левосторонняя сенсоневральная тугоухость I степени. OU катаракта вторичная начальная, смешанный астигматизм. OD начальная стадия помутнения роговиц. Недостаточность митрального и трикуспидального клапанов. ХСН 0ст. Состояние после надмыщелковой корригирующей остеотомии обеих бедренных костей от 2012г., медиального гемиэпифизеодеза дистальной зоны роста левой бедренной кости от 2015г.
В зависимости от первичного генетического дефекта, приводящего к снижению активность лизосомных ферментов выделяют несколько типов мукополисахаридозов.(табл. 1).
Таблица 1 - Классификация (номенклатура) МПС.
Этиология и патогенез
МПС IVА, ген GALNS локализован в хромосомной области 16q24.3. МПС IVВ -, ген GBS локализован в хромосомной области 3q21.33. Важно отметить, что мутация гена, кодирующего β-галактозидазу, вызывает также ганглиозидоз типа I.
Тип наследования: Наследуется по аутосомно-рецессивному типу.
Эпидемиология
Распространенность МПС IVА 1:250 000 новорожденных, МПС IVВ встречается еще реже.
Клиническая картина
Cимптомы, течение
Основные клинические проявления: значительные деформации скелета, особенно конечностей и грудной клетки 1.
Внешний вид: дети рождаются без признаков болезни. Первые симптомы появляются в возрасте 1-3 года; к 7-8 годам клиническая картина уже полностью выражена. Отмечается отставание в росте и физическом развитии. Кожа утолщена, ее тургор и эластичность снижены. Могут наблюдаться широкий рот, короткий нос, редкие зубы, возможно истончение зубной эмали. Аномалия грудной клетки, общая слабость мышц, Х-образная деформация ног, дисплазия тазобедренных суставов, укорочение шеи. Интеллект сохранён.
Костная система: болезнь характеризуется карликовостью (рост взрослого больного около 80-115 см), непропорциональным телосложением (относительно короткое туловище, микроцефалия, короткая шея). Выражена деформация скелета, особенно грудной клетки (куриная, бочкообразная, килевидная). Отмечается кифосколиоз грудного и поясничного отделов позвоночника, при тугоподвижности крупных суставов определяется расслабление связочного аппарата в мелких суставах, что приводит к их гипермобильности. Выявляются контрактуры в локтевых, плечевых, коленных суставах; вальгусная деформация нижних конечностей, плоскостопие.
Центральная нервная система: наиболее частым неврологическим осложнением у пациентов с МПС IV типа является шейная миелопатия. Причиной сдавления спинного мозга у этой группы пациентов является нестабильность шейных и грудных (реже) позвонков. В случае компрессии спинного мозга в шейном отделе отмечается поражение пирамидной системы, что может привести верхнему вялому и нижнему спастическому парапарезу. При грубых изменениях возникают условия для компрессии каудального отдела спинного мозга, что приводит к развитию вялого парапареза ног. Также могут иметь место нарушения тазовых функций. Интеллект обычно не нарушен или умеренно снижен. Характерно снижение слуха, с возрастом развивается глухота. Синдром запястного канала для пациентов с МПС IV типа не характерен.
Сердечно-сосудистая система: поражение сердца наблюдается довольно редко у детей. Типична недостаточность аортального клапана, реже митрального. Кардиомегалия обычно носит вторичный характер. На позднем сроке болезни проявления более выраженные.
Желудочно-кишечная система: отсутствует гепатоспленомегалия. Часто выявляются пупочные и паховые грыжи, расхождение прямых мышц живота.
Диагностика
Диагноз МПС IV устанавливается на основании совокупности клинических данных, результатов лабораторного исследования и молекулярно-генетического анализа (Приложение Г1).
В отличие от других типов, мукополисахаридозов IV тип характеризуется отсутствием снижения интеллекта, помутнения роговицы, гепатоспленомегалии 2.
• При сборе анамнеза и жалоб следует обратить внимание на следующие жалобы и анамнестические события 5:
Выраженность клинических проявлений в зависимости от возраста дебюта различных подтипов МПС IV, представлены в Приложении Г2.
Основные физикальные проявления - значительные деформации скелета, особенно конечностей и грудной клетки 3.
• Рекомендовано исследование активности галактозамин-6-сульфат-сульфатазы (в случае МПС IVA), β-галактозидазы (в случае МПС IVB) в культуре фибробластов, изолированных лейкоцитов, либо в пятнах крови, высушенных на фильтровальной бумаге.
Комментарий: у пациентов МПС IV типа выявляется снижение активности ферментов в зависимости от подтипа
• Рекомендовано молекулярно-генетическое исследование: выявление мутаций в генах GALNS (для МПС IVA) и GBS (для МПС IVB).
• Рекомендовано проведение ультразвукового исследования (УЗИ) органов брюшной полости, селезенки, почек.
Комментарии: выявляются деформации скелета, особенно грудной клетки (куриная, бочкообразная, килевидная), кифосколиоз грудного и поясничного отделов позвоночник. Выявляются уплощение и расширение тел позвонков, чем объясняется характерное укорочение туловища и необычно короткая шея. Выраженный углообразный кифоз. Рентген длинных трубчатых костей выявляет недоразвитость эпифизов, укорочение костей предплечья: локтевая кость не достигает лучезапястного сустава. Изменены кости таза: вертлужные впадины плоские и широкие, их крыша скошена, крылья подвздошных костей неправильной формы; контуры всех костей неровные; головки бедренных костей уплощены.
Комментарии: исследование позволяет оценить функциональное состояние мышечных тканей, нервов и нервно-мышечной передачи; стимуляционная электронейромиография (ЭНМГ) позволяет определить сдавление срединного нерва даже до появления симптомов и должна проводиться, начиная с возраста 4-5 лет ежегодно.
Комментарии: осуществляют для контроля изменений функции коры головного мозга и глубинных мозговых структур, своевременной диагностики эпилепсии.
Комментарии: для диагностики обструктивного апноэ сна проводится полисомнография, которая позволяет определить характер дыхательных нарушений (исключить центральный генез, связь с гипертрофией аденоидов, сердечной недостаточностью или комплекс причин).
Комментарии: регулярное проведение ЭКГ, Эхо-КГ, холтеровского мониторирования ЭКГ, суточного мониторинга артериального давления необходимо пациентам с данной патологией, так как с раннего возраста у них отмечаются сердечно-сосудистые нарушения.
Комментарии: клиническая картина неврологических проявлений и результаты объективных методов обследования не всегда коррелируют. Результаты магнитно-резонансной томографии (МРТ) головного мозга пациентов с МПС не являются диагностически значимыми для определения когнитивного дефицита.
Скрининг на клинические и визуализационные признаки компрессии спинного мозга. Нестабильность атлантоаксиального сустава может быть выявлена при рентгенографии шейного отдела позвоночника с нагрузкой, однако для подтверждения компрессии спинного мозга вследствие утолщения его оболочек требуется проведение МРТ.
• Рекомендовано проведение компьютерной томографии (КТ) головного мозга
Проводится с различными вариантами нанизма, наследственными заболеваниями скелета, сопровождающимися спондилоэпифизарными нарушениям,, 3.
Лечение
Очень важно симптоматическое лечение 4.
• Рекомендована коррекция нарушения осанки, контрактур суставов с использованием нехирургических методов включает физиопроцедуры и применение ортопедических устройств.
• При симптоматической эпилепсии рекомендовано назначение антиконвульсантов, однако дозировки рекомендуется использовать меньше среднетерапевтических для снижения риска развития возможных нежелательных эффектов.
Комментарии: подбор антиконвульсанта осуществляется психоневрологом в зависимости от вида приступов, локализации очага патологической активности.
• Коррекцию сердечно-сосудистой недостаточности, артериальной гипертензии рекомендуется проводить стандартными методами консервативного лечения, принятыми в детской кардиологии.
• Рекомендовано при рецидивирующих отитах, частых респираторных заболеваниях верхних дыхательных путей проведение симптоматической, по показаниям - антибактериальной терапии при отсутствии показаний к хирургическому вмешательству. При снижении слуха - подбор и ношение слуховых аппаратов.
• При офтальмологических нарушениях рекомендовано проведение лечения по показаниям, подбор терапии осуществляется на основании общепринятых рекомендаций по лечению соответствующих нозологий.
• При развитии сдавления спинного мозга, нестабильности атланто-аксиального сочленения рекомендовано хирургическое вмешательство.
• При наличии показаний к оперативному лечению шейной миелопатии и деформаций конечностей, первой рекомендовано производить оперативную декомпрессию спинного мозга. Только при благоприятном исходе данного вмешательства рекомендована оперативная коррекция деформаций конечностей. По показаниям рекомендовано осуществлять хирургическую коррекцию деформаций конечностей, исправление оси нижней конечности
• Несмотря на то, что нестабильность краниоцервикального перехода играет ведущую роль в патологии спинного мозга, рекомендовано рассмотреть вопрос проведения оперативной декомпрессии без окципитоцервикальной стабилизации, что может обусловить хорошие постоперационные результаты. Успешные результаты изолированной ламинэктомии с непродолжительном периодом наблюдения указывают на роль перманентного повреждения спинного мозга на фоне утолщения экстрадуральных мягких тканей, доказывая сочетанный патогенез изменений краниоцервикального перехода.
• В случае проведения спондилодеза после проведение оперативного вмешательства в течение длительного времени рекомендовано ношение гало-аппарата.
• При наличии показаний рекомендовано проведение других хирургических вмешательств: аденотомии, тонзиллэктомии, грыжесечения.
Медицинская реабилитация
Пациенту с мукополисахаридозом IV типа физиотерапевтом и врачом-ЛФК разрабатывается индивидуальный курс реабилитации, включающий массаж, лечебную физкультуру, физиотерапевтические процедуры (магнитотерапию, термотерапию, ударно-волновую терапию, метод биологической обратной связи и другие процедуры).
Реабилитационные курсы (массаж, ЛФК, физиопроцедуры, психолого-педагогическая помощь) желательно проводить в условиях дневного стационара проводится с частотой 3-4 раза в год, длительность - определяется тяжестью состояния и ответом на проводимые мероприятия.
Психолого-педагогическая помощь
Проводится в комплексе реабилитационных мероприятий. Коррекционно-педагогическое воздействие определяется в зависимости от тяжести и длительности течения болезни, структуры нарушений здоровья, степени недоразвития познавательной деятельности, типа эмоционального реагирования, особенностей поведения ребенка. Включение коррекционно-педагогического сопровождения в комплекс восстановительных мероприятий обеспечивает дополнительную оценку динамики психического развития как одного из важных показателей состояния здоровья, повышает эффективность терапевтических вмешательств, снижает экономическое бремя данной патологии за счет социализации пациентов и сохранения психологического потенциала трудоспособных членов семьи.
Необходимо оказание всесторонней помощи (медицинской, психосоциальной и материальной) детям с неизлечимыми ограничивающими срок жизни заболеваниями. В состав паллиативных служб входят врачи, медицинские сестры, психологи и социальные работники. Несмотря на тяжелое состояние и постоянную потребность в мониторинге, все пациенты преимущественно находятся дома в кругу своей семьи и друзей. Основной целью работы паллиативных служб является создание всех необходимых условий для обеспечения нахождения больных в домашних условиях, а не в стенах лечебного учреждения, что позволяет не только улучшить качество жизни больных и их семей, но и существенно снизить государственные затраты на постоянное стационарное лечение таких пациентов.
При проведении общей анестезии необходимо помнить о высоком риске компрессии спинного мозга вследствие нестабильности атлантоаксиального сустава. Короткая шея, ограничение подвижности нижней челюсти, увеличение языка, выраженная гипертрофия аденоидов и миндалин создают проблемы при проведении анестезиологического пособия, поэтому предпочтение следует отдавать местному или региональному обезболиванию. Пациент предварительно консультируется кардиологом, оториноларингологом, анестезиологом, невропатологом. Обязательно проведение полного кардиологического обследования, полисомнографии (для выявления степени дыхательных нарушений), при необходимости - эндоскопии носоглотки и компьютерной томографии легких. Оперативное вмешательство с анестезией необходимо проводить в крупных медицинских центрах, имеющих ОРИТ, так как интубация и последующая экстубация у таких пациентов может вызвать затруднения.
Прогноз
Летальный исход наступает до достижения возраста 20 лет вследствие сердечно-легочной недостаточности, развивающейся на фоне интеркуррентных заболеваний. Возможна внезапная смерть в результате смещения атланто-окципитального сочленения и повреждения ствола мозга.
Профилактика
Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью разъяснения генетического риска. Как и при других аутосомно-рецессивных заболеваниях при МПС тип IV, для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, существует возможность проведения пренатальной и преимплантационной диагностики. С этой целью генетик рекомендует родителям соответствующие диагностические лаборатории и медицинские центры.
Пренатальная диагностика возможна путем измерения активности ферментов в биоптате ворсин хориона на 9-11 неделе беременности. Для семей с известным генотипом возможно проведение ДНК-диагностики.
Заболевание имеет мультисистемную природу и необратимые, прогрессирующие клинические проявления, что обусловливает необходимость наблюдения не только узкими специалистами (оториноларингологами, хирургами-ортопедами, офтальмологами, кардиологами, пульмонологами, невропатологами, стоматологами), но и физиотерапевтами, логопедами, психологами и работниками паллиативных служб 1.
Пациенты с данной нозологией должны постоянно находиться под наблюдением; 1 раз в 6-12 мес. (в соответствии с тяжестью состояния) показано комплексное обследование в многопрофильных стационарах. Длительность нахождения в стационаре / дневном стационаре 21-28 дней.
Наблюдение больных по месту жительства (в амбулаторно-поликлинических условиях) должно осуществляться постоянно. Лабораторные и инструментальные обследования и рекомендуемая частота их проведения представлена в Приложении Г3.
Мукополисахаридоз: орфанное заболевание в практике педиатра
Редкие (орфанные) заболевания - это встречающиеся с определенной частотой жизнеугрожающие или хронические прогрессирующие заболевания, приводящие без лечения к смерти или пожизненной инвалидизации пациентов. Сегодня в мире насчитывается около 7 тыс. редких заболеваний. Примерно половина из них обусловлена генетическими отклонениями. Симптомы могут быть очевидны с рождения или проявляться в детском и более старшем возрасте.
В разных странах определение и перечень орфанных болезней принимаются на государственном уровне, единого определения для них не существует, так же как нет единого критерия отнесения заболеваний к этой группе.
В нашей стране к орфанным относятся заболевания, которые имеют распространенность не более 10 случаев заболевания на 100000 населения. С 1 января 2012 года в силу вступил новый Федеральный закон от 21.11.2011 №323-ФЗ «Об основах охраны здоровья граждан в Российской Федерации», в котором впервые на государственном уровне введено понятие редких (орфанных) заболеваний. В список орфанных болезней внесено 230 наименований. Постановление Правительства РФ от 26 апреля 2012 г. № 403 определило перечень из 24 из них, согласно которому пациенты должны быть обеспечены за сч ет государства необходимыми лекарственными препаратами и лечебным питанием. Одним из заболеваний, включ енным в группу орфанных, является мукополисаха-ридоз (код по МКБ-10 Е76.1 - «нарушение обмена гликозаминогликанов»).
Мукополисахаридозы (МПС) - это группа наследственных болезней соединительной ткани, обусловленных нарушением обмена гликозаминогликанов (ГАГ) (кислых мукопо-лисахаридов) в результате генетически обусловленной неполноценности 1 из 11 известных ферментов, участвующих в их расщеплении. МПС относится к лизосомным болезням накопления (табл. 1). Таблица 1. Типы мукополисахаридозов
Тип МПС
Название
Дефицит фермента
Синдром Гурлер (Hurler)
Синдром Шейе (Scheie)
Синдром Гурлер-Шейе (Hurler-Scheie)
Синдром Хантера (Hunter)
Синдром Санфилиппо (Sanfilippo)
ацетил-КоA альфа-глюкозаминидин ацетилтрансфераза
Синдром Моркио (Morquio)
Синдром Марото-Лами (Maroteaux-Lamy)
Синдром Слая (Sly)
Синдром дефицита гиалуронидазы (Natowicz)
Подавляющее большинство МПС наследуются по аутосомно-рецессивному типу. Исключение составляет болезнь Хантера (МПС II типа), которая наследуется по X-сцепленному рецессивному механизму.
Для МПС характерно полисистемное поражение. Формируются множественные деформации скелета и задержка физического развития. При некоторых формах наблюдается умственная отсталость. Выявляются нарушения сердечной деятельности и функции бронхол егочной системы, патология органов зрения и слуха, образование грыж, неврологические расстройства, увеличение печени и селезенки 2.
Представляется клиническая и лабораторная характеристика заболевания на примере МПС II типа. На сегодняшний день в Московской области наблюдается 9 детей с МПС, из них 7 - с МПС II типа.
Мукополисахаридоз II типа (синонимы: недостаточность фермента лизосом-ной идуронат-2-сульфатазы (a-L-идуро-носульфатсульфатазы), синдром Гунтера (Хантера) - сцепленное с Х-хромосомой рецессивное заболевание, возникающее в результате снижения активности лизосомной идуронат-2-сульфатазы, участвующей в метаболизме ГАГ. Возникает почти исключительно у мальчиков (XY). У гетерозиготных женщин клинических проявлений синдрома Хантера, как правило, не наблюдается («носители»). К настоящему моменту описано лишь 2 случая заболевания у девочек, связанных с инактивацией второй, нормальной, Х-хромосомы [2].
Это панэтническое заболевание, частота встречаемости в мире - около 1 на 75 000 живых новорожд енных мальчиков. Частота заболевания в популяции варьирует от 1 на 165 000 (Австралия) до 1 на 34 000 (Израиль). Развитие МПС II типа обусловлено мутациями в структурном гене лизосомной идуронат-2-сульфатазы - IDS, расположенном на длинном плече Х-хромосомы в локусе Xq28. В настоящее время описано более трехсот различных мутаций в гене IDS. Более 50% мутаций составляют точечные мутации, около 26% - мелкие делеции и вставки, 11% - крупные де-леции и перестройки гена IDS. У пациентов из России ДНК-анализ гена IDS показал, что крупные делеции и перестройки гена IDS составляют только 5,4% числа найденных мутаций [2].
Клинический фенотип гетерогенен и довольно условно подразделяется на тяж елую и л егкую формы, различающиеся по тяжести клинических фенотипов. У пациентов с тяж елой формой наблюдают сходные с МПС I типа (синдромом Гурлер) клинические симптомы, но заболевание прогрессирует медленнее (рис. 1).
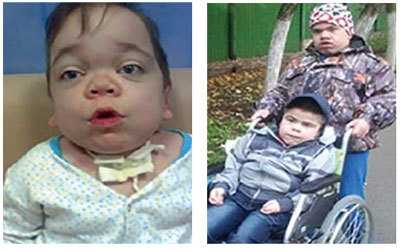
Рис. 1. У девочки (слева) диагноз МПС I типа (синдром Гурлер-Шейе). У мальчика (справа) диагноз МПС II типа (синдром Хантера).
(Фото из личного архива автора и Межрегиональной Благотворительной Общественной Организации «Хантер-синдром»)
Заболевание манифестирует в возрасте от 1 до 3 лет. Отмечается изменение черт лица по типу гарголоилизма, задержка роста, признаки множественного костного дизостоза, огрубление и утолщение кожи, прогрессирующее снижение интеллекта. Специфичными для данного типа МПС являются изменения на коже спины, груди, шеи цвета слоновой кости, «монголоидные пятна» в пояснично-крестцовой области. Нарушения функции органов пищеварения проявляются в виде гепатомегалии, хронической диареи. Среди неврологических нарушений преобладают симптомы прогрессирующей сообщающейся гидроцефалии, спастическая параплегия в результате компрессии спинного мозга и прогрессирующая тугоухость. У детей с синдромом Хантера отмечается тугоподвижность крупных и мелких суставов. Прогрессируют сердечно-легочные расстройства. Летальный исход обычно наступает на втором десятилетии жизни.
Для подтверждения МПС II типа проводится определение уровня экскреции ГАГ с мочой и измерение активности лизосомной идуронат-2-сульфатазы в лейкоцитах или культуре кожных фибробластов. В случае заболевания в моче возрастает суммарная экскреция ГАГ.
Проведение ДНК-анализа - длительная и сложная диагностическая процедура, позволяющая определить молекулярные дефекты, приводящие к болезни Хантера, наиболее часто используется для определения носительства и пренатальной диагностике в семьях, имеющих больного ребенка. Применяются методы косвенной ДНК-диагностики, основанной на исследовании локусов Х-хромосомы, расположенных близко к гену IDS. Кроме того, пренатальная диагностика проводится пут ем измерения активности идуронат-2-сульфатазы в биоптате ворсин хориона на 9-11-й неделе беременности или определения спектра ГАГ в амниотической жидкости на 20-22-й неделе беременности.
Наиболее часто дифференциальная диагностика проводится внутри группы МПС, а также с другими лизосомными болезнями накопления (муколипидозами, галактосиалидозом, ганглиозидозом и др.).
В качестве клинического примера приводится выписка из истории болезни Никиты Б., 11.12. 2011 г.р.
Анамнез жизни. Ребенок от II беременности (I беременность окончилась выкидышем на раннем сроке), протекавшей на фоне маловодия, гипоксии плода, угрозы прерывания, отеков. Роды срочные, самостоятельные. Вес при рождении 4040 г, рост 58 см. С рождения на грудном вскармливании. Формула развития: голову держит с 2 мес., переворачивается с 5 мес., сидит с 9 мес., ходит с 1 г. 1 мес. К году - 5 слов. Перенесенные заболевания - ОРВИ 2 раза, трахеит.
Анамнез заболевания. С рождения - водянка оболочек яичек, с 5 мес. - пахово-мошоночная грыжа, в 7 мес. диагностирован кифоз поясничного отдела позвоночника, в 12 мес. по данным ЭКГ - синдром ранней реполяри-зации желудочков НБПНГ, в 1 г. 3 мес. проведена Эхо КГ, выявлена аневризма МПП. В 1 г. 4 мес. ребенок обследован в МГНЦ РАМН, заподозрен и подтвержден биохимическим и молекулярно-генетическим методами МПС II типа.
При поступлении в клинику МОНИКИ (2013 г.) состояние реб енка средней тяжести. Вес 15 кг. Рост 93 см. Волосы жесткие, тусклые, макростомия, макроглоссия, короткая шея, легкие черты гарголоидизма, камподактилия 4 и 5 пальцев справа, широкое пупочное кольцо, кифоз поясничного отдела позвоночника, ки-левидная деформация грудной клетки, пахо-во-мошоночная грыжа слева, кожа плотная на ощупь. Гипертрофия миндалин 2-3 ст. Дыхание через нос не затруднено. Дыхание везикулярное. Тоны сердца ритмичные, акцент второго тона. ЧСС 120 уд/мин. Живот увеличен в объеме, печень +3,5см, +4, см верхняя треть. Селезенка не пальпируется. Стул оформленный. Дизурических явлений нет.
Неврологический статус: в сознании, гиперактивен, быстро возбудим, говорит отдельные слова. Общемозговых, менингеальных знаков нет. ОГ=50 см. глазные щели OD=OS. Зрачки OD=OS. Движение глазных яблок в полном объеме. Низкий тембр голоса. Быстро истощается, ходит самостоятельно, легкая атаксия. Мышечная гипотония. Сухожильные рефлексы живые.
Общий анализ крови и мочи без патологических изменений.
Биохимический анализ крови - умеренное повышение уровня АЛТ (42 Ед/л), АСТ (49 Ед/л).
ЭКГ: вертикальное положение ЭОС, выраженная синусовая тахикардия 182-200 на фоне беспокойства. В ортостазе ЧСС 111-166 уд/мин.
УЗИ брюшной полости: печень увеличена, правая доля 100 мм, левая - 45 мм. Структура однородная, стенки сосудов и протоков уплотнены. Эхогенность повышена. Поджелудочная железа 14×8×15 мм, структура неоднородная, эхогенность не изменена. Желчный пузырь - форма обычная, просвет чистый, стенки не утолщены. Селезенка не увеличена 62×32 мм. Структура однородная. Почки без патологических изменений. Яички в мошонке, вагинальный отросток брюшины расширен с обеих сторон до 3 мм.
ЭЭГ: диффузные общемозговые изменения биоэлектрической активности по органическому типу в виде доминирования дельта-активности частотой 3-4 Гц по всем отделам конвекса безградиентно в сочетании с отдельными группами низкочастотных тета-колебаний. Эпилептиформная активность, фотопа-роксизмальная реакция не выявлены.
На основании комплексного исследования установлен диагноз: мукополисахаридоз II типа (синдром Хантера). Дегенеративное заболевание нервной системы. Задержка речевого развития. Синдром гипервозбудимости. Множественный костный дизостоз. Порок развития L1-L2 позвонков, врожденный кифоз. 2-х стороннее гидроцеле. Пупочная грыжа.
По жизненным показания ребенку назначено внутривенное введение «Элапразы» в дозе 0,5 мг/кг еженедельно (15 кг×0,5=7,5 мг на введение). В комплексную терапию включены занятия с логопедом, дефектологом, курсы физиотерапии, ЛФК, L-карнитин (Элькар), Кудесан, Глицин, Церебролизин, МагнеВ6. В настоящее время ферментнозаместительную терапию ребенок получает по месту жительства.
Список литературы:
1. Neufeld E. F., Muenzer J. The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, et al (eds). The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill; 2001: 3421-3452.
3. Meikle P.J. et al. Prevalence of lysosomal storage disorders/ JAMA. - 1999. - 281: 249-254.
Синдром Моркио: причины, симптомы, лечение
Существует отдельная группа болезней обмена веществ - мукополисахаридозы. Связаны они с нарушением в организме обмена особых соединений - гликозаминогликанов, которые характеризуются поражением разных органов и тканей. Выделяют несколько типов мукополисахаридозов, IV из которых является синдром Моркио.
Синдром Моркио - наследственная болезнь, которая обусловлена дефицитом в организме ферментов галактозамин-6-сульфат-сульфатазы или β-галактозидазы и характеризуется отложением в тканях гликозаминогликана (кератансульфата), что приводит к выраженным деформациям костной системы и отставанию больного в росте, а в итоге - к его инвалидизации и даже смерти.
Синонимами мукополисахаридоза IV типа, или синдрома Моркио, являются спондило-эпифизарная дисплазия, болезнь Моркио, синдром Моркио-Ульриха или Моркио-Брайлсфорда, хондроостеодистрофия, К-мукополисахаридоз, синдром Дугве-Мелхиора-Клаузена, эксцентрохондроплазия.
Впервые болезнь описана в 1929 году уругвайским врачом Л. Моркио и английским Дж. Брайлсфордом. Регистрируется с частотой 1 случай на 250 тысяч новорожденных или еще реже.
О том, почему возникает это заболевание, о симптомах, принципах его диагностики и направлениях лечения вы узнаете из нашей статьи.
Причины и механизм развития
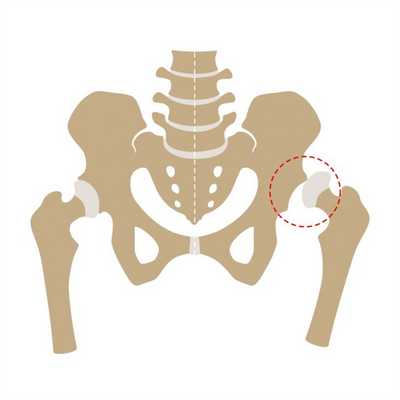
У многих лиц, страдающих синдромом Моркио, обнаруживается дисплазия тазобедренных суставов.
Мукополисахаридоз IV типа возникает в результате мутации в гене, отвечающем за выработку особого вещества - галактозамин-6-сульфат-сульфатазы. Это наследственная патология, которая передается из поколения в поколение по аутосомно-рецессивному типу. Это означает, что у родителей, являющихся носителями болезни (они имеют по 1 мутантному гену), с 25%-й вероятностью ребенок будет здоров, с 50%-й - носителем поврежденного гена и еще с 25%-й - заболеет мукополисахаридозом IV типа. Если же мутантный ген имеется лишь у одного из родителей, их потомство не заболеет, но 1 из 4 детей также получит патологический ген и будет являться носителем.
Галактозамин-6-сульфат-сульфатаза (GALNS) в здоровом организме участвует в расщеплении кислых гликозаминогликанов. Генная мутация приводит к тому, что GALNS вырабатывается мало и гликозаминогликаны не расщепляются, а накапливаются в тканях организма, вызывая соответствующую клиническую симптоматику.
Стоит отметить, что синдром Моркио - гетерогенная болезнь. Существует порядка 250 мутаций указанного выше гена, что обусловливает у разных больных разную клиническую картину и тяжесть течения патологии.
Симптомы
Основные проявления болезни связаны с изменениями скелета, но поражаются и другие органы и системы организма человека.
Изменения внешнего вида
Новорожденные дети выглядят здоровыми. Патологические изменения становятся заметны визуально лишь через 1-3 года, а к 7-8 годам клиническая картина синдрома Моркио выражена максимально.
- Ребенок низкорослый, отстает от сверстников в физическом развитии.
- Кожа его утолщена, слабо эластичная, со сниженным тургором.
- Может иметь место короткий нос, широкий рот с редкими зубами, эмаль которых, вероятно, будет истончена.
- Шея короткая.
- Грудная клетка деформирована.
- Мышечная сила в той или иной степени снижена.
- Ноги деформированы - сходятся к коленям, имеют вид буквы Х. .
- Изменения со стороны интеллекта отсутствуют.
Изменения со стороны костной системы
- Больные низкорослы - рост взрослого, страдающего данной патологией, варьируется в пределах от 80 до 115 см.
- Телосложение их диспропорционально: шея и туловище короткие, голова маленького размера.
- Грудная клетка деформирована - бочкообразная, куриная, килевидная.
- Грудной и поясничный отделы позвоночника искривлены в сторону и кзади (то есть имеет место кифосколиоз).
- Крупные суставы тугоподвижны, мелкие - напротив, гипермобильны (объем движений в них превышает таковой у здоровых лиц).
- В крупных суставах конечностей (как правило, коленных, плечевых и локтевых) обнаруживаются контрактуры.
- Вальгусная (х-образная) деформация нижних конечностей.
- Разной степени плоскостопие.
Центральная нервная система
Наиболее распространенное проявление мукополисахаридоза IV типа со стороны ЦНС - шейная миелопатия. Нестабильность позвонков шейного и грудного отдела позвоночника приводит к компрессии (сдавлению) спинного мозга.
- Компрессия шейного отдела спинного мозга характеризуется поражением пирамидной системы, развитием верхнего вялого и нижнего спастического парапареза.
- Результатом сдавления каудального (хвостового, нижнего) отдела спинного мозга становится вялый парапарез нижних конечностей.
- У ряда больных нарушаются функции тазовых органов.
- Слух снижается с возрастом вплоть до глухоты.
- Интеллект сохранен или незначительно снижен.
Сердце и сосуды
У детей сердце, как правило, в патологический процесс не вовлечено. По мере прогрессирования болезни может возникнуть недостаточность аортального или митрального клапанов и вторичная кардиомегалия (увеличение сердца в размерах), что проявляется соответствующей симптоматикой.
Орган зрения
У некоторых больных имеет место помутнение роговицы.
Изменения со стороны органов пищеварения
При мукополисахаридозе IV типа зачастую обнаруживаются паховые, пупочные грыжи и расхождение прямых мышц живота.
Печень и селезенка не увеличены.
Принципы диагностики
В анализе крови таких больных обнаружится дефицит ферментов галактозамин-6-сульфат-сульфатазы или β-галактозидазы.
Диагноз «синдром Моркио» очень серьезный. Выставляют его на основе жалоб, данных анамнеза жизни и заболевания, объективного обследования пациента, результатов лабораторных и инструментальных методов исследования.
Общаясь с пациентом, врач обратит внимание на такие жалобы:
- слабость в конечностях;
- ухудшение переносимости нагрузок;
- появление деформаций грудной клетки и других частей скелета;
- боли в спине и нижних конечностях, чувство онемения, покалывания, «ползания мурашек» в них;
- ухудшение, неловкость мелкой моторики;
- непроизвольное мочеиспускание и другие нарушения функций тазовых органов;
- синдром обструктивного сонного апноэ.
Важную роль в диагностике играет указание больного или его родителей на сходные симптомы у кого-то из родственников, что является намеком на генетическую природу диагностируемого заболевания.
Среди лабораторных методов диагностики значение имеют следующие:
- анализ мочи на спектр в ней гликозаминогликанов и их концентрацию (при легких формах количество этих веществ может быть в пределах нормы);
- анализ компонентов крови на степень активности галактозамин-6-сульфат-сульфатазы или β-галактозидазы (в зависимости от подтипа синдрома Моркио - А или В - активность того или иного фермента будет снижена);
- молекулярно-генетическая диагностика (в указанных ранее генах будут выявлены мутации).
В зависимости от особенностей клинической картины мукополисахаридоза IV типа больному могут быть назначены следующие методы инструментальной диагностики:
- УЗИ органов брюшной полости и забрюшинного пространства (почек);
- рентгенография разных отделов скелета (выявляется множество разнообразных изменений: недоразвитость эпифизов длинных трубчатых костей (бедренной, плечевой и прочих), укорочение локтевой кости (она не достигает лучезапястного сустава), уплощение головок бедренных костей и вертлужных впадин, изменение формы крыльев подвздошных костей, изменения грудной клетки и позвоночного столба, описанные выше - в разделе «симптомы»);
- аудиометрия (может быть диагностировано снижение слуха); (позволяет обнаружить нарушения функции внешнего дыхания, которые могут стать следствием деформаций грудной клетки и позвоночника);
- полисомнография (может быть диагностировано сонное апноэ и выявлена его природа); и УЗИ сердца, холтеровское мониторирование (позволяют обнаружить изменения функционирования сердца и сосудов, пороки сердца, а также оценить их динамику с течением времени);
- электронейромиография, электромиография (обнаруживают слабость мышц и сдавление нервов даже в бессимптомную стадию); (позволяет верифицировать нарушения функции головного мозга, вовремя обнаружить очаги эпилептической активности);
- КТ и/или МРТ головного мозга, шейного отдела позвоночника (чтобы более детально рассмотреть изменения, обнаруженные иными методами диагностики, визуализировать компрессию спинного мозга и так далее).
Дифференциальная диагностика
В связи со сходными клиническими проявлениями синдром Моркио дифференцируют с такими заболеваниями:
- (никорослостью) разнообразной природы;
- генетическими болезнями костной системы, сопровождающимися деформациями его отделов.
Принципы лечения
Практически все больные получают симптоматическую консервативную терапию, нередко возникают показания и для хирургического лечения.
Консервативное лечение включает в себя:
- ортопедическую и физиотерапевтическую коррекцию нарушений осанки и контрактур суставов;
- противосудорожные препараты - если диагностирована симптоматическая эпилепсия; подбор лекарственного средства осуществляет врач-психоневролог в зависимости от особенностей течения эпилепсии конкретного пациента; , сердечные гликозиды и прочие лекарственные средства для коррекции сердечно-сосудистой патологии;
- антибиотики - в случае частых ОРВИ, рецидивирующих отитов и других инфекционных заболеваний; средства симптоматической терапии их;
- подбор слухового аппарата - в случае патологии слуха;
- при патологии со стороны органа зрения - соответствующее лечение у врача-офтальмолога.
Показаниями к хирургическому лечению являются:
- нестабильность атланто-аксиального сочленения;
- сдавление спинного мозга;
- деформации конечностей, особенно неправильная ось нижней конечности.
В зависимости от сопутствующих заболеваний пациенту могут быть проведены тонзилэктомия, операция по удалению аденоидов, грыжесечение и прочие вмешательства.
Реабилитация

Реабилитация лиц, страдающих синдромом Моркио, помимо прочих мероприятий включает в себя массаж.
Каждому больному синдромом Моркио специалисты ЛФК и физиотерапии индивидуально разрабатывают курс реабилитации, который включает в себя процедуры магнито-, термо-, ударно-волновой терапии, массаж, упражнения ЛФК. Кроме того, эти пациенты нуждаются в психолого-педагогической помощи.
Проводят курсы реабилитации в дневном стационаре, длительность их индивидуальна, периодичность - каждые 3-4 месяца.
Диспансерное наблюдение
Ребенок, страдающий мукополисахаридозом IV типа, должен состоять на диспансерном учете у педиатра и в связи с мультисистемностью природы его патологии и прогрессирующего характера ее течения регулярно наблюдаться у целого ряда специалистов узкого профиля - хирурга-ортопеда, отоларинголога, кардиолога, невролога, пульмонолога, офтальмолога, стоматолога, физиотерапевта, логопеда, психолога и прочих.
1 раз в полгода-год такие больные должны проходить всестороннее обследование в многопрофильном стационаре, длительность которого обычно составляет 3-4 недели.
Профилактика и прогноз
Родителям, имеющим ребенка с синдромом Моркио, при планировании следующих беременностей необходимо проходить медико-генетическое консультирование. Целью его является выяснить степень генетического риска. Как было сказано выше, для каждой беременности существует 25 % риска рождения больного ребенка.
В современных условиях существует возможность проведения преимплантационной диагностики. Поэтому в рискованных случаях родителям рекомендуют на этапе планирования ребенка посетить генетика.
Синдром Моркио - неизлечимое и, к сожалению, неуклонно прогрессирующее заболевание. Прогноз относительно выздоровления неблагоприятный. Практически во всех случаях до достижения 20 лет больные погибают от недостаточности функций сердца и легких. Смещение атланто-окципитального сочленения приводит к повреждению ствола головного мозга, исходом которого становится внезапная смерть человека.
Заключение
Синдром Моркио - заболевание из группы мукополисахаридозов, которое наследуется аутосомно-рецессивным путем, характеризуется поражением преимущественно костно-мышечной, а также других систем организма, неуклонно прогрессирует и в большинстве случаев к 20 годам приводит к летальному исходу.
В основе диагностики лежат лабораторные методы исследования, а именно определение в моче вида и концентрации гликозаминогликанов, в крови - галактозамин-6-сульфат-сульфатазы или β-галактозидазы, а также молекулярно-генетическая диагностика.
Этиологическое и патогенетическое лечение отсутствует. Больному назначают препараты и немедикаментозные методы лечения, позволяющие облегчить течение того или иного проявления болезни. В ряде случаев проводят хирургические вмешательства, направленные преимущественно на декомпрессию сдавленного ранее спинного мозга и коррекцию деформаций опорно-двигательного аппарата.
Больные находятся на диспансерном учете у специалистов разного профиля и раз в 6-12 месяцев проходят всестороннее обследование в многопрофильном стационаре.
Прогноз неблагоприятный - заболевание прогрессирует, со временем добавляются новые и новые симптомы, которые приводят к сердечно-легочной недостаточности - ведущей причине смерти таких больных.
Предупредить рождение детей с синдромом Моркио родителям, в семье которых уже есть больной данной патологией ребенок, поможет медико-генетическое консультирование и пренатальная диагностика.
Мукополисахаридоз
Мукополисахаридозы - это группа генетически обусловленных заболеваний, возникающих вследствие нарушения обмена кислых мукополисахаридов (гликозаминогликанов). Характерны системные поражения скелета и задержка физического развития. При некоторых формах наблюдается умственная отсталость. Возможны нарушения сердечной деятельности, патология органов зрения, образование грыж, неврологические нарушения, гипертрихоз, увеличение печени и селезенки. Диагноз выставляется на основании клинических признаков, данных рентгенографии и других исследований. Лечение симптоматическое.
МКБ-10
Общие сведения
Мукополисахаридозы - группа генетических заболеваний, сопровождающихся накоплением кислых мукополисахаридов в органах и тканях. Причиной развития является передающаяся по наследству неполноценность лизосомных ферментов. Впервые мукополисахаридоз был описан Гурлер в 1917 году. Лечение мукополисахаридоза осуществляют травматологи-ортопеды при участии кардиологов, офтальмологов, неврологов, отоларингологов и других специалистов.
Виды мукополисахаридоза
Мукополисахаридоз типа IH
Мукополисахаридоз типа IH или синдром Гурлер встречается у 1 новорожденного из 20-25 тысяч. Симптомы мукополисахаридоза появляются в течение первого года жизни, полная клиническая картина формируется к 1-2 годам. Для данной формы мукополисахаридоза характерны грубые черты лица и деформация черепа в форме лодочного киля (скароцефалия). Из-за увеличенных аденоидов и пороков развития в области лица и носа больные дышат ртом. Отмечаются прогрессирующие деформации конечностей и других частей скелета, отставание в росте.
Позвоночник пациентов с мукополисахаридозом искривлен, из-за чего в положении сидя возникает симптом «кошачьей спины». Отмечается укорочение шеи, высокое расположение лопаток и выстояние нижних ребер. Кисти широкие, напоминающие когтистую лапу. Со временем у больных мукополисахаридозом формируются контрактуры суставов. Вначале поражаются локтевые и плечевые суставы, затем - коленные, тазобедренные и голеностопные. Из-за ограничения движений возникает характерная походка - на цыпочках с полусогнутыми ногами.
Тоны сердца приглушены, границы расширены. При аускультации определяются систолические шумы, на ЭКГ выявляется диффузное поражение миокарда. Передняя брюшная стенка пациентов с мукополисахаридозом ослаблена, живот увеличен, часто выявляются гидроцеле, пупочные и паховые грыжи. Печень и селезенка увеличены. Характерны нарушения зрения и слуха. Возможно помутнение и увеличение роговицы, пигментная дистрофия сетчатки, глаукома, атрофия зрительных нервов и застойные явления в области глазного дна. У больных мукополисахаридозом этого типа развивается прогрессирующая умственная отсталость. Могут возникать нарушения координации движений, парезы и параличи. Выявляется избыточный рост пушковых волос.
Рентгенография позвоночника пациентов с мукополисахаридозом свидетельствует о характерной деформации позвонков. Позвонки имеют кубовидную форму, их контуры закруглены, в переходном отделе выявляется скошенность передневерхних углов, углообразный кифоз, утолщение и укорочение отростков. На рентгенографии грудной клетки определяется утолщение передних и истончение задних отделов ребер, сопровождающиеся их лопатовидной или саблевидной деформацией, укорочение и деформация ключиц, уменьшение и смещение головок плечевых костей.
Рентгенография таза подтверждает сужение тазового кольца, скошенность вертлужных впадин и уменьшение головок бедренных костей. На рентгенограммах трубчатых костей выявляется истончение кортикального слоя и расширение костномозгового канала. Рентгенография костей кисти свидетельствует о недоразвитии ногтевых фаланг, укорочении и расширении пястных костей, проксимальных и средних фаланг. На рентгенограммах черепа определяется недоразвитие костей лицевого черепа, краниостеноз и макроцефалия.
Мукополисахаридоз типа I-S
Мукополисахаридоз типа I-S или болезнь Шейе (описана в 1962 г. американским офтальмологом Шейе) является более поздним, относительно благоприятно протекающим вариантом мукополисахаридоза типа IH (синдрома Гурлер). До 3-6 лет развитие детей соответствует норме. Первым признаком мукополисахаридоза становятся сгибательные контрактуры пальцев рук. В последующем ограничивается разгибания в лучезапястных, локтевых и плечевых суставах. Контрактуры нижних конечностей, как правило, слабо выражены. Полная клиническая картина мукополисахаридоза формируется к началу подросткового возраста.
Пациенты с мукополисахаридозом коренастые, невысокие, с грубыми чертами лица и хорошо развитой мускулатурой. Отмечается повышенное оволосение (гипертрихоз). Часто возникают паховые или пупочные грыжи. Кожа на пальцах натянута и утолщена. Из-за сдавления срединного нерва возможно развитие синдрома запястного канала, сопровождающееся атрофией мышц области тенара и парестезиями в области III-IV пальцев. У некоторых больных мукополисахаридозом выявляется аортальный стеноз, недостаточность клапанов аорты, пигментная дистрофия сетчатки, глаукома и помутнение роговицы. Интеллект в норме, увеличение селезенки и печени нехарактерно. На рентгенограммах определяется картина, аналогичная мукополисахаридозу типа IH, но патологические изменения выражены менее резко.
Мукополисахаридоз типа II
Мукополисахаридоз типа II или синдром Хантера чаще выявляется у мальчиков и обычно развивается на 2-3 году жизни. Как и при мукополисахаридозе типа IH наблюдается скафоцефалия, огрубление черт лица, низкий голос и затруднения дыхания из-за деформации лицевого скелета. При этом характерный для мукополисахаридоза типа IH кифоз обычно не выявляется, симптом «кошачьей спины» отрицательный. Пациенты часто страдают от респираторных инфекций (бронхитов, трахеитов, пневмоний). Постепенно развиваются нарушения координации движений, возрастает агрессивность. Настроение неустойчивое, с резкими перепадами.
Также при данной форме мукополисахаридоза наблюдается незначительное увеличение селезенки и печени, прогрессирующая тугоухость, узелки на коже спины и некоторое снижение интеллекта. В последующем может развиться помутнение роговицы. Рентгенологическая картина - как при мукополисахаридозе типа I-S. Возможны два варианта развития болезни: благоприятный (вариант В) и неблагоприятный (вариант А). При благоприятном варианте симптомы выражены нерезко, иногда возникает незначительная умственная отсталость, пациенты с мукополисахаридозом могут доживать до 30 и более лет. Для неблагоприятного варианта характерна яркая клиническая картина и серьезные нарушения интеллекта. Летальный исход наступает в подростковом возрасте.
Мукополисахаридоз типа III
Мукополисахаридоз типа III или болезнь Санфилиппо (описана в 1963 г. американским педиатром Санфилиппо) развивается у 1 новорожденного из 100-200 тысяч. В первые годы жизни развитие соответствует возрасту, иногда наблюдаются затруднения глотания и неуклюжая походка. В возрасте 3-5 лет ребенок с мукополисахаридозом становится апатичным и начинает отставать в развитии. Возникают нарушения речи, огрубление черт лица, недержание мочи и кала. Со временем умственная отсталость прогрессирует и занимает центральное положение в клинической картине этого типа мукополисахаридоза.
Наряду с нарушениями интеллекта наблюдается умеренное увеличение печени и селезенки, повышенное оволосение, контрактуры и задержка роста. Патология со стороны глаз и сердечно-сосудистой системы для этой формы мукополисахаридоза нехарактерна. Рентгенологическая картина - без изменений или как при мукополисахаридозе типа I-S, но менее выраженная. Больные мукополисахаридозом третьего типа, как правило, погибают в возрасте 10-20 лет от инфекционных осложнений.
Мукополисахаридоз типа IV
Мукополисахаридоз типа IV или болезнь Моркио (описана в 1929 г. уругвайским педиатром Моркио) наблюдается у 1 новорожденного из 40 тысяч. До 1-3 лет дети развиваются нормально. В последующем возникает значительное отставание в росте, укорочение шеи и туловища, контрактуры и вальгусная деформация конечностей, сколиоз или кифоз, разнообразные деформации грудной клетки, снижение силы мышц, утолщение кожи и огрубление черт лица. При этом типе мукополисахаридоза часто развиваются паховые и пупочные грыжи, дистрофия роговицы и тугоухость. Интеллект сохранен.
На рентгенограммах позвоночника определяется кифоз, сколиоз, расширение и уплощение тел позвонков. При проведении рентгенографии таза и конечностей выявляются множественные деформации, неровность контуров, уплощение головок бедренных костей, укорочение костей предплечья и деформации стоп. Средняя продолжительность жизни больных мукополисахаридозом - менее 20 лет. Смерть при данной форме мукополисахаридоза наступает из-за сопутствующих заболеваний, осложняющихся сердечно-легочной недостаточностью.
Другие типы мукополисахаридоза
Мукополисахаридоз типа VI или болезнь Марото-Лами (описана в 1960 г. французами Лами и Марото) развивается в возрасте 2 года и старше. Возникает огрубление черт лица, отставание в росте, укорочение шеи, контрактуры суставов и бочкообразная деформация грудной клетки. Характерны частые простуды. Возможны грыжи, увеличение печени и селезенки. Интеллект не страдает. Наблюдается два варианта течения: классический и мукополисахаридоз со слабой выраженностью клинических проявлений. На рентгенограммах больных мукополисахаридозом определяется кубовидная или клиновидная деформация позвонков, треугольная деформация таза, недоразвитие и деформация головок бедренных костей, укорочение малоберцовых костей.
Мукополисахаридоз типа VII протекает, как мупоколисахаридоз типа III, различия выявляются только при проведении биохимических исследований. Мукополисахаридоз типа VIII по симптомам напоминает мукополисахаридоз типа IV, но, в отличие от него, сопровождается задержкой умственного развития.
Диагноз мукополисахаридоза устанавливается на основании характерной клинической и рентгенологической картины, выявления гликозаминогликанов в моче, изучения активности ферментов в клеточных культурах, генетического секвенирования. В ходе обследования больным мукополисахаридозом назначаются консультации различных специалистов: кардиолога, гастроэнтеролога, офтальмолога, отоларинголога, невролога, психиатра и т. д., проводятся инструментальные исследования для оценки состояния различных органов и систем.
Лечение мукополисахаридоза
Патогенетическая терапия не разработана. Лечение симптоматическое, может быть как консервативным, так и оперативным. Осуществляется профилактика и лечение респираторных инфекций, проводится коррекция нарушений зрения и слуха. При необходимости выполняются грыжесечение и герниопластика, операции по устранению контрактур и коррекции деформаций скелета.
Прогноз и профилактика
Прогноз при всех типах мукополисахаридоза неблагоприятный - продолжающееся накопление продуктов обмена в тканях приводит к усугублению патологических изменений со стороны всех органов и систем. Использование любых лечебных средств (переливания крови, введение гормонов и т. д.) при мукополисахаридозе обеспечивает лишь временное улучшение. Рекомендуется пренатальная профилактика.
1. Руководство по педиатрии. Врожденные и наследственные заболевания/ Новиков П.В., Баранов А.А., Каганов Б.С., Шиляев Р.Р - 2007
Читайте также: