Синдром Странского-Ригела (Stransky-Regala) - синонимы, авторы, клиника
Добавил пользователь Валентин П. Обновлено: 22.01.2026
Неврология:
Популярные разделы сайта:
Альтернирующие синдромы моста мозга: Мийяра — Гюблера, Фовилля, Бриссо — Сикара, Раймона — Сестана, Гасперини, Грене
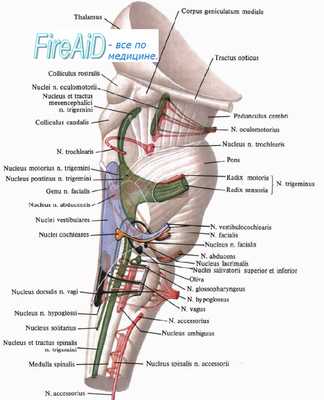
Среди альтернирующих синдромов варолиевого моста чаще всего встречаются синдромы Мийяра — Гюблера, Фовилля, Бриссо -Сикара, Раймона — Сестана, Гасперини, Грене.
Альтернирующий синдром Мийяра — Гюблера. Описан французскими врачами A. Millard (1856 г.) и A. Gubler (1859 г.). Патологический очаг локализуется в основании вентрокаудальной части моста, повреждает двигательное ядро или внутристволовые корешковые волокна VII нерва, а также пирамидный путь.
Синдром встречается при нарушениях кровообращения в парамедианных артериях, отходящих от основной артерии мозга. Гораздо реже он наблюдается при закупорке длинных огибающих артерий и чаще вызывается кровоизлиянием, чем тромбозом. При медленном, постепенном развитии синдрома речь идет обычно об опухолевом процессе — чаще всего о глиоме моста, значительно реже о солитарных бугорках, метастазах рака или саркоме.
Ипсилатеральный симптом — периферический прозопарез (прозоплегия).
Контралатеральный симптом — центральный гемипарез.
Альтернирующий синдром Фовилля
Описан французским неврологом и психиатром L. Foville в 1858 г. Патологический очаг локализуется в вентральной части моста и имеет большую протяженность в рострокаудальном направлении (по сравнению с синдромом Мийяра — Гюблера). Повреждает ядро VI нерва, корешковые волокна VII нерва, пирамидный путь. Генез синдрома Фовилля тот же, что и синдрома Мийяра — Гюблера.
Ипсилатеральные симптомы:
— парез наружной прямой мышцы глаза (сходящееся косоглазие, недоведение глазного яблока при взгляде в сторону очага, диплопия при взгляде в сторону очага), возможен парез взора в сторону очага;
— возможен также периферический прозопарез. Контралатеральный симптом — центральный гемипарез.
Альтернирующий синдром Бриссо — Сикара
Описан в 1895 г. французскими врачами — патологом A. Brissaund и неврологом R. Sicard. Патологический очаг локализуется в вентрокаудальной части основания моста или на основании мозга, раздражая двигательный корешок VII нерва и сдавливая пирамидный путь. Синдром возникает при инфекционно-токсических и очаговых ишемических поражениях базилярной артерии или ее ветвей, объемных процессах на основании мозга.
Ипсилатеральный симптом — лицевой гемиспазм, который со временем может трансформироваться в периферический прозопарез.
Контралатеральный симптом — центральный гемипарез.
Альтернирующий синдром Раймона — Сестана
Описан в 1903 г. французскими неврологами F. Raumond и М. R. Cestan. Очаг локализуется в покрышке и основании моста. Он повреждает следующие анатомические структуры: мостовой центр взора, медиальный продольный пучок, понтоцеребеллярный путь (средняя ножка мозжечка), пирамидный путь, а также (нередко) медиальную и спинальную петли.
Этиологическими факторами синдрома являются ишемический инсульт в бассейне сосудов покрышки и основания моста, опухоли, туберкуломы, очаги демиелинизации и воспаления.
Ипсилатеральные симптомы:
— парез взора в сторону очага;
— мозжечковая гемиатаксия. Контралатеральные симптомы:
— центральный гемипарез;
— гемипарезу может сопутствовать гемигипестезия.
Альтернирующий синдром Гасперини
Описан итальянским неврологом М. Gasperini в 1906 г. Патологический очаг локализуется в покрышке моста. Он повреждает ядра V—VIII черепных нервов, медиальный и задний продольный пучок, медиальную и спинальную петлю (пирамидный путь, как правило, не страдает). Наиболее частой причиной синдрома Гасперини является ишемический инсульт в бассейне передней нижней мозжечковой артерии (ветвь основной артерии).
Синдром может также встречаться при опухолях и воспалительных процессах, захватывающих каудальную часть покрышки моста.
Ипсилатеральные симптомы:
— диссоциированные расстройства поверхностной чувствительности на всей половине лица (преимущественно в оральных и средних зонах Зельдера);
— парез наружной прямой мышцы глаза или парез взора в сторону очага;
— периферический прозопарез;
— центральные кохлеовестибулярные расстройства;
— симптом Горнера.
Контралатеральный симптом — гемигипестезия (исключая лицо).
Альтернирующий синдром Грене
Описан в 1907 г. Очаг локализуется в каудальных отделах нижней трети покрышки моста. Повреждает корешок спинномозгового тракта тройничного нерва, медиальную и спинальную петли. Наиболее частой причиной развития синдрома являются ишемические нарушения в бассейне ветвей базилярной артерии.
Ипсилатеральные симптомы: диссоциированные расстройства поверхностной чувствительности на лице, преимущественно в оральных и средних зонах Зельдера.
Контралатеральный симптом: гемигипестезия поверхностной чувствительности (исключая лицо).
Синдром острого понтинного миелинолиза
Синдром острого понтинного миелинолиза характеризуется прогрессирующей демиелинизацией проводящих путей в центральной части основания моста (за исключением латеральных областей). Вначале развиваются прогрессирующий тетрапарез (дебютирует чаще со слабости в ногах) и псевдобульбарный паралич.
Затем быстро присоединяются качественные и количественные расстройства сознания, офтальмопарезы, генерализованные судороги. Возникновение синдрома описано при голодании, хронических интоксикациях, инфекционных заболеваниях, почечной недостаточности на фоне прогрессирующей уремии. В половине случаев он развивается у больных с хроническим алкоголизмом, причем чаще на фоне воздержания от приема алкоголя.
Странского — Регалы болезнь
общее название различных форм семейной гемолитической анемии, наблюдаемых у населения Малайских островов.
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
Смотреть что такое "Странского — Регалы болезнь" в других словарях:
Странского-Регалы болезнь — (истор.; Е. Stransky, род. в 1891 г., амер. педиатр; А. С. Regala, амер. педиатр 20 в.; син. Странского Регалы синдром) общее название различных форм семейной гемолитической анемии, наблюдаемых у населения Малайских островов … Большой медицинский словарь
Странского-Регалы синдром — (истор., Е. Stransky; А. С. Regala) см. Странского Регалы болезнь … Большой медицинский словарь
Стра́нского — Ре́галы синдро́м — (истор., Е. Stransky; А.С. Regala) см. Странского Регалы болезнь … Медицинская энциклопедия
Странского — Регалы синдром
Смотреть что такое "Странского — Регалы синдром" в других словарях:
Стра́нского — Ре́галы боле́знь — (истор.; Е. Stransky, р. 1891 г., американский педиатр; А.С. Regala, американский педиатр XX в.; син. Странского Регалы синдром) общее название различных форм семейной гемолитической анемии, наблюдаемых у населения Малайских островов … Медицинская энциклопедия
Синдром Картагенера ( Двигательная цилиопатия , Первичная цилиарная дискинезия , Синдром Зиверта-Картагенера , Синдром неподвижных ресничек )
Синдром Картагенера - это генетическая патология цилиарного аппарата, ведущая к развитию хронических риносинуситов, бронхитов, бронхоэктазов, сочетающаяся с обратным расположением органокомплекса «сердце-лёгкие». Заболевание дебютирует в младенческом возрасте и характеризуется частыми гнойно-воспалительными процессами верхних и нижних дыхательных путей. Диагностируется с помощью лучевых методов исследования органов грудной клетки, биопсии слизистых оболочек бронхов или носа. В консервативной терапии используются антибиотики, кортикостероиды, бронхолитики. При необходимости выполняются хирургические операции в области назальных синусов, частичная резекция лёгких.
МКБ-10
Общие сведения
Синдром Картагенера (триада Зиверта-Картагенера, синдром неподвижных ресничек) относится к наследственным болезням из группы первичных цилиарных дискинезий. Одним из первых в 1902 году заболевание описал российский врач А.К. Зиверт. В 1933 году швейцарский терапевт М. Картагенер детально изучил триаду и доказал её наследственную природу. Синдром Зиверта-Картагенера является редкой генетической патологией и встречается у 1 новорождённого на 25 000 - 50 000 родившихся живыми детей. У 50% пациентов с данным пороком встречается полная транспозиция (зеркальное расположение) внутренних органов. Патология нередко сочетается с другими врождёнными аномалиями (полидактилия, «заячья губа», глухонемота и прочие).
Причины
Причиной возникновения триады Картагенера являются мутации генов, отвечающих за нормальное функционирование ресничек и жгутиков различных клеток человеческого организма. Генетические дефекты передаются по аутосомно-рецессивному типу наследования. Первичная цилиарная дискинезия наблюдается у нескольких членов одной семьи. Заболевание проявляется у половины носителей мутантных генов. У родственников больного могут отсутствовать один или несколько признаков классической триады.
Патогенез
Из-за генетического дефекта нарушается синтез структурных белков жгутиков и ресничек. Цилиарный аппарат больного неподвижен или колеблется асинхронно. Во внутриутробном периоде из-за неправильного движения реснитчатого эпителия эмбриона должным образом не выполняется поворот внутренних органов, что приводит к их полному или частичному обратному расположению.
Неспособность мерцательного эпителия дыхательных путей к синхронному движению резко снижает дренажную функцию респираторной системы. Мокрота застаивается. При присоединении вторичной инфекции легко возникают очаги воспаления, формируются бронхоэктазы. Неподвижность или аномальное колебание ресничек эпителия, выстилающего придаточные пазухи носа и евстахиеву трубу, провоцирует рецидивирующие синуситы, евстахииты и отиты. Отсутствие или дисфункция жгутиков сперматозоидов затрудняет их передвижение и является причиной снижения способности к оплодотворению у мужчин.
Симптомы
С первых месяцев жизни у детей с синдромом Картагенера неподвижных ресничек возникают частые рецидивирующие эпизоды насморка и кашля, сопровождающиеся подъёмом температуры до фебрильных цифр. Выделения из носа обычно бывают гнойного характера. Нередко к явлениям ринита присоединяются признаки евстахиита и отита. Дети испытывают головные боли распирающего характера, пульсирующую «стреляющую» боль в ушах.
К 2-3 летнему возрасту у ребёнка происходит хронизация бронхита, кашель становится постоянным. Отделяется слизисто-гнойная (жёлто-зелёная) мокрота. Присоединяется синдром обструкции верхних дыхательных путей. Больного периодически беспокоят приступы мучительного непродуктивного кашля, одышка при физической нагрузке. Рецидивирующие пневмонии носят затяжной характер. Увеличивается количество госпитализаций, удлиняются сроки лечения в стационаре. Синусит также приобретает хроническое течение. В полости носа и придаточных пазухах нередко разрастаются полипы. Появляется постоянная заложенность носа.
Из-за хронического кислородного голодания и частых респираторных инфекций страдает общее развитие ребёнка. У таких пациентов снижается аппетит, выявляется недостаточная масса тела, отставание в росте. Наблюдается общая слабость, повышенная утомляемость, ухудшаются способности к обучению. Синдром Картагенера часто является причиной бесплодия у взрослых мужчин.
Осложнения
Острые респираторные заболевания часто встречаются в младшей возрастной группе. По этой причине синдром Зиверта во всём мире выявляется у детей в основном в 3,5-4-летнем возрасте. К этому времени успевают сформироваться бронхоэктазы. Последствия болезни разнообразны. Хроническое воспаление среднего уха приводит к тугоухости или глухоте. Риносинуситы, полипозные разрастания в носу и его придаточных пазухах вызывают резкое снижение обоняния. Бронхоэктазы являются очагом хронической инфекции и провоцируют развитие грозных осложнений. У пациентов с триадой Картагенера часто развивается системный амилоидоз, формируется лёгочно-сердечная и почечная недостаточность. Нередко случаются эпизоды кровохарканья.
Диагностика
Врачи-педиатры первыми сталкиваются с проявлениями триады Картагенера у маленьких пациентов. При осмотре таких детей можно выявить признаки хронической гипоксии. Губы, носогубный треугольник, кончики пальцев приобретают синюшный оттенок. Дистальные фаланги пальцев рук утолщаются подобно барабанным палочкам, ногти приобретают вид часовых стёкол (синдром Мари-Бамбергера). Для окончательного подтверждения диагноза выполняются:
- Физикальное исследование. Перкуторно выявляется смещение кардиальной тупости вправо. При аускультации лёгких выслушиваются жёсткое дыхание и обилие сухих и влажных мелко- и среднепузырчатых хрипов, которые могут исчезать после откашливания. Сердечный толчок и верхушечный тон определяются в 5-ом межрёберном промежутке справа от грудины.
- Лучевая диагностика. На рентгенограмме грудной клетки наблюдаются правостороннее расположение тени сердца, зеркальная транспозиция лёгких. При исследовании назальных синусов часто выявляется недоразвитие фронтальных пазух. Компьютерная томография грудной полости помогает уточнить локализацию и распространённость бронхоэктазий.
- Функциональные методы исследования. Носят вспомогательный характер. На ЭКГ определяется декстрокардия - противоположное направление основных зубцов, признаки лёгочного сердца. При спирометрии (выполняется у детей от 5 лет и старше) выявляется снижение функции внешнего дыхания преимущественно по обструктивному типу.
- Биопсия. Забор материала производится в период ремиссии, не ранее 4-6 недель после купирования обострения. Биоптат слизистой оболочки носа или бронха исследуется с помощью электронной и световой микроскопии. Выявляются аномалии строения цилиарного аппарата, анализируются частота и синхронность колебания ресничек.
Возможно генетическое подтверждение диагноза, однако для синдрома неподвижных ресничек такой метод исследования считается нецелесообразным. Детские пульмонологи дифференцируют синдром Картагенера с муковисцидозом, кистозной гипоплазией лёгких, бронхоэктазами другой этиологии, бронхиальной астмой и первичными иммунодефицитными состояниями.
Лечение синдрома Картагенера
Заболевание генетической природы полностью излечить невозможно. Терапевтические мероприятия выполняются для улучшения качества жизни пациента, сохранения трудоспособности и минимизации последствий. Наследственный синдром является полиорганной патологией. В лечебном процессе также принимают участие оториноларингологи, при необходимости - и другие специалисты. Для первичного подбора базисной терапии лёгочных проявлений показана госпитализация в отделение пульмонологии. Осуществляется длительное консервативное ведение пациента. Используются следующие основные группы лекарственных средств:
- Антибактериальные препараты. При обострении бронхолёгочной патологии и гнойно-воспалительных процессов назальных придаточных пазух необходимы антибиотики. Препарат выбирают с учётом чувствительности микроорганизмов и применяют в зависимости от состояния пациента внутрь или парентерально.
- Кортикостероиды и бронхолитики. Показанием для назначения ингаляционных кортикостероидов, бета-адреномиметиков и холинолитиков является бронхоспастический синдром. При тяжёлой обструкции дыхательных путей применяют препараты системного действия. Топические назальные кортикостероидные гормоны рекомендуются при сочетании полипозного синусита с аллергическим ринитом.
- Муколитики. Назначают профилактическими курсами и при лечении обострений. Предпочтение отдаётся препаратам карбоцистеина, ацетилцистеина, амброксола. Рекомендуется пероральный приём. Исследования использования муколитиков при лечении цилиарных дискинезий в педиатрии доказали неэффективность их ингаляционного введения.
В лечении триады Зиверта-Картагенера широко применяются кинезитерапия, массаж грудной клетки, при необходимости выполняется бронхоальвеолярный лаваж. Иногда для улучшения носового дыхания, аэрации и дренажа назального синуса необходима хирургическая коррекция. В современной оториноларингологии такие операции выполняются преимущественно малоинвазивным эндоскопическим методом. Редко, при выраженных нагноительных процессах осуществляется резекция участка лёгочной ткани. При тяжёлой лёгочно-сердечной недостаточности возможна одномоментная трансплантация комплекса сердце-лёгкие.
Прогноз и профилактика
Прогноз заболевания зависит от распространённости бронхоэктазий, наличия лёгочного сердца и других осложнений. Полного выздоровления не происходит, но своевременная диагностика, чёткое выполнение врачебных рекомендаций позволяют значительно продлить жизнь пациента, улучшить её качество, полностью или частично сохранить трудоспособность.
В качестве первичной профилактики родителям больного ребёнка рекомендуется генетическое обследование перед планированием новой беременности. Пациенту необходимо получать полноценное высококалорийное питание, вести здоровый образ жизни. Для предупреждения обострений показана ежедневная ирригационная терапия - промывание носа и горла солевым раствором. Назначаются курсовые реабилитационные мероприятия. Желательна сезонная профилактическая вакцинация против пневмококка и гриппа.
1. Случай синдрома Картагенера у новорожденного ребенка/ Лебеденко А.А., Козырева Т.Б., Шокарев А.В., Тараканова Т.Д., Касьян М.С., Носова Е.В., Козырева Н.О.// Журнал фундаментальной медицины и биологии. - 2013 - №4.
4. Компьютерно томографическая характеристика легких и околоносовых пазух при синдроме Зиверта-Картагенера/ Маряшева Ю.А., Соколина И.А., Леонович А.Е., Кокина Н.И., Шехтер А.И.// Радиология - практика. - 2005 - №3.
Синдром Черджа-Стросс
Синдром Черджа-Стросс - воспалительно-аллергическое поражение мелких и средних сосудов (капилляров, венул, артериол), протекающее с образованием некротизирующих эозинофильных гранулем. Для синдрома Черджа-Стросс характерны гиперэозинофилия, поражение бронхо-легочной системы, сердца, ЖКТ, центральной и периферической нервной системы, кожи и суставов. Диагноз синдрома Черджа-Стросс основан на данных анамнеза, клинической картины, лабораторных исследований, рентгенографии органов грудной клетки, биопсии легких. В качестве основной терапии синдрома Черджа-Стросс показано назначение системных глюкокортикостероидов и цитостатиков.
Синдром Черджа-Стросс - разновидность системного васкулита с гранулематозным воспалением сосудов среднего и мелкого калибра и преимущественным поражением респираторного тракта. Синдром Черджа-Стросс относится к полисистемным нарушениям, чаще всего затрагивающим органы с богатым кровоснабжением - кожу, легкие, сердце, нервную систему, ЖКТ, почки. Синдром Черджа-Стросс во многом напоминает узелковый периартериит, но в отличие от него поражает не только мелкие и средние артерии, но и капилляры, вены и венулы; характеризуется эозинофилией и гранулематозным воспалением, преимущественным поражением легких. В ревматологии синдром Черджа-Стросс встречается редко, ежегодная заболеваемость составляет 0,42 случая на 100 тыс. населения. Синдромом Черджа-Стросс страдают люди от 15 до 70 лет, средний возраст пациентов составляет 40-50 лет; у женщин заболевание выявляется несколько чаще, чем у мужчин.
Причины синдрома Черджа-Стросс неизвестны. Патогенез связан с иммунным воспалением, пролиферативно-деструктивными изменениями и повышением проницаемости сосудистой стенки, тромбообразованием, кровоизлияниями и ишемией в зоне повреждения сосудов. Важную роль в развитии синдрома Черджа-Стросс играет повышенный титр антинейтрофильных цитоплазматических антител (ANCA), антигенными мишенями которых являются ферменты нейтрофилов (главным образом, протеиназа-3 и миелопероксидаза). ANCA вызывают преждевременную дегрануляцию и нарушение трансэндотелиальной миграции активированных гранулоцитов. Сосудистые изменения приводят к появлению многочисленных эозинофильных инфильтратов в тканях и органах с образованием некротизирующих воспалительных гранулем.
На первый план при синдроме Черджа-Стросс выходит поражение легких. При гистологическом исследовании выявляются интерстициальные и периваскулярные эозинофильные инфильтраты в стенках легочных капилляров, бронхов, бронхиол и альвеол, перивазальных и перилимфатических тканях. Инфильтраты имеют разнообразную форму, обычно локализуются в нескольких сегментах легкого, но могут распространяться на всю легочную долю. Кроме острофазных воспалительных реакций, отмечаются рубцовые склеротические изменения в сосудах и легочной ткани.
Спровоцировать развитие синдрома Черджа-Стросс могут вирусная или бактериальная инфекция (например, гепатит В, стафилококковое поражение носоглотки), вакцинация, сенсибилизация организма (аллергические заболевания, лекарственная непереносимость), стрессы, охлаждение, инсоляция, беременность и роды.
В своем развитии синдром Черджа-Стросс проходит три стадии.
Продромальная стадия может длиться несколько лет. При типичном течении синдром Черджа-Стросс начинается с поражения респираторного тракта. Появляются аллергический ринит, симптомы носовой обструкции, полипозные разрастания слизистой носа, рецидивирующие синуситы, затяжные бронхиты с астматическим компонентом, бронхиальная астма.
Вторая стадия синдрома Черджа-Стросс характеризуется повышением уровня эозинофилов в периферической крови и тканях; проявляется тяжелыми формами бронхиальной астмы с сильными приступами кашля и экспираторного удушья, кровохарканьем. Приступы бронхоспазма сопровождаются выраженной слабостью, длительной лихорадкой, миалгией, похуданием. Хроническая эозинофильная инфильтрация легких может привести к развитию бронхоэктатической болезни, эозинофильной пневмонии, эозинофильного плеврита. При появлении плеврального выпота отмечаются боли в грудной клетке при дыхании, усиление одышки.
Третья стадия синдрома Черджа-Стросс характеризуется развитием и доминированием признаков системного васкулита с полиорганным поражением. При генерализации синдрома Черджа-Стросс степень тяжести бронхиальной астмы уменьшается. Период между появлением симптомов бронхиальной астмы и васкулита составляет в среднем 2-3 года (чем короче промежуток, тем неблагоприятнее прогноз заболевания). Отмечается высокая эозинофилия (35-85%). Со стороны сердечно-сосудистой системы возможно развитие миокардита, коронарита, констриктивного перикардита, недостаточности митрального и трехстворчатого клапанов, инфаркта миокарда, пристеночного фибропластического эндокардита Леффлера. Поражение коронарных сосудов может стать причиной внезапной смерти больных синдромом Черджа-Стросс.
Для поражения нервной системы характерны периферическая нейропатия (мононейропатия, дистальная полинейропатия «по типу перчаток или чулок»; радикулопатии, нейропатия зрительного нерва), патология ЦНС (геморрагический инсульт, эпилептические приступы, эмоциональные расстройства). Со стороны ЖКТ отмечается развитие эозинофильного гастроэнтерита (абдоминальные боли, тошнота, рвота, диарея), реже - кровотечения, перфорация желудка или кишечника, перитонит, кишечная непроходимость.
При синдроме Черджа-Стросс возникает полиморфное поражение кожи в виде болезненной геморрагической пурпуры на нижних конечностях, подкожных узелков, эритемы, крапивницы и некротических пузырьков. Часто наблюдаются полиартралгии и непрогрессирующий мигрирующий артрит. Поражение почек встречается редко, носит невыраженный характер, протекает в форме сегментарного гломерулонефрита и не сопровождается ХПН.
Больные синдромом Черджа-Строcс за первичной помощью обычно обращаются к различным специалистам - отоларингологу, пульмонологу, аллергологу, неврологу, кардиологу, гастроэнтерологу и поздно попадают к ревматологу. Диагностика синдрома Черджа-Стросс основана на клинико-лабораторных данных и результатах инструментальных исследований. Диагностическими критериями синдрома Черджа-Стросс считаются: гиперэозинофилия (>10% от общего числа лейкоцитов), бронхиальная астма, моно- или полинейропатия, синусит, эозинофильные инфильтраты в легких, экстраваскулярные некротизирующие гранулемы. Наличие не менее 4-х критериев подтверждает диагноз в 85% случаев.
При синдроме Черджа-Стросс также выявляется анемия, лейкоцитоз, повышение СОЭ и уровня общего IgE. Для более половины случаев синдрома Черджа-Стросс характерно обнаружение перинуклеарных антител с антимиелопероксидазной активностью (рANCA).
Рентгенография органов грудной клетки при синдроме Черджа-Стросс позволяет обнаружить быстро исчезающие, ограниченные затемнения и очаговые тени в легких, наличие плеврального выпота. При биопсии легкого определяется гранулематозное воспаление мелких сосудов, инфильтраты в околососудистом пространстве, содержащие эозинофилы. Дифференциальную диагностику синдрома Черджа-Стросс следует проводить с узелковым полиартериитом, гранулематозом Вегенера, хронической эозинофильной пневмонией, идиопатическим гиперэозинофильным синдромом, микроскопическим полиангиитом.
Лечение синдрома Черджа-Стросс
Лечение предполагает длительное назначение высоких доз системных глюкокортикостероидов. По мере улучшения состояния дозу препаратов снижают. При наличии поражений сердечно-сосудистой системы, легких, множественного мононеврита возможно применение пульс-терапии метилпреднизолоном. При неэффективности глюкокортикостероидов используются цитостатики (циклофосфамид, азатиоприн, хлорбутин), которые способствуют более быстрой ремиссии и снижению риска рецидивов, но создают высокий риск инфекционных осложнений. Перед началом терапии отменяются все лекарственные препараты, к которым у больного выявлена сенсибилизация.
Прогноз
Без лечения прогноз синдрома Черджа-Стросс неблагоприятный. При полиорганном поражении происходит быстрое прогрессирование синдрома Черджа-Стросс с высоким риском смертельного исхода от сердечно-легочных нарушений. При адекватном лечении 5-летняя выживаемость составляет 60-80%.
Читайте также:
- Диффузное расширение общего ствола легочной артерии и незаращение овального окна. Болезнь Лютембахера
- Параноидное расстройство личности: причины, симптомы и лечение
- Синдром Гельмгольца-Харрингтона (Helmholz-Harrington)
- Симптомы синдрома Когана на голове и шее
- Эпигенетика и механизмы влияния задержки внутриутробного развития на нее