Синдром Улльриха (Ullrich) - синонимы, авторы, клиника
Добавил пользователь Дмитрий К. Обновлено: 01.02.2026
Мукополисахаридозы (МПС) - группа наследственных болезней обмена веществ, связанных с нарушением метаболизма гликозаминогликанов (ГАГ), приводящее к поражению органов и тканей. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул.
Мукополисахаридоз IV типа (Синдром Моркио) - (синонимы: болезнь Моркио, спондило-эпифизарная дисплазия, хондроостеодистрофия, деформирующая остеохондродистрофия, Моркио - Брайлсфорда синдром, Моркио - Ульриха синдром, К - мукополисахаридоз, эксцентрохондроплазия, Дугве - Мелхиора - Клаузена синдром) - наследственная болезнь накопления, обусловленная дефицитом лизосомных гидролаз: галактозамин-6-сульфат-сульфатазы (МПС IVА) или β-галактозидазы (МПС IVВ), обусловлена отложением в соединительной ткани кератансульфата и характеризуется значительной деформацией скелета и отставанием в росте. Все вышеперечисленные признаки приводят к инвалидизации, а при тяжелом течении болезни - к летальному исходу 4.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
Классификация
• Мукополисахаридоз IVА типа. Стеноз шейного и верхнегрудного отдела позвоночного канала. Миелопатия шейного отдела спинного мозга. Вальгусная деформация нижних конечностей, состояние после оперативного лечения. Правосторонний экссудативный отит. Аденоиды I степени. Правосторонняя кондуктивная тугоухость II степени. Двусторонний экссудативный отит. Левосторонняя сенсоневральная тугоухость I степени. OU катаракта вторичная начальная, смешанный астигматизм. OD начальная стадия помутнения роговиц. Недостаточность митрального и трикуспидального клапанов. ХСН 0ст. Состояние после надмыщелковой корригирующей остеотомии обеих бедренных костей от 2012г., медиального гемиэпифизеодеза дистальной зоны роста левой бедренной кости от 2015г.
В зависимости от первичного генетического дефекта, приводящего к снижению активность лизосомных ферментов выделяют несколько типов мукополисахаридозов.(табл. 1).
Таблица 1 - Классификация (номенклатура) МПС.
Этиология и патогенез
МПС IVА, ген GALNS локализован в хромосомной области 16q24.3. МПС IVВ -, ген GBS локализован в хромосомной области 3q21.33. Важно отметить, что мутация гена, кодирующего β-галактозидазу, вызывает также ганглиозидоз типа I.
Тип наследования: Наследуется по аутосомно-рецессивному типу.
Эпидемиология
Распространенность МПС IVА 1:250 000 новорожденных, МПС IVВ встречается еще реже.
Клиническая картина
Cимптомы, течение
Основные клинические проявления: значительные деформации скелета, особенно конечностей и грудной клетки 4.
Внешний вид: дети рождаются без признаков болезни. Первые симптомы появляются в возрасте 1-3 года; к 7-8 годам клиническая картина уже полностью выражена. Отмечается отставание в росте и физическом развитии. Кожа утолщена, ее тургор и эластичность снижены. Могут наблюдаться широкий рот, короткий нос, редкие зубы, возможно истончение зубной эмали. Аномалия грудной клетки, общая слабость мышц, Х-образная деформация ног, дисплазия тазобедренных суставов, укорочение шеи. Интеллект сохранён.
Костная система: болезнь характеризуется карликовостью (рост взрослого больного около 80-115 см), непропорциональным телосложением (относительно короткое туловище, микроцефалия, короткая шея). Выражена деформация скелета, особенно грудной клетки (куриная, бочкообразная, килевидная). Отмечается кифосколиоз грудного и поясничного отделов позвоночника, при тугоподвижности крупных суставов определяется расслабление связочного аппарата в мелких суставах, что приводит к их гипермобильности. Выявляются контрактуры в локтевых, плечевых, коленных суставах; вальгусная деформация нижних конечностей, плоскостопие.
Центральная нервная система: наиболее частым неврологическим осложнением у пациентов с МПС IV типа является шейная миелопатия. Причиной сдавления спинного мозга у этой группы пациентов является нестабильность шейных и грудных (реже) позвонков. В случае компрессии спинного мозга в шейном отделе отмечается поражение пирамидной системы, что может привести верхнему вялому и нижнему спастическому парапарезу. При грубых изменениях возникают условия для компрессии каудального отдела спинного мозга, что приводит к развитию вялого парапареза ног. Также могут иметь место нарушения тазовых функций. Интеллект обычно не нарушен или умеренно снижен. Характерно снижение слуха, с возрастом развивается глухота. Синдром запястного канала для пациентов с МПС IV типа не характерен.
Сердечно-сосудистая система: поражение сердца наблюдается довольно редко у детей. Типична недостаточность аортального клапана, реже митрального. Кардиомегалия обычно носит вторичный характер. На позднем сроке болезни проявления более выраженные.
Желудочно-кишечная система: отсутствует гепатоспленомегалия. Часто выявляются пупочные и паховые грыжи, расхождение прямых мышц живота.
Диагностика
Диагноз МПС IV устанавливается на основании совокупности клинических данных, результатов лабораторного исследования и молекулярно-генетического анализа (Приложение Г1).
В отличие от других типов, мукополисахаридозов IV тип характеризуется отсутствием снижения интеллекта, помутнения роговицы, гепатоспленомегалии 3.
• При сборе анамнеза и жалоб следует обратить внимание на следующие жалобы и анамнестические события 3:
Выраженность клинических проявлений в зависимости от возраста дебюта различных подтипов МПС IV, представлены в Приложении Г2.
Основные физикальные проявления - значительные деформации скелета, особенно конечностей и грудной клетки 4.
• Рекомендовано исследование активности галактозамин-6-сульфат-сульфатазы (в случае МПС IVA), β-галактозидазы (в случае МПС IVB) в культуре фибробластов, изолированных лейкоцитов, либо в пятнах крови, высушенных на фильтровальной бумаге.
Комментарий: у пациентов МПС IV типа выявляется снижение активности ферментов в зависимости от подтипа
• Рекомендовано молекулярно-генетическое исследование: выявление мутаций в генах GALNS (для МПС IVA) и GBS (для МПС IVB).
• Рекомендовано проведение ультразвукового исследования (УЗИ) органов брюшной полости, селезенки, почек.
Комментарии: выявляются деформации скелета, особенно грудной клетки (куриная, бочкообразная, килевидная), кифосколиоз грудного и поясничного отделов позвоночник. Выявляются уплощение и расширение тел позвонков, чем объясняется характерное укорочение туловища и необычно короткая шея. Выраженный углообразный кифоз. Рентген длинных трубчатых костей выявляет недоразвитость эпифизов, укорочение костей предплечья: локтевая кость не достигает лучезапястного сустава. Изменены кости таза: вертлужные впадины плоские и широкие, их крыша скошена, крылья подвздошных костей неправильной формы; контуры всех костей неровные; головки бедренных костей уплощены.
Комментарии: исследование позволяет оценить функциональное состояние мышечных тканей, нервов и нервно-мышечной передачи; стимуляционная электронейромиография (ЭНМГ) позволяет определить сдавление срединного нерва даже до появления симптомов и должна проводиться, начиная с возраста 4-5 лет ежегодно.
Комментарии: осуществляют для контроля изменений функции коры головного мозга и глубинных мозговых структур, своевременной диагностики эпилепсии.
Комментарии: для диагностики обструктивного апноэ сна проводится полисомнография, которая позволяет определить характер дыхательных нарушений (исключить центральный генез, связь с гипертрофией аденоидов, сердечной недостаточностью или комплекс причин).
Комментарии: регулярное проведение ЭКГ, Эхо-КГ, холтеровского мониторирования ЭКГ, суточного мониторинга артериального давления необходимо пациентам с данной патологией, так как с раннего возраста у них отмечаются сердечно-сосудистые нарушения.
Комментарии: клиническая картина неврологических проявлений и результаты объективных методов обследования не всегда коррелируют. Результаты магнитно-резонансной томографии (МРТ) головного мозга пациентов с МПС не являются диагностически значимыми для определения когнитивного дефицита.
Скрининг на клинические и визуализационные признаки компрессии спинного мозга. Нестабильность атлантоаксиального сустава может быть выявлена при рентгенографии шейного отдела позвоночника с нагрузкой, однако для подтверждения компрессии спинного мозга вследствие утолщения его оболочек требуется проведение МРТ.
• Рекомендовано проведение компьютерной томографии (КТ) головного мозга
Проводится с различными вариантами нанизма, наследственными заболеваниями скелета, сопровождающимися спондилоэпифизарными нарушениям,, 1.
Лечение
Очень важно симптоматическое лечение 6.
• Рекомендована коррекция нарушения осанки, контрактур суставов с использованием нехирургических методов включает физиопроцедуры и применение ортопедических устройств.
• При симптоматической эпилепсии рекомендовано назначение антиконвульсантов, однако дозировки рекомендуется использовать меньше среднетерапевтических для снижения риска развития возможных нежелательных эффектов.
Комментарии: подбор антиконвульсанта осуществляется психоневрологом в зависимости от вида приступов, локализации очага патологической активности.
• Коррекцию сердечно-сосудистой недостаточности, артериальной гипертензии рекомендуется проводить стандартными методами консервативного лечения, принятыми в детской кардиологии.
• Рекомендовано при рецидивирующих отитах, частых респираторных заболеваниях верхних дыхательных путей проведение симптоматической, по показаниям - антибактериальной терапии при отсутствии показаний к хирургическому вмешательству. При снижении слуха - подбор и ношение слуховых аппаратов.
• При офтальмологических нарушениях рекомендовано проведение лечения по показаниям, подбор терапии осуществляется на основании общепринятых рекомендаций по лечению соответствующих нозологий.
• При развитии сдавления спинного мозга, нестабильности атланто-аксиального сочленения рекомендовано хирургическое вмешательство.
• При наличии показаний к оперативному лечению шейной миелопатии и деформаций конечностей, первой рекомендовано производить оперативную декомпрессию спинного мозга. Только при благоприятном исходе данного вмешательства рекомендована оперативная коррекция деформаций конечностей. По показаниям рекомендовано осуществлять хирургическую коррекцию деформаций конечностей, исправление оси нижней конечности
• Несмотря на то, что нестабильность краниоцервикального перехода играет ведущую роль в патологии спинного мозга, рекомендовано рассмотреть вопрос проведения оперативной декомпрессии без окципитоцервикальной стабилизации, что может обусловить хорошие постоперационные результаты. Успешные результаты изолированной ламинэктомии с непродолжительном периодом наблюдения указывают на роль перманентного повреждения спинного мозга на фоне утолщения экстрадуральных мягких тканей, доказывая сочетанный патогенез изменений краниоцервикального перехода.
• В случае проведения спондилодеза после проведение оперативного вмешательства в течение длительного времени рекомендовано ношение гало-аппарата.
• При наличии показаний рекомендовано проведение других хирургических вмешательств: аденотомии, тонзиллэктомии, грыжесечения.
Медицинская реабилитация
Пациенту с мукополисахаридозом IV типа физиотерапевтом и врачом-ЛФК разрабатывается индивидуальный курс реабилитации, включающий массаж, лечебную физкультуру, физиотерапевтические процедуры (магнитотерапию, термотерапию, ударно-волновую терапию, метод биологической обратной связи и другие процедуры).
Реабилитационные курсы (массаж, ЛФК, физиопроцедуры, психолого-педагогическая помощь) желательно проводить в условиях дневного стационара проводится с частотой 3-4 раза в год, длительность - определяется тяжестью состояния и ответом на проводимые мероприятия.
Психолого-педагогическая помощь
Проводится в комплексе реабилитационных мероприятий. Коррекционно-педагогическое воздействие определяется в зависимости от тяжести и длительности течения болезни, структуры нарушений здоровья, степени недоразвития познавательной деятельности, типа эмоционального реагирования, особенностей поведения ребенка. Включение коррекционно-педагогического сопровождения в комплекс восстановительных мероприятий обеспечивает дополнительную оценку динамики психического развития как одного из важных показателей состояния здоровья, повышает эффективность терапевтических вмешательств, снижает экономическое бремя данной патологии за счет социализации пациентов и сохранения психологического потенциала трудоспособных членов семьи.
Необходимо оказание всесторонней помощи (медицинской, психосоциальной и материальной) детям с неизлечимыми ограничивающими срок жизни заболеваниями. В состав паллиативных служб входят врачи, медицинские сестры, психологи и социальные работники. Несмотря на тяжелое состояние и постоянную потребность в мониторинге, все пациенты преимущественно находятся дома в кругу своей семьи и друзей. Основной целью работы паллиативных служб является создание всех необходимых условий для обеспечения нахождения больных в домашних условиях, а не в стенах лечебного учреждения, что позволяет не только улучшить качество жизни больных и их семей, но и существенно снизить государственные затраты на постоянное стационарное лечение таких пациентов.
При проведении общей анестезии необходимо помнить о высоком риске компрессии спинного мозга вследствие нестабильности атлантоаксиального сустава. Короткая шея, ограничение подвижности нижней челюсти, увеличение языка, выраженная гипертрофия аденоидов и миндалин создают проблемы при проведении анестезиологического пособия, поэтому предпочтение следует отдавать местному или региональному обезболиванию. Пациент предварительно консультируется кардиологом, оториноларингологом, анестезиологом, невропатологом. Обязательно проведение полного кардиологического обследования, полисомнографии (для выявления степени дыхательных нарушений), при необходимости - эндоскопии носоглотки и компьютерной томографии легких. Оперативное вмешательство с анестезией необходимо проводить в крупных медицинских центрах, имеющих ОРИТ, так как интубация и последующая экстубация у таких пациентов может вызвать затруднения.
Прогноз
Летальный исход наступает до достижения возраста 20 лет вследствие сердечно-легочной недостаточности, развивающейся на фоне интеркуррентных заболеваний. Возможна внезапная смерть в результате смещения атланто-окципитального сочленения и повреждения ствола мозга.
Профилактика
Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью разъяснения генетического риска. Как и при других аутосомно-рецессивных заболеваниях при МПС тип IV, для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, существует возможность проведения пренатальной и преимплантационной диагностики. С этой целью генетик рекомендует родителям соответствующие диагностические лаборатории и медицинские центры.
Пренатальная диагностика возможна путем измерения активности ферментов в биоптате ворсин хориона на 9-11 неделе беременности. Для семей с известным генотипом возможно проведение ДНК-диагностики.
Заболевание имеет мультисистемную природу и необратимые, прогрессирующие клинические проявления, что обусловливает необходимость наблюдения не только узкими специалистами (оториноларингологами, хирургами-ортопедами, офтальмологами, кардиологами, пульмонологами, невропатологами, стоматологами), но и физиотерапевтами, логопедами, психологами и работниками паллиативных служб 4.
Пациенты с данной нозологией должны постоянно находиться под наблюдением; 1 раз в 6-12 мес. (в соответствии с тяжестью состояния) показано комплексное обследование в многопрофильных стационарах. Длительность нахождения в стационаре / дневном стационаре 21-28 дней.
Наблюдение больных по месту жительства (в амбулаторно-поликлинических условиях) должно осуществляться постоянно. Лабораторные и инструментальные обследования и рекомендуемая частота их проведения представлена в Приложении Г3.
Врожденная мышечная дистрофия Ульриха - Ullrich congenital muscular dystrophy

врожденная мышечная дистрофия Ульриха - это форма врожденной мышечной дистрофии. Это связано с вариантами типа VI коллаген, обычно это связано с мышечной слабостью и респираторными проблемами, хотя проблемы с сердцем не связаны с этим типом CMD. Он назван в честь Отто Ульриха, который также известен своим синдромом Ульриха-Тернера.
Содержание
- 1 Признаки и симптомы
- 2 Генетика
- 3 Диагноз
- 3.1 Дифференциальный диагноз
- 4.1 Прогноз
Признаки и симптомы
Презентация Врожденная мышечная дистрофия Ульриха у пострадавшего выглядит следующим образом:
- слабость
- Затруднения при ходьбе (преимущественно проксимальных мышц, например шеи)
- Слабость в суставах (преимущественно в дистальных суставах)
Генетика
С точки зрения генетики врожденной мышечной дистрофии Ульриха, есть мутации в генах COL6A1, COL6A2, и COL6A3. Этот подтип мышечной дистрофии имеет аутосомно-рецессивный характер.
COL6A1 играет важную роль в поддержании целостности различных тканей человеческого тела. Субъединица альфа 1 типа VI коллаген представляет собой кодируемый белок.
Диагноз
С точки зрения диагностики врожденной мышечной дистрофии Ульриха при осмотре фолликулярного гиперкератоза, может быть дерматологическим индикатором, кроме того, уровень креатинкиназы сыворотки может быть несколько выше нормы. Другие исследования / методы для определения наличия у человека врожденной мышечной дистрофии Ульриха:
Дифференциальный диагноз
Лечение врожденной мышечной дистрофии Ульриха может включать физиотерапия и регулярная растяжка для предотвращения и уменьшения контрактур. В какой-то момент пациенту может потребоваться респираторная поддержка.
Хотя сердечные осложнения не являются проблемой при этом типе ВМД, в отношении респираторных проблем возможна вентиляция через трахеостомию в некоторых случаях.
Прогноз
Прогноз этого подтипа MD указывает на то, что пострадавший человек может в конечном итоге иметь трудности с кормлением. В какой-то момент хирургическое вмешательство может быть вариантом лечения сколиоза.
Сколиоз, который представляет собой искривление позвоночного человека в сторону, определяется множеством факторов, включая степень (легкая или тяжелая), и в этом случае если возможно, человек может использовать бандаж.
Исследование
Что касается возможного исследования врожденной мышечной дистрофии Ульриха, один источник указывает, что циклоспорин A может быть полезным для людей с этим типом CMD.
Согласно обзору Bernardi, et al., циклоспорин A (CsA), используемый для лечения мышечной дистрофии коллагена VI, демонстрирует нормализацию митохондриальной реакции на ротенон..
Врождённые мышечные дистрофии и структурные миопатии
Врождённые (конгенитальные) миопатии — группа генетически обусловленных миопатий, характеризующихся ранним началом (обычно с рождения до 1 года), характерным симптомокомплексом (синдромом «вялого ребёнка» ) и непрогрессирующим или медленно прогрессирующим течением. Среди конгенитальных миопатий выделяют конгенитальные мышечные дистрофии и конгенитальные миопатии (структурные миопатии).
КЛИНИЧЕСКАЯ КАРТИНА
Большинство конгенитальных миопатий и дистрофий проявляется синдромом «вялого ребёнка». Клинические проявления синдрома «вялого ребёнка» сводятся к выраженной мышечной гипотонии, слабому сопротивлению мышц при пассивных движениях, недержанию головы, задержке моторного развития, гипермобильности суставов. Синдром «вялого ребёнка» также наблюдают при спинальных мышечных атрофиях и других врождённых заболеваниях ЦНС, болезнях обмена (гликогенозы, аминоацидурии) и др. Приблизительно 80% случаев синдрома «вялого ребёнка» обусловлены первичным поражением ЦНС. Основной метод диагностики врождённых миопатий — морфологическое исследование мышц; ЭМГ лишь подтверждает первично-мышечный характер поражения на начальном этапе дифференциальной диагностики. Наряду с синдромом «вялого ребёнка» отмечаютслабость лицевой мускулатуры, мышц туловища и дыхательной мускулатуры. У большинства развиваются Koнтpaктypы тазобедренных, коленных и локтевых суставов, мышц-разгибателей шеи, сколиоз, позже может присоединиться наружная офтальмоплегия. Несмотря на задержку моторного развития, большинство детей способны самостоятельно сидеть, некоторые могут самостоятельно ходить (при частичном дефиците мерозина). При ЭМГ выявляют первично-мышечный тип изменений, при этом спонтанная активность мышечных волокон либо отсутствует, либо незначительна.
СИМПТОМЫ
Для конгенитальной мышечной дистрофии Фукуямы типичны выраженная мышечная слабость (дети либо вообще не способны самостоятельно ходить, либо начинают ходить только в возрасте 2-8 лет), симптоматическая эпилепсия (у 50%), умственная отсталость, офтальмологическая патология (микрофтальмия, гипоплазия сетчатки, катаракта, близорукость, косоглазие) . Характерны множественные и разнообразные изменения на МРТ (дисплазия, агирия, расширение желудочков, кисты). Для конгенитальной мышечной дистрофии Ульриха, помимо синдрома “вялого ребёнка”, характерны кифоз, гипермобильность дистальных суставов, дисплазия тазобедренных суставов, гиперкератоз. Способность к самостоятельной ходьбе зависит от тяжести течения, однако к 2- 10 годам, как правило, утрачивается из-за выраженных контрактур. Синдром Уолкера-Варбурга — одно из самых тяжёлых конгенитальных нервномышечных заболеваний (средняя продолжительность жизни — 9 мес). Наблюдают многочисленные врождённые аномалии: менингоцеле, агирию, агенезию мозолистого тела, гипоплазию пирамидного тракта, расширение желудочков, микроцефалию, микрофтальмию, гипоплазию зрительных нервов, катаракту, глаукому и другие изменения, приводящие к слепоте, синдром «вялого ребёнка» и бульбарные нарушения.
ЛЕЧЕНИЕ
Лечение должно производиться исключительно врачом-неврологом. Самолечение недопустимо. Специфической терапии не существует, цель лечения — коррекция ортопедических нарушений (дисплазия тазобедренных суставов, сколиоз), профилактика контрактур, поддержание мышечной силы, терапия кардиомиопатии и симптоматической эпилепсии.
Врожденная мышечная дистрофия Ульриха это форма врожденная мышечная дистрофия. Это связано с вариантами типа VI. коллаген, это обычно связано с мышечной слабостью и респираторными проблемами, хотя проблемы с сердцем не связаны с этим типом ВМД. [4] [5] Он назван в честь Отто Ульриха, который также известен благодаря Синдром Ульриха-Тернера. [6]
Врожденная мышечная дистрофия Ульриха у пострадавшего выглядит следующим образом: [2] [7]
- слабое место
- Трудности при ходьбе (преимущественно проксимальных мышц, например шеи)
- Расшатанность суставов (преимущественно в дистальных суставах)
С точки зрения генетики врожденной мышечной дистрофии Ульриха, есть мутации в генах COL6A1, COL6A2, и COL6A3. Этот подтип мышечной дистрофии аутосомно-рецессивный в природе. [1] [8]
COL6A1 играет важную роль в поддержании целостности различных тканей человеческого тела. Субъединица альфа-1 типа VI коллаген кодируемый белок. [9]
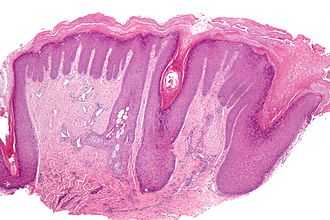
С точки зрения диагностики врожденной мышечной дистрофии Ульриха при осмотре фолликулярной гиперкератоз, может быть дерматологическим индикатором, кроме того, уровень креатинкиназы в сыворотке может быть несколько выше нормы. [5] К другим исследованиям / методам определения наличия врожденной мышечной дистрофии Ульриха относятся: [ требуется медицинская цитата ]
Дифференциальная диагностика
Это включает в себя [10]
Лечение врожденной мышечной дистрофии Ульриха может состоять из физиотерапии и регулярных растяжек для предотвращения и уменьшения контрактуры. В какой-то момент пациенту может потребоваться респираторная поддержка. [3]
Хотя сердечные осложнения не вызывают беспокойства при этом типе ВМД, в отношении респираторных проблем вентиляция через трахеостомия это возможность в некоторых случаях. [5] [11]
Прогноз для этого подтипа MD указывает на то, что у пораженного человека в конечном итоге могут возникнуть трудности с кормлением. В какой-то момент операция может стать вариантом для сколиоз. [3]
Сколиоз, который представляет собой изгиб позвоночника вбок, определяется множеством факторов, включая степень (легкая или тяжелая), и в этом случае, если возможно, человек может использовать скобу. [12]
Что касается возможных исследований врожденной мышечной дистрофии Ульриха, один источник указывает, что циклоспорин А может быть полезным для людей с этим типом CMD. [13]
Согласно обзору Бернарди и др., Циклоспорин A (CsA), используемый для лечения мышечных дистрофий коллагена VI, демонстрирует нормализацию митохондриальной реакции на ротенон. [14]
Синдром Ульриха-Фейхтигера

Клиническая картина. При данном лор синдроме наблюдается аномалия развития наружного уха. Раннее снижение слуха обусловлено пороком развития внутреннего уха.
Со стороны лицевого скелета отмечается седловидная деформация носа, лицо имеет «восковой» (безэмоциональный) вид, за счёт нарушения иннервации мимической мускулатуры. Лобные кости увеличены, за счёт чего лоб имеет выпуклый вид. Рост у таких пациентов, как правило, выше среднего. Возможно расщепление твёрдого и мягкого нёба и верхней челюсти (заячья губа).
Со стороны органа зрения отмечается помутнение роговицы, катаракта глаза. Глазные щели сужены и уменьшены в размере (микроофтальмия). Реже данный порок сопровождается пороком сердца и поликистозом почечно-лоханочной системы.
Диагностика. Консультация лор врача, отомикроскопическое исследование, акуметрическое исследование слуха, аудиометрическое исследование слуха, исследование вызванными потенциалами мозга (в детском возрасте), тимпанометрическое исследование, исследование в позе Ромберга, воздушная нистагмометрия, консультация отоневролога, консультация медицинского генетика, консультация уролога, консультация нефролога, ультразвуковое исследование почек, общий анализ мочи, анализ мочи по Земницкому, экскреторное урографическое исследование, магнитно-резонансное томографическое исследование головного мозга, компьютерное томографическое исследование околоносовых пазух и височных костей, консультация офтальмолога, определение остроты зрения и осмотр глазного дна, консультация кардиолога, электрокардиографическое исследование.
Информацию из данного раздела нельзя использовать для самодиагностики и самолечения. В случае боли или иного обострения заболевания диагностические исследования должен назначать только лечащий врач. Для постановки диагноза и правильного назначения лечения следует обращаться к Вашему лечащему врачу.
Читайте также: