Синдром Мошковиц (Moschcowitz) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Мошкович болезнь I Мошко́вич боле́знь (Е. Moschcowitz, американский врач, 1879—1964; синоним: тромботическая пурпура, тромбоцитопенический акроангиотромбоз, тромботическая микроангиопатия, синдром Мошкович — Сингера — Симмерса)
заболевание, характеризующееся множественным тромбообразованием вследствие спонтанной агрегации тромбоцитов и диссеминированной закупоркой мелких артерий и артериол гиалиновыми тромбами; сопровождается тромбоцитопенией, а также ишемическим поражением головного мозга, почек, печени, сердца и других органов. Встречается в любом возрасте, чаще у детей.
Этиология и патогенез. Заболевание может возникнуть после инфицирования Mukoplasma pneumoniae, введения вакцины (противогриппозной, комбинированной и др.), приема некоторых лекарственных препаратов (например, пенициллина, дифенина). Состояния, напоминающие М.б, могут наблюдаться при менингококковой инфекции, злокачественных новообразованиях, а также при системной красной волчанке, ревматоидном артрите, синдроме Шегрена. Одна из наиболее вероятных причин возникновения М.б — остро возникающий (например, на фоне инфекции) дефицит ингибитора фактора агрегации тромбоцитов, в результате чего происходит спонтанное тромбообразование.
Основным звеном патогенеза является интенсивное тромбирование мелких артерий и артериол гиалиновыми тромбами, состоящими из гранул тромбоцитов и компонентов их цитоплазмы с незначительным содержанием фибрина. Гемолитическая анемия и тромбоцитопения при М.б обусловлены механическим разрушением эритроцитов и потреблением тромбоцитов. Нередко встречаются микроаневризмы пораженных артериол.
Клиническая картина. Развернутой стадии заболевания обычно предшествуют слабость, головная боль, тошнота, рвота, боли в животе (вплоть до картины, напоминающей острый живот), нарушения зрения, появление синяков и петехий на коже, в редких случаях возможны маточные, желудочные и другие кровотечения. Для развернутой стадии характерны лихорадка, разнообразные непостоянные неврологические расстройства (атаксия, гемипарезы и гемиплегии, нарушения зрения, судороги), иногда возникают психические нарушения. Печень и селезенка нередко увеличены. Поражение почек сопровождается появлением в моче белка, эритроцитов и цилиндров, часто наблюдается олигурия. Возможна надпочечниковая недостаточность (см. Надпочечники).
Большая часть клинических проявлений — следствие множественного тромбирования (см. Тромбоз) артериального русла различных органов с последующей их ишемией. Одно из основных проявлений заболевания — Гемолиз, который сопровождается желтухой, появлением в крови шизоцитов (фрагментированных эритроцитов), нередко содержащих остатки ядер эритрокариоцитов, а также повышением числа ретикулоцитов. Содержание гемоглобина снижается до 40—80 г/л, гематокритное число — около 20—30%. В плазме крови резко увеличивается содержание свободного гемоглобина и неконъюгированного билирубина. Число тромбоцитов колеблется от 5 до 100․10 9 /л, лейкоцитов — от 15 до 25․10 9 /л. Протромбиновое время, парциальное тромбопластиновое время, содержание фибриногена и продуктов его деградации могут быть незначительно изменены. Обнаружение выраженных сдвигов в системе свертывания крови свидетельствует о коагулопатии потребления или ассоциированном с гемолизом внутрисосудистым свертывании. При исследовании костного мозга обнаруживаются гиперплазия эритроидных элементов и увеличение числа мегакариоцитов. Примерно у 20% больных выявляются антинуклеарные антитела.
У большинства больных заболевание длится от нескольких дней до нескольких недель. Основной причиной смерти является прогрессирующее поражение сосудов головного мозга, сердца и почек.
Диагноз устанавливают на основании клинической картины, данных лабораторного исследования. С помощью трепанобиопсии, биопсии участка кожи или слизистой оболочки в местах кровоизлияний выявляют гиалиновые тромбы в мелких артериях и артериолах без выраженных признаков васкулита. Исследуют плазму больного, которая вызывает агрегацию отмытых нормальных донорских тромбоцитов, причем агрегация не предотвращается добавлением ацетилсалициловой кислоты; она может быть блокирована добавлением нормальной бестромбоцитной плазмы.
Дифференциальный диагноз проводят с идиопатической тромбоцитопенической пурпурой (см. Пурпура тромбоцитопеническая), при которой тромбоцитопения обусловлена повышенным разрушением тромбоцитов вследствие иммунных механизмов; с тромбоцитопениями, связанными с пониженной продукцией тромбоцитов, в частности при метастазах злокачественных опухолей в костный мозг, апластической анемии, поражением костного мозга, обусловленном, например, воздействием ионизирующего излучения. М. б. следует отличать от болезни Шенлейна — Геноха, при которой повреждаются в основном капилляры, артериолы и венулы, а также от макроглобулинемии Вальденстрема, множественной миеломы, криоглобулинемии и других моноклональных диспротеинемий, характеризующихся повышенной вязкостью крови. По клиническим проявлениям близок к М.б гемолитико-уремический синдром, встречающийся, как правило, у детей, чаще в возрасте до 3 лет; он приводит к поражению почечных клубочков и развитию почечной недостаточности в результате образования гиалиново-тромбоцитарных тромбов.
Лечение проводят в отделении интенсивной терапии. Основным методом лечения является плазмообмен, который осуществляется с помощью плазмафереза (см. Плазмаферез, Цитаферез). При этом объем удаленной плазмы (от 1,5 до 3 л) обязательно восполняется свежезамороженной донорской плазмой, содержащей ингибитор фактора агрегации тромбоцитов. Частота проведения плазмообмена зависит от клинического эффекта. Ряд авторов предлагает наряду с плазмаферезом вводить преднизолон (от 100 до 1000 мг в сутки).
Прогноз зависит от своевременного установления диагноза и оперативности проведения лечебных мероприятий (плазмафереза, трансфузий свежезамороженной плазмы). Обычно удается спасти 60—70% больных.
Библиогр.: Баркаган З.С. Геморрагические заболевания и синдромы, с. 435, 1988; Лабораторные методы исследования системы гомеостаза под ред. Е.Д. Гольдберга, Томск, 1981); Руководство по гематологии, под ред. А.И. Воробьева, т. 2, с. 189. М., 1985.
острая болезнь, вероятно аутоиммунной природы, характеризующаяся сочетанием гемолитической анемии с тромбоцитопенией и геморрагическим синдромом.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
МОШКОВИЧ БОЛЕЗНЬ
Мошкович болезнь (E. Moschcowitz, амер. врач, 1879—1964; син.; микроангиопатическая гемолитическая анемия, тромботическая тромбоцитопеническая пурпура, тромботический микроангиотромбоз, микроангиопатия тромботическая, гемолитическая анемия с микроангиотромбозом, синдром Мошкович) — редкое заболевание, проявляющееся лихорадкой, гемолитической анемией, тромбоцитопенической пурпурой, поражением почек и центральной нервной системы.
В 1925 г. Мошкович описала заболевание у 16-летней девушки, сопровождающееся высокой лихорадкой, острым внутрисосудистым гемолизом и геморрагической пурпурой; больная умерла через две недели от начала заболевания. В отечественной литературе Мошкович болезнь описана М. И. Теодори и другими. Поданным Хилла и Купера (J. В. Hill, W. H. Cooper), к 1968 г. было описано более 300 случаев Мошкович болезни Заболевание может развиться в любом возрасте.
Содержание
Этиология и патогенез
Этиология и патогенез не известны; существуют различные теории о происхождении и развитии заболевания (вирусная, бактериальноаллергическая, токсическая, аутоиммунная). По своим гематологическим и патогистологическим особенностям Мошкович болезнь близка к системной красной волчанке, синдрому Фишера — Эванса, гемолитико-уремическому синдрому и, по-видимому, может быть отнесена к группе иммунопатий. Однако И. Е. Тареева (1972) указывает, что при наблюдении за 268 больными системной красной волчанкой ни разу не удалось отметить развития Мошкович болезни. Антитела у больных Мошкович болезнью обнаруживаются очень редко. В основе заболевания лежит распространенное поражение мелких сосудов с пролиферацией эндотелия, фибриноидным некрозом, образованием в участках повреждения пристеночных и обтурирующих фибриновых и гиалиновых тромбов. Фибрин откладывается сначала субэндотелиально, что сопровождается набуханием эндотелия, сужением просвета сосудов. Прохождение эритроцитов через суженный просвет капилляров, повторный контакт эритроцитов с патологически измененными сосудами приводят к фрагментации эритроцитов и к внутрисосудистому гемолизу (см.). Даси (J. V. Dacie, 1967) считает, что повреждение эндотелия сосудов и разрушение эритроцитов сопровождается высвобождением тканевого и клеточного тромбопластина с последующим повышением содержания тромбина, агрегацией тромбоцитов, диссеминированным внутрисосудистым свертыванием, тромбоцитопенией.
Отсутствие комплемента в стенках пораженных сосудов, в клубочковой базальной мембране почек и во внутрисосудистых тромбах, частое обнаружение в участках поражения только фибрина без иммуноглобулина свидетельствуют, по-видимому, о том, что реакция антиген — антитело не играет роли в пусковом механизме болезни. Однако полностью исключить значение иммунных комплексов в патогенезе болезни Мошкович не представляется возможным .
Патологическая анатомия
Патологическая анатомия характеризуется микроангиопатией, дистрофическими и некротическими изменениями в различных органах, обусловленными сосудистыми нарушениями, общим гемосидерозом вследствие гемолиза эритроцитов и изменений в органах иммунитета. Макроскопически выявляется геморрагическая пурпура кожи и слизистых оболочек, малокровие органов и тканей, на разрезе — буроватый оттенок ткани печени, селезенки, лимф, узлов и костного мозга вследствие отложений в них гемосидерина. Отмечается увеличение в 1,5—2 раза всех лимф, узлов и селезенки с небольшим соскобом пульпы, гиперплазия костного мозга. Инфаркты могут встречаться наиболее часто в головном мозге и почках. На створках клапанов сердца нередко находят бородавчатые наложения с развитием картины эндокардита Либмана — Сакса (см. Красная волчанка). Характерны изменения почек: они увеличены, капсула легко снимается, обнажая гладкую поверхность с обильным красным крапом.
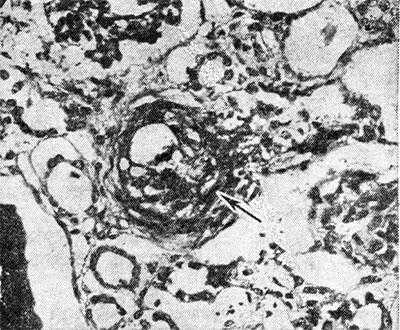
Рис. 1. Микропрепарат почки при болезни Мошкович: стрелкой указано фибриноидное набухание и плазматическое пропитывание стенки мелкой артерии; окраска гематоксилин-эозином, х 200.
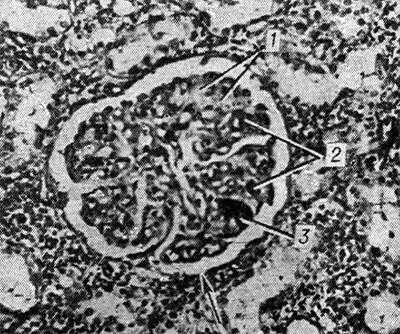
Рис. 2. Микропрепарат почки при болезни Мошкович: почечный клубочек (указан стрелкой) с участками фибриноидного некроза (1), окклюзией просветов капиллярных петель (2) эозинофильными массами и скоплением гематоксилинофильных ложноизвестковых зерен (3); окраска гематоксилин-эозином, X 200.
Микроскопически во всех органах обнаруживают характерное поражение капилляров, артериол и мелких артерий, к-рое наиболее выражено в почках и зависит от длительности течения заболевания. При кратковременном течении (до 1 месяца) и обострении процесса преобладают фибриноидное набухание с плазматическим пропитыванием (рис. 1) с последующим фибриноидным некрозом сосудов и окклюзией просвета эозинофильными PAS-положительными массами, дающими положительную реакцию на фибрин. При длительности заболевания более 2 мес. наряду с фибриноидными изменениями выражены пролиферативные и склеротические. Гематоксилинофильные зернистые массы в сосудах, дающие отрицательную реакцию Коссы на известь и названные Прокшом и Томси (P. Proks, F. Tomsi, 1955) ложноизвестковыми зернами, могут быть скоплением микробов или продуктами гемолиза. Они часто встречаются в капиллярах почечных клубочков наряду с фибриноидным некрозом капиллярных петель и окклюзией их эозинофильными массами (рис, 2). В печени, почках, селезенке, костном мозге, лимф, узлах с помощью реакции Перльса обнаруживают скопления гемосидерина. В костном мозге и селезенке количество плазматических клеток увеличено.
Клиническая картина
Начало заболевания острое, иногда ему предшествует острое респираторное заболевание, непереносимость лекарственных средств. Первыми признаками обычно являются лихорадка, боли в животе, рвота, головные боли. Вскоре появляется резкая бледность и умеренная иктеричность кожных покровов, на коже отмечаются петехии и экхимозы. Может быть мелена, гематурия, маточные кровотечения. Затем присоединяются симптомы поражения головного мозга в виде головных болей, возбуждения, бреда, галлюцинаций, клонических судорог, локальной неврологической симптоматики, спутанного сознания, комы. Почки в связи со множественным поражением сосудов и острым внутрисосудистым гемолизом вовлекаются в процесс особенно часто. Почечный синдром проявляется гипертензией, протеинурией, гематурией, цилиндрурией. Характерно развитие острой почечной недостаточности (см.) с появлением олигурии и даже анурии, азотемии, нарушений водно-электролитного баланса.
В крови отмечается лейкоцитоз со сдвигом влево в лейкоцитарной формуле (см.), выраженная тромбоцитопения, анемия с ретикулоцитозом. Особенно характерно резкое изменение формы эритроцитов— анизоцитоз, пойкилоцитоз; встречаются обломки эритроцитов и так наз. шлемовидные эритроциты. Содержание билирубина повышено незначительно или нормальное. Прямая проба Кумбса (см. Кумбса реакция), как правило, отрицательная.
Течение заболевания может быть бурное, острое или подострое. Различия в клинических проявлениях Мошкович болезни у взрослых и детей отсутствуют. Продолжительность заболевания от 1—2 недель до нескольких месяцев. Описаны случаи, когда болезнь Мошкович длилась несколько лет.
Диагноз
Диагноз устанавливают на основании характерной клин, картины и результатов биопсии почек, кожи, слизистой оболочки десен, мышц, лимф, узлов и трепанобиопсии костного мозга, позволяющих обнаружить характерную морфологическую картину.
Дифференциальный диагноз следует проводить с узелковым периартериитом (см. Периартериит узелковый), системной красной волчанкой (см.), гемолитико-уремическим синдромом (см.), затяжным септическим эндокардитом (см.), болезнью Верльгофа (см. Пурпура тромбоцитопеническая), синдромом Фишера — Эванса (см. Фишера — Эванса синдром).
Лечение
Считают целесообразным раннее назначение антикоагулянтной терапии, поскольку в патогенезе заболевания ведущая роль отводится диссеминированному внутрисосудистому свертыванию. Бернсток и Хирсон (L. Bernstock, С. Hirson, 1960) отмечают ремиссии при лечении гепарином, причем чем раньше начато лечение, тем благоприятнее результаты. Кваан (H. С. Kwaan, 1968) рекомендует использовать стрептокиназу. Широкое распространение нашли дезагреганты (ацетилсалициловая к-та, дипиридомол).
Применяется также ацетилсалициловая к-та в сочетании с преднизолоном, хотя многие гематологи сдержанно относятся к назначению преднизолона. Наиболее эффективным методом терапии считают плазмаферез (см.). Гемотрансфузии следует назначать только по жизненным показаниям.
Лечение острой почечной недостаточности проводят с использованием гемодиализа (см.) или перитонеального диализа (см.). Показания к гемодиализу устанавливаются индивидуально, в зависимости от клинических симптомов уремической интоксикации, нарушений азотистого, водного и электролитного обмена. В ряде случаев рекомендуют спленэктомию (см.).
Прогноз неблагоприятный.
Библиография: Болезни почек в детском возрасте, под ред. М. Я. Студеникина, с. 312, М., 1976; Дрозд Т. Н. К морфологии и патогенезу синдрома Мошковица, Арх. патол., т. 32, № 10, с. 30, 1970, библиогр.; Игнатова М. С. и Вельтищев Ю. Е. Болезни почек у детей, с. 224, М., 1973; Савкив Б. Т. и Голотенко Н. И. О болезни Мошковица, Тер. арх., т. 41, № 10, с. 120, 1969; Теодори М. И. и Лихачев Ю. П. К клинике и патологии так называемой тромботической тромбоцитопенической пурпуры, там же, т. 32, № 3, с. 76,1960; Уранова Е. В. и Штырен М. Я. К патогенезу и морфологической характеристике тромботической тромбоцитопенической пурпуры (болезнь Мошковица), Арх. патол., т. 28, № п, с. 54, 1966; Hill J. В. a. Cooper W. H. Thrombotic thrombocytopenic purpura, Arch, intern. Med., v. 122, p. 353, 1968; Mosсhcowitz E. An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries, ibid., v. 36, p. 89, 1925; Zacharski L. R., Walworth C. a. McIntyre O. R. Antiplatelet therapy for thrombotic thrombocytopenic purpura, New Engl. J. Med., V. 285, p. 408, 1971.
Болезнь Мошковица ( Гемолитическая анемия с микроангиотромбозом , Тромботическая микроангиопатия , Тромботическая тромбоцитопеническая пурпура )
Болезнь Мошковица - это полисиндромная патология, сочетающая в себе тромбоцитопению, гемолитическую анемию, окклюзию артериол с развитием ишемических поражений органов. Клинически протекает с лихорадкой, кровотечениями, неврологическими нарушениями, абдоминальным синдромом. Возможно развитие ТИА, инфаркта миокарда и почек, гепатита, мезентериальной ишемии. Диагностика основана на исследовании крови (ОАК, тромбоциты, ЛДГ, активность металлопротеиназы), биоптатов почек, кожи, костного мозга. Лечение предполагает использование дезагрегантов, антикоагулянтов, глюкокортикоидов; проведение плазмафереза, переливание плазмы, по показаниям - гемодиализ.
МКБ-10
Общие сведения
Клинический случай неизвестного заболевания, включающего анемию, лихорадку, геморрагическую пурпуру, ишемический инсульт, острую сердечную недостаточность и множественные тромбозы, впервые был описан в 1924 г. американским патологом Е. Мошковицем. В дальнейшем болезнь была названа его именем, позднее в клиническую практику были введены другие термины: тромботическая тромбоцитопеническая пурпура (ТТП), тромботическая микроангиопатия, гемолитическая анемия с микроангиотромбозом. Частота синдрома - 1,5-6 случаев на 1 млн. населения. Заболевают преимущественно лица 30-40 лет, из них женщины в 2 раза чаще.
Причины
Этиология болезни Мошковица остается предметом исследования в гематологии. Важным прорывом последних десятилетий в этом вопросе служит выявление связи ТТП с недостаточностью фермента ADAMTS-13 - металлопротеазы, расщепляющей фактор Виллебранда. Нарушение ферментной активности может быть вызвано как врожденными, так и приобретенными причинами:
- Врожденные. Включают мутации в гене ADAMTS-13, приводящие к дефициту дизинтегрин-подобной металлопротеазы-13 с повтором тромбоспондина 1 типа. К настоящему времени изучено свыше 80 мутаций, обусловливающих развитие наследственной формы болезни Мошковица.
- Приобретенные. В основном связаны с наличием антител, угнетающих ADAMTS-13. Такие состояния могут возникать при аутоиммунных болезнях (АФС, СКВ, склеродермии), HELLP-синдроме, после перенесенной вирусной инфекции, вакцинации, трансплантации костного мозга. Также снижение активности ADAMTS-13 отмечается при ДВС-синдроме, уремии, циррозе печени, сепсисе, онкопатологии. Примерно половина случаев болезни Мошковица расцениваются как идиопатические.
Патогенез
Дефицит или ингибирование ADAMTS-13 приводит к повышенной активности VWF (фактора фон Виллебранда), который начинает стимулировать неконтролируемую внутрисосудистую агрегацию тромбоцитов и тромбообразование в микроциркуляторном русле. Количество циркулирующих тромбоцитов уменьшается, развивается тромбоцитопения.
Одновременно происходит пролиферация эндотелия мелких сосудов, что вызывает дополнительное сужение их просвета. Механическое затруднение для тока крови и высвобождение различных эндотелиальных факторов способствует фрагментации эритроцитов с развитием внутрисосудистого гемолиза.
Агрегаты тромбоцитов вызывают закупорку артериол и капилляров головного мозга, сердца, почек, легких. На коже и слизистых возникает геморрагическая пурпура, внутренние органы увеличиваются, в них обнаруживаются отложения гемосидерина. На фоне микроангиопатии формируются ишемические и некротические изменения тканей, возникают инфаркты сердечной мышцы, церебральной ткани, почечной паренхимы.
Классификация
С точки зрения влияния этиологических факторов, болезнь Мошковица подразделяется на наследственные и приобретенные варианты. Эти группы (особенно приобретенная форма) крайне неоднородны, отличаются по времени дебюта, доброкачественности течения, прогнозу:
1. Наследственная. Представлена синдромом Апшоу-Шульмана с аутосомно-рецессивным механизмом наследственной передачи. Чаще манифестирует в детском возрасте, однако известны эпизоды начала заболевания в 35 лет. Течение хроническое, рецидивирующее.
2. Приобретенные формы ТТП.
- Идиопатическая ТТП. Может быть острой (с молниеносным течением) и рецидивирующей. Во втором случае рецидивы могут быть как однократными, так и множественными.
- Вторичные ТТП. Могут быть сопряжены с беременностью и родами, приемом ЛС (сульфаниламидов, антибиотиков, КОК, иммунодепрессантов). Нередко провоцируются аутоиммунной патологией (волчанка, системная склеродермия, АФС, РА, синдром Шегрена, узелковый периартериит), инфекциями (кишечные, ВИЧ), злокачественными опухолями (чаще аденокарциномой желудка). Отмечена ассоциация болезни Мошковица с трансплантаций органов и костного мозга.
Симптомы болезни Мошковица
В большинстве случаев заболевание манифестирует остро и внезапно. Часто отмечается простудоподобное продромальное состояние. Клиника развивается стремительно. Классическая форма болезни Мошковица характеризуется пентадой признаков: тяжелой тробоцитопенией, гемолитической анемией, неврологическими нарушениями, лихорадкой и почечным синдромом.
В дебюте ТТП у 35% больных возникает абдоминальный синдром: рвота, выраженные боли в животе, свидетельствующие о мезентериальной ишемии. Типична лихорадка. Может выявляться гепатит, панкреатит, желтуха, спленомегалия. Иногда заболевание манифестирует с инфаркта миокарда, ОРДС, рабдомиолиза, гангрены.
Тромбоцитопения сопровождается появлением петехиальной сыпи на коже, кровотечений различной степени выраженности и локализации (десневые, носовые, меноррагии, мелена, кровохарканье, кровоизлияние в подпаутинное пространство). Церебральные и почечные нарушения отражают ишемические поражения органов. В неврологическом статусе отмечаются цефалгии, судороги, нарушения сознания. Могут возникать парезы и гемиплегия, атаксия, афатические расстройства. В тяжелых случаях возможна кома.
Признаками почечного синдрома служат массивные отеки, артериальная гипертензия, гематурия. Нарастание олигурии, электролитного дисбаланса и азотемии свидетельствует о развитии острой почечной недостаточности. Чаще болезнь Мошковица протекает остро или подостро. Возможно рецидивирующее течение.
Осложнения
Без экстренного назначения адекватного лечения смертность от болезни Мошковица составляет 85-100%. Летальные исходы обусловлены ишемическим поражением органов-мишеней (острый инфаркт миокарда, ОНМК, инфаркт почек), геморрагическим синдромом (церебральные кровоизлияния, внутренние кровотечения), органной дисфункций (ОПН, СН). Описаны случаи внезапной сердечной смерти, связанные с фатальными аритмиями, кардиогенным шоком. В поздних стадиях развивается ДВС-синдром.
Диагностика
Сложность распознавания болезни Мошковица объясняется отсутствием патогномоничной клинической картины. Больные часто поступают в стационар с клиникой инсульта, острой коронарной недостаточности, абдоминальной ишемии и безуспешно лечатся от соответствующих синдромов. Между тем, наряду с осмотром врача-невролога, кардиолога, нефролога, пациентам необходима консультация гематолога:
- Объективный статус. При осмотре поступившего больного отмечается бледность и желтушность кожных покровов, мелкопятнистые кровоизлияния на коже (пурпура), иногда - спленомегалия, повышение АД. Пациент может быть дизориентирован, возбужден, нередки бред и галлюцинации, спутанность сознания. Типичные жалобы - на боли в животе, головные боли.
- Клиническая лабораторная диагностика. Характерными изменениями гемограммы при болезни Мошковица выступают анемия, ретикулоцитоз, выраженная тромбоцитопения (микрогематурия, цилиндрурия, протеинурия.
- Биохимическая лабораторная диагностика. Проба Кумбса отрицательная. Характерно обнаружение в крови макромолекул фактора Виллебранда. Резкое повышение уровня ЛДГ указывает на ишемию тканей и гемолиз. При специальном исследовании определяется снижение активности металлопротеиназы, наличие антител к ADAMTS-13, антифосфолипидных Ат.
- Биопсия. Для уточнения морфологических изменений в тканях выполняется биопсия кожи, мышц, слизистой десен, почек, костного мозга. Для миелограммы характерно нормобластное кроветворение с увеличением мегалобластов, угнетение белого и раздражение эритроидного ростка.
- Инструментальная диагностика. При КТ либо МРТ-сканировании головы обнаруживаются церебральные ишемические очаги, кровоизлияния. При кровохарканьи показано проведении КТ легких, фибробронхоскопии; при желудочно-кишечном кровотечении ‒ ФГДС. Ишемические изменения органов диагностируются с помощью ЭКГ, УЗИ органов брюшной полости, почек.
Дифференциальный диагноз
На основании лабораторно-инструментальных данных дифференциальная диагностика болезни Мошковица осуществляется с:
- синдромом Эванса-Фишера (аутоиммунная гемолитическая анемия + тромбоцитопения);
- гемолитико-уремическим синдромом;
- болезнью Верльгофа;
- пароксизмальной ночной гемоглобинурией;
- коллагенозами и др.
Лечение болезни Мошковица
При малейшем подозрении на ТТП рекомендуется незамедлительно начинать патогенетическое лечение. Пациенты, как правило, госпитализируются в ОРИТ, где проводится постоянный мониторинг клинико-лабораторных показателей. Терапия болезни Мошковица включает несколько направлений:
- Базисная терапия. Должна быть начата в первые сутки после появления клинических симптомов. Она включает инфузии больших доз свежезамороженной или криосупернатантной плазмы, назначение антиагрегантов (аспирин, дипиридамол), антикоагулянтов (гепарин), глюкокортикостероидов (преднизолон, дексаметазон).
- Экстракорпоральная гемокоррекция.Плазмаферез считается наиболее эффективным методом терапии болезни Мошковица, поскольку позволяет удалить из крови ингибиторы металлопротеазы. Пациенты с ПН нуждаются в заместительной почечной терапии - гемодиализе, перитонеальном диализе.
- Вспомогательное лечение. При выраженном анемическом синдроме используются трансфузии эритроцитов. Переливание тромбоцитов осуществляется только при жизнеугрожающих кровотечениях. Ряд клиницистов рекомендует всем больным назначать фолиевую кислоту.
При рефрактерных к стандартной терапевтической схеме формах болезни Мошковица применяются цитостатики, иммунодепрессанты, моноклональные антитела. В целях снижения риска рецидивов показана спленэктомия.
Прогноз и профилактика
Болезнь Мошковица ‒ тяжелая, сложно диагностируемая патология с агрессивным течением. Без своевременного лечения летальность очень высока. При быстрой постановке диагноза и раннем начале лечения (проведении экстракорпоральной гемокоррекции) удается спасти свыше 80% больных. В дальнейшем для пролонгирования ремиссии необходима постоянная поддерживающая терапия. Часто сохраняются остаточные изменения в виде энцефалопатии, ХПН. Профилактика ТТП не разработана. При наследственных формах проводится генетическое консультирование.
2. Клинический случай своевременной диагностики и успешной терапии больной с болезнью Мошковица/ Кузнецова Е.Ю., Соколова-Попова Т.А., Ольховик Т.И., Михалев М.А., Бахтина В.И., Кузнецова-Подзолкова Е.П.// Современные проблемы науки и образования. - 2020. - №4.
3. Особенности диагностики и лечения тромботической тромбоцитопенической пурпуры, развившейся во время беременности: обзор литературы и собственное наблюдение/ Войцеховский В.В., Филатов Л.Б., Пивник А.В., Авдонин П.В., Есенина Т.В., Судаков А.Г. // Клиническая онкогематология. -2014. - No7(4).
4. Случай врожденной тромботической тромбоцитопенической пурпуры, диагностированный с помощью определения активности ADAMTS13/ Фроленко А.Л., Афанасьева Е.И., Резник Н.В., Каган М.Ю.// Оренбургский медицинский вестник. ‒ 2017. ‒ Том V, No2 (18).
Болезнь Мошковица
Болезнь Мошковица. Это полисиндромная патология, сочетающая тромбоцитопению, гемолитическую анемию, окклюзию артериол с развитием ишемического поражения органа. Клинически протекает при лихорадке, кровотечениях, неврологических расстройствах, абдоминальном синдроме. Возможно развитие ТИА, инфаркта миокарда и почек, гепатита, ишемии брыжейки. Диагноз ставится на основании анализов крови (ОАК, тромбоциты, ЛДГ, активность металлопротеиназы), биопсии почек, кожи и костного мозга. Лечение предполагает использование антиагрегантов, антикоагулянтов, глюкокортикоидов; проведение плазмафереза, переливания плазмы, по показаниям - гемодиализ.
Болезнь Мошковица
Дополнительные факты
Клинический случай неизвестного заболевания, в том числе анемии, лихорадки, геморрагической пурпуры, ишемического инсульта, острой сердечной недостаточности и множественного тромбоза, был впервые описан в 1924 г. Американским патологом Э. Мошковицем. Позже болезнь была названа его именем, позже в клиническую практику были введены другие термины: тромботическая тромбоцитопеническая пурпура (ТТП), тромботическая микроангиопатия, гемолитическая анемия с микроангиотромбозом. Заболеваемость синдромом составляет 1,5-6 случаев на миллион населения. Чаще всего страдают люди в возрасте 30-40 лет, в два раза чаще - женщины.
Этиология болезни Мошковица остается предметом исследования в гематологии. Основным прорывом в последние десятилетия в этой области стало выявление взаимосвязи между ТТП и дефицитом фермента ADAMTS-13, металлопротеиназы, расщепляющей фактор фон Виллебранда. Нарушение активности фермента может быть вызвано как врожденными, так и приобретенными причинами:
• Врожденный. Включает мутации в гене ADAMTS-13, приводящие к дефициту металлопротеазы-13 дезинтегринового типа с повторением тромбоспондина типа 1. На сегодняшний день изучено более 80 мутаций, вызывающих развитие наследственной формы болезни Мошковица.
• Куплен. В основном связано с наличием антител, ингибирующих ADAMTS-13. Такие состояния могут возникать при аутоиммунных заболеваниях (APS, SLE, склеродермия), HELLP-синдроме, после вирусной инфекции, вакцинации, трансплантации костного мозга. Кроме того, снижение активности ADAMTS-13 наблюдается при диссеминированном внутрисосудистом свертывании, уремии, циррозе печени, сепсисе и онкопатологии. Около половины случаев болезни Мошковица считаются идиопатическими.
Дефицит или ингибирование ADAMTS-13 приводит к увеличению активности VWF (фактора фон Виллебранда), который начинает стимулировать неконтролируемую внутрисосудистую агрегацию тромбоцитов и образование тромбов в микроваскуляризации. Уменьшается количество циркулирующих тромбоцитов, развивается тромбоцитопения.
При этом происходит разрастание эндотелия мелких сосудов, что вызывает дальнейшее сужение их просвета. Механическая обструкция кровообращения и выброс различных эндотелиальных факторов способствуют фрагментации эритроцитов с развитием внутрисосудистого гемолиза.
Агрегаты тромбоцитов вызывают закупорку артериол и капилляров в головном мозге, сердце, почках, легких. На коже и слизистых оболочках появляется геморрагическая пурпура, внутренние органы увеличиваются, там обнаруживаются отложения гемосидерина. На фоне микроангиопатии образуются ишемические и некротические изменения тканей, возникают инфаркты сердечной мышцы, ткани головного мозга и почечной паренхимы.
С точки зрения влияния этиологических факторов болезнь Мошковица делится на наследственные и приобретенные варианты. Эти группы (особенно приобретенная форма) крайне разнородны, различаются временем дебюта, хорошим качеством течения и прогнозом:
1. Наследственный. Он представлен синдромом Апшоу-Шульман с аутосомно-рецессивным наследственным механизмом передачи. Чаще проявляется в детстве, однако известны эпизоды заболевания в 35 лет. Течение хроническое, рецидивирующее.
2. Приобретенные формы ТПП.
• Идиопатическая ТТП. Он может быть острым (с молниеносным течением) и рецидивирующим. Во втором случае рецидивы могут быть как единичными, так и множественными.
• Вторичная ТЭЦ. Они могут быть связаны с беременностью и родами, приемом лекарств (сульфаниламиды, антибиотики, КОК, иммунодепрессанты). Часто вызывается аутоиммунной патологией (волчанка, системный склероз, APS, RA, синдром Шегрена, узелковый полиартериит), инфекциями (кишечными, ВИЧ), злокачественными новообразованиями (чаще аденокарцинома желудка). Болезнь Мошковица связана с трансплантацией органов и костного мозга.
В большинстве случаев болезнь проявляется остро и внезапно. Часто отмечается простудное продромальное состояние. Клиника стремительно развивается. Классическая форма болезни Мошковица характеризуется пентадой симптомов: тяжелой тробоцитопенией, гемолитической анемией, неврологическими расстройствами, лихорадкой и почечным синдромом.
В начале ТТП у 35% пациентов развивается абдоминальный синдром: рвота, сильная боль в животе, указывающая на ишемию брыжейки. Лихорадка типична. Могут обнаруживаться гепатит, панкреатит, желтуха, спленомегалия. Иногда заболевание проявляется инфарктом миокарда, ОРДС, рабдомиолизом, гангреной.
Тромбоцитопения сопровождается появлением петехиальной сыпи на коже, кровотечениями разной степени тяжести и локализации (десневое, носовое, меноррагия, мелена, кровохарканье, кровоизлияние в субарахноидальное пространство). Заболевания головного мозга и почек отражают ишемическое поражение органов. В неврологическом состоянии возникают головные боли, судороги и нарушение сознания. Могут возникать парез и гемиплегия, атаксия и афтические расстройства. В тяжелых случаях возможна кома.
Признаки почечного синдрома - массивные отеки, повышенное артериальное давление, гематурия. Повышение олигурии, электролитного дисбаланса и азотемии свидетельствует о развитии острой почечной недостаточности. Чаще всего болезнь Мошковица протекает в острой или подострой форме. Возможен повторный курс.
Ассоциированные симптомы: Боль в животе. Боль в сердце. Боль во внутренних органах. Боль за грудиной. Гематурия. Изменения в моче. Отек. Отеки ног. Отеки рук. Отсутствие речи. Протеинурия. Ретикулоцитоз. Судороги. Судороги в ногах.
Возможные осложнения
Без срочного назначения соответствующего лечения летальность от болезни Мошковица составляет 85-100%. Летальные исходы обусловлены ишемическим поражением органов-мишеней (острый инфаркт миокарда, инсульт, инфаркт почек), геморрагическим синдромом (кровоизлияние в мозг, внутреннее кровотечение), органной дисфункцией (ОПН, СН). Описаны случаи внезапной сердечной смерти, связанной со фатальными аритмиями и кардиогенным шоком. На более поздних стадиях развивается диссеминированное внутрисосудистое свертывание (ДВС-синдром).
Сложность диагностики болезни Мошковица связана с отсутствием патогномоничной клинической картины. Пациенты часто поступают в больницу с клиникой инсульта, острой коронарной недостаточностью, ишемией брюшной полости и безуспешно проходят лечение по поводу соответствующих синдромов. Между тем, наряду с осмотром невролога, кардиолога, нефролога, пациентам необходима консультация гематолога:
• Целевой статус. При осмотре пациента обнаруживаются бледно-желтые кожные покровы, мелкие синяки на коже (пурпура), иногда - увеличение селезенки, повышение артериального давления. Пациент может быть в замешательстве, возбуждении, бреду и галлюцинациях, нередко бывает дезориентация. Типичные жалобы - боли в животе и головные боли.
• Клиническая лабораторная диагностика. Характерные изменения гемограммы при болезни Мошковица - анемия, ретикулоцитоз, тяжелая тромбоцитопения (
При малейшем подозрении на ТТП рекомендуется немедленно начать патогенетическое лечение. Пациенты, как правило, поступают в отделения интенсивной терапии, где постоянно контролируются клинические и лабораторные показатели. Терапия болезни Мошковица включает несколько направлений:
• Базовая терапия. Следует начать в первый день после появления клинических симптомов. Оно включает в себя инфузию больших доз свежезамороженной или криосупернатантной плазмы, назначение антиагрегантов (аспирин, дипиридамол), антикоагулянтов (гепарин), глюкокортикостероидов (преднизолон, дексаметазон).
• Экстракорпоральная гемокоррекция. Плазмаферез считается наиболее эффективным методом лечения болезни Мошковица, поскольку он удаляет из крови ингибиторы металлопротеаз. Пациентам с ПП требуется заместительная почечная терапия - гемодиализ, перитонеальный диализ.
• Сопутствующее лечение. При тяжелом анемическом синдроме используются переливания эритроцитов. Переливание тромбоцитов проводится только в том случае, если кровотечение опасно для жизни. Ряд врачей рекомендуют всем пациентам назначать фолиевую кислоту.
Когда формы болезни Мошковица не поддаются стандартному режиму лечения, используются цитостатики, иммунодепрессанты и моноклональные антитела. Для снижения риска рецидива показана спленэктомия.
Прогноз
Болезнь Мошковица - серьезная патология, трудно диагностируемая, с агрессивным течением. Без своевременного лечения смертность очень высока. Благодаря быстрой диагностике и раннему началу лечения (экстракорпоральная гемокоррекция) можно спасти более 80% пациентов. В дальнейшем для продления ремиссии требуется постоянная поддерживающая терапия. Остаточные изменения в виде энцефалопатии, хронической почечной недостаточности часто сохраняются. Профилактика ТТП не разработана. При наследственных формах проводится генетическое консультирование.
Список литературы
1. Тромботическая тромбоцитопеническая пурпура (болезнь Мошковица)/ Филатов Л.Б. - 2006.
2. Клинический случай своевременной диагностики и успешной терапии больной с болезнью Мошковица/ Кузнецова Е.Ю., Соколова-Попова Т.А., Ольховик Т.И., Михалев М.А., Бахтина В.И., Кузнецова-Подзолкова Е.П.// Современные проблемы науки и образования. - 2020. - №4.
3. Особенности диагностики и лечения тромботической тромбоцитопенической пурпуры, развившейся во время беременности: обзор литературы и собственное наблюдение/ Войцеховский В.В., Филатов Л.Б., Пивник А.В., Авдонин П.В., Есенина Т.В., Судаков А.Г. // Клиническая онкогематология. -2014. - No7(4).
4. Случай врожденной тромботической тромбоцитопенической пурпуры, диагностированный с помощью определения активности ADAMTS13/ Фроленко А.Л., Афанасьева Е.И., Резник Н.В., Каган М.Ю.// Оренбургский медицинский вестник. ‒ 2017. ‒ Том V, No2 (18).
Детский врач
Медицинский сайт для студентов, интернов и практикующих врачей педиатров из России, Украины! Шпаргалки, статьи, лекции по педиатрии, конспекты, книги по медицине!
Синдром Мошковица
- Вы студент медик? Интерн? Детский врач? Добавьте наш сайт в социальные сети!

В 1925 году Б. Мошковиц описал тяжелый острый синдром, характеризующийся постоянным сочетанием тромбоцито пенической пурпуры с гемолитической анемией и неврологическими проявлениями.
Этиопатогенез синдрома Мошковица.
Причина синдрома не уточнена, но, по-видимому, ряд исследований доказал инфекционную природу синдрома, возбудителем которого является менингококк. Ввиду клинико-биологического сходства, синдром Мошковица приближается к коллагенозам, с которыми он часто сочетается (в особенности с рассеянной эритематозной волчанкой и узелковым периартериитом).
Частота (статистика) синдрома Мошковица.
Синдром Мошковица встречается в любом возрасте (даже у грудного и новорожденного ребенка), но чаще всего у людей между 25 и 50 годами. Он одинаково поражает оба пола. Число описанных случаев не велико. Согласно статистике опубликованной в 1968 г., отмечалось 300 случаев. Но с тех пор число диагностированных случаев значительно возросло.
Множество синонимов обозначает ту же клиническую картину синдрома Мошковица:
- тромботическая тромбоцитопеническая пурпура;
- симптоматический комплекс Мошковица - Сингера;
- тромботическая микроангиопатия;
- тромбоцитопения Саммерса;
- острая плейохромная анемия;
- синдром Мошковица - Сингера - Саммерса;
- тромбогемолитическая тромбоцитопеническая пурпура;
- общий капиллярный и артериальный тромбоз;
- лихорадочная острая анемия с тромбоцитопенической пурпурой и сосудистым тромбозом;
- тромбозирующий акроангиотромбоз;
- рассеянный тромбоцитопенический тромбоз;
- тромбозирующий тромбоцитный синдром;
- тромбозирующая микроангиотромботическая гемолитическая анемия.
Симптоматология синдрома Мошковица.
Клиническое начало обычно проявляется внезапно, вне зависимости от возраста.
Вначале появляются неспецифические общие клинические признаки:
- невысокая температура,
- анорексия,
- астения,
- миалгии,
- артралгии.
Позже, устанавливаются специфические признаки синдрома:
Сердечно-сосудистые проявления:
- тахикардия,
- галопообразный ритм и систолический шум в очаге выслушивания митрального клапана.
Геморрагические признаки:
- пурпура;
- кожные петехии или кровоподтеки;
- кровоточащие десны;
- гематурия;
- метроррагии;
- мелена или гематемезис.
Дигестивные проявления:
- рвота желчью или кровью;
- гепато- и спленомегалия;
- легкая желтуха.
Почечные признаки:
- олигурия,
- острая почечная недостаточность с задержкой азота.
Неврологические признаки.
Они появляются рано и протекают очень тяжело. Своим тяжелым течением приводят к мысли о наличии какого-нибудь менингита или подарахноидального кровоизлияния. Постоянно неврологические нарушения сочетаются или даже обусловливают появление некоторых значительных психических расстройств.
Так у больных могут проявляться:
- головные боли;
- атаксия мозжечка;
- афазия;
- парезы или спастические параличи, обычно односторонние;
- нарушения поля зрения или слепота;
- тонико-клонические конвульсии;
- нарушения сознания;
- летаргия;
- ступор;
- туманное состояние;
- бред;
- кома.
Диагностика синдрома Мошковица.
Лабораторные обследования предоставляют важные данные для точного диагноза, а именно:
- значительную тромбоцитопению (число тромбоцитов обычно ниже 100 000/мм3); число их снижается во время острого приступа, а затем снова возрастают (но никогда до нормальной величины) в периоды ремиссии;
- время кровотечения — повышенное;
- гемолитическую анемию; в периферической крови, при пониженном числе эритроцитов, появляется множество скизоцитов и сфероцитов;
- миелограмма выявляет присутствие множество мегакариоцитов и гиперплазию эритроцитов;
- дисглобулинемию (непостоянно), всегда с нарастанием плазматических гаммаглобулинов;
- задержку азота (мочевина в крови увеличивается, совместно с установлением острой почечной недостаточности); альбуминурию; время свертывания и время протромбина всегда нормальное.
Электроэнцефалограмма (ЭЭГ) выявляет чаще всего рассеянное поражение мозга и только изредка его очаговые поражения. Но описаны и случаи, при которых мозговая электроактивность нормальная.
Анатомопатологическое обследование при синдроме Мошковица.
Систематический патологоанатомический анализ часто способствует распознаванию случаев синдрома Мошковица, диагноз которых не был поставлен прижизненно.
Основное поражение - это микроангиопатии на месте соединения атериол с капиллярами, с подэндотелиальным отложением гиалинового, ацидофильного материала. Недавно проведенные исследования установили природу этого вещества, доказав, что оно принадлежит к мукополисахаридам и состоит из сочетания гиалуроновой и хондроитинсерной кислоты с некоторыми протеинами.
Пораженный эндотелий создает гнезда, в которых откладываются тромбоциты, способствуя, таким образом, выключению из кровообращения.
Тромботические поражения имеются во всей сосудистой системе, но локализуются предпочтительно на уровне миокарда, почек, поджелудочной железы, надпочечников и серого вещества мозга.
Течение и прогноз синдрома Мошковица.
Приблизительно в 80% случаев течение быстро ведет к смертельному исходу, в течение нескольких дней или недель от начала заболевания. В остальных 20% случаев, течение подострое или даже хроническое с периодами ремиссии. В этих случаях смерть наступает обычно через 6 месяцев после начала болезни.
Прогноз синдрома Мошковица — неблагоприятный. Исключительно редко больные могут прожить несколько лет.
Лечение синдрома Мошковица.
Лечение — в общем, неудовлетворительное и все же, подходящее лечение, быстро и правильно примененное, может привести к ободряющим результатам, хотя во всех случаях они только временные.
В зависимости от случая, рекомендуется обыкновенная кортикотерапия, сочетающаяся с переливанием крови или с удалением селезенки. В некоторых случаях, гепарин совместно с обменозамещающим переливанием крови.
Кортикотерапия в дозе 2 мг/кг веса тела/день перорально, благоприятно влияет на гематологические и геморрагические проявления, без того, однако, чтобы повлиять на смертельный исход.
Переливание крови и даже обменнозамещающее переливание крови не оказывает длительного влияния на существующую анемию.
Удаление селезенки, произведенное как можно ближе к началу болезни, влияет положительно на гемолитический компонент анемии.
Читайте также: