Синдром Унны-Тоста (Unna-Thost) - синонимы, авторы, клиника
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
В статье освещены сведения о кератодермиях - гетерогенной группе состояний, характеризующихся аномальным утолщением кожи ладоней и подошв. Традиционно выделяют приобретенные и наследственные формы.
Резюме. В статье освещены сведения о кератодермиях - гетерогенной группе состояний, характеризующихся аномальным утолщением кожи ладоней и подошв. Традиционно выделяют приобретенные и наследственные формы. В клинической практике наиболее часто встречается гиперкератоз ладоней и подошв как одно из проявлений псориаза, экземы, дерматомикозов и многих других заболеваний. К развитию гиперкератоза ладоней и подошв могут также привести механические и токсические факторы (в том числе прием лекарственных препаратов), поступление с пищей токсических веществ, приводящих к изменениям слизистой кишечника, современные требования моды и красоты могут способствовать развитию множественного дефицита витаминов. Значительно реже встречаются наследственные формы кератодермий, являющиеся самостоятельными заболеваниями. Раннее начало и семейный анамнез предполагают генетическую природу кератодермии. Отличительными особенностями наследственных форм служат характер наследования, степень поражения эпидермиса, наличие/отсутствие распространения очагов за пределы кожи ладоней и подошв, сопутствующая патология. В основе развития наследственных форм лежат мутации различных генов, кодирующих белки (например, кератин, десмосомы, лорикрин, катепсин С, белки щелевых контактов), которые принимают участие в процессе кератинизации. Наследственные ладонно-подошвенные кератодермии имеют большую генетическую и фенотипическую неоднородность, вследствие чего постановка точного диагноза на основе одних лишь клинических проявлений, когда нет возможности выполнить молекулярно-генетическое исследование, является весьма сложной задачей. Благодаря секвенированию нового поколения был достигнут значительный прогресс в расшифровке генетической основы кератодермий. В данном обзоре рассмотрены патогенетические, клинические, диагностические особенности диффузных форм кератодермий, варианты симптоматической терапии, учитывая торпидность и резистентность патологического процесса.
Кератодермии - гетерогенная группа заболеваний, характеризующаяся стойким утолщением эпидермиса кожи ладоней и подошв. После ихтиозиформных поражений кожи кератодермии занимают второе место в общей структуре генодерматозов. Заболевание чаще встречается у женщин, наследственная отягощенность колеб-лется от 1:1000 до 1:4000-9000 [1]. Выделяют наследственные и приобретенные ладонно-подошвенные кератодермии.
В клинической практике чаще всего встречаются приобретенные формы при псориазе, красном плоском лишае, питириазе, красном волосяном отрубевидном (болезни Девержи), экземе, ихтиозе, дерматомикозах, злокачественных новообразованиях и ряде других заболеваний. В ряде случаев присоединяется патологическая сухость кожи (чаще у женщин), нередко кератодермии имеют генетическое происхождение [2]. Также к развитию гиперкератоза ладоней и подошв могут привести механические и токсические факторы (в том числе прием лекарственных препаратов), поступление с пищей токсических веществ, приводящих к изменениям слизистой кишечника, современные требования моды и красоты могут способствовать развитию множественного дефицита витаминов, возможны и иные повреждающие факторы [3, 4]. Исключив вероятность развития приобретенной формы, необходимо задуматься о наследственной.
В настоящее время описано более 20 различных видов кератодермий, являющихся самостоятельными заболеваниями [3]. В основе развития наследственных форм лежат мутации различных генов, кодирующих белки (например, кератин, десмосомы, лорикрин, катепсин С, белки щелевых контактов), которые принимают участие в процессе кератинизации [5].
Варианты мутаций обуславливают клинический полиморфизм. Ряд кератодермий характеризуется лишь ограниченным поражением кожи ладоней и подошв, в то время как при других типах наблюдаются генерализованные нарушения кератинизации. Патологическим изменениям могут подвергаться придатки кожи (ногти и волосы), зубы, возможна потеря слуха, поражение глаз, развитие кардиомиопатии, злокачественных новообразований [6]. Вследствие хронического прогредиентного течения у пациентов формируется физический и психоэмоциональный дискомфорт.
Наследственные ладонно-подошвенные кератодермии имеют большую генетическую и фенотипическую неоднородность, вследствие чего постановка точного диагноза на основе одних лишь клинических проявлений, когда нет возможности выполнить молекулярно-генетическое исследование, является весьма сложной задачей для клиницистов [5].
Секвенирование нового поколения помогло обнаружить гены, неизвестные ранее. Выяснение механизмов развития заболевания открывает новые возможности для разработки альтернативных методов лечения, все больше вызывая интерес к данной патологии. Новые открытия стали предпосылкой к реклассификации некоторых ладонно-подошвенных кератодермий, которые учитывают не только клинические особенности, но и молекулярную основу.
Кератодермия Унны - Тоста (Unna - Thost palmoplantar keratoderma) наследуется по аутосомно-доминантному типу и является наиболее распространенной наследственной кератодермией с частотой развития 1:100 000 [6]. Описаны мутации в гене KRT9, расположенном на хромосоме 17q12-21, которые обуславливают тяжелый эпидермолиз. Ген KRT9 специфичен только для кожи ладоней и подошв, поэтому патологический процесс при данной кератодермии не распространяется за пределы ладонно-подошвенной области. Реже обнаруживается мутация в гене KRT1, при этом явления эпидермолиза минимальны либо отсутствуют совсем. Считается, что нарушение целостности промежуточных филаментов в результате этих мутаций уменьшает устойчивость цитоскелета к незначительной внешней травме, что приводит к образованию пузырей, гиперкератозу, а также к эпидермолизу.
В 2002 г. Кюстер и другие исследователи обнаружили эпидермолитический гиперкератоз по данным гистологии, что характерно для эпидермолитической кератодермии Вернера [7]. Генетическое тестирование выявило мутацию p.R162W в KRT9 в исходном семействе, отмеченную Унной и Тостом, и мутацию p.N160I в KRT9 в исходном семействе, отмеченную Вернером. Обе мутации были расположены на одном сегменте coli-1A в начале домена центрального стержня KRT9. Согласно данному открытию стало очевидно, что данные кератодермии являются одним и тем же типом [8]. Клинически такой тип кератодермии характеризуется избыточным ороговением кожи ладоней и подошв, при этом не наблюдается перехода на другие области. У детей на первом году жизни можно заметить легкое утолщения кожи, далее отмечается постепенное нарастание гиперкератоза. Как правило, к 4-5 годам патологический процесс приобретает вид грубых, толстых гиперкератотических наслоений желтого цвета, с резко ограниченным краем, окружен эритематозным венчиком шириной 1-3 мм. Процесс сопровождается глубокими трещинами, болезненностью, локальным гипергидрозом, нередко появлением пузырей [1].
Ряд авторов выявили рентгенологическую картину остеопороза и атрофии фаланг, наличие подвывихов и деформирующего артроза межфаланговых суставов кистей и стоп [5]. Ногти обычно подвергаются незначительным изменениям, могут иметь деформацию в форме песочных часов. Часто встречаются грибковые суперинфекции.
При гистологическом исследовании выявляется эпидермолитический гиперкератоз (акантокератолизис и дегенерация гранулярного слоя) [3].
Кератодермия Меледа (болезнь острова Меледа (Млета), акрокератома врожденная прогрессирующая, наследственная преходящая ладонно-подошвенная кератодермия, кератоз наследственный трансгредиентный и прогредиентный) является редкой формой кератодермий с предполагаемой распространенностью 1:100 000 [9]. Большинство случаев происходит в родственных кланах, это семьи из Хорватии, Алжира, Израиля и Туниса. Заболевание распространено по всему миру, особенно в регионах, которые исторически были торговыми путями Дубровницкой республики. Мутация распространилась в результате миграции и сохранилась только потому, что она не смертельна и не влияет на воспроизводство потомства [10]. Впервые о данном заболевании стало известно от дубровницкого врача Луки Стулли (Стулича), который описал его в итальянском журнале Antologia в 1826 г. Он наблюдал кератодермию у ряда своих островных пациентов, охарактеризовал ее как «ненатуральную структуру кожи». Незаразное наследственное кожное заболевание было названо mal de Meleda (млетская болезнь) в честь острова Меледа (Млет) в Адриатическом море. Остров сотни лет использовался как изолятор для больных чумой и лепрой, что способствовало созданию условий для кровного родства [11].
Болезнь острова Меледа характеризуется аутосомно-рецессивным типом наследования. Кератодермия обусловлена биаллельными мутациями в ARS-гене (расположен на хромосоме 8q24.3), кодирующем протеин SLURP1 (секретируемый белок Ly-6/PLAUR 1) [3]. Ген SLURP1 локализуется в зернистом слое эпидермиса и выполняет роль проаптотического белка. Арредондо и другие доказали, что белок SLURр1 связывается через никотиновые рецепторы ацетилхолина с кератиноцитами и приводит к уменьшению количества этих клеток. Соответственно, мутация гена, кодирующего данный белок при кератодермии Меледа, приводит к нарушению его функции, регуляция апоптоза кератиноцитов меняется, и возникает гиперкератоз. Наличие хронического воспалительного инфильтрата можно объяснить еще одной функцией белка SLURP1. Считается, что он ингибирует высвобождение макрофагами и кератиноцитами фактора некроза опухоли альфа (ФНО-α). Из-за отсутствия функционального белка SLURP1 процесс не регулируется должным образом. Высвобождение ФНО-α в эпидермисе в конечном итоге приводит к хемотаксису дендритных клеток и клеток памяти, поддерживающих воспаление [11]. Клинические признаки, как правило, включают двустороннюю ладонно-подошвенную кератодермию, развитие диффузного гиперкератоза в форме перчаток или носков с резкими границами и желтым оттенком. Начало болезни характеризуется появлением стойкой эритемы с шелушением кожи ладоней и подошв, как правило, в раннем детском возрасте. Далее, обычно к 15-20 годам, наблюдается усиление ороговения. Характерны участки желто-коричневого гиперкератоза, которые с возрастом переходят на тыльную поверхность кистей и стоп, область локтевых и коленных суставов. Края очага поражения очерчены каймой с фиолетовым оттенком, ширина которой составляет несколько миллиметров. Выражен локальный гипергид-роз, в результате чего поверхность пораженных участков становится слегка влажной, с черными точками (вывод-ные протоки потовых желез). В периоральной области наблюдаются стойкая шелушащаяся эритема, небольшая инфильтрация. Кератодермия часто сопровождается дистрофией ногтей (койлонихия, подногтевой гиперкератоз, поперечная или продольная исчерченность, вдавления, онихогрифоз), характерны сочетания болезни острова Меледа с атопией, возможно присоединение бактериальной и грибковой инфекции. В некоторых случаях кератоз сочетается с умственным или физическим недоразвитием, у отдельных больных наблюдались изменения на электроэнцефалограмме [9]. Гистологически в эпидермисе выявляют гиперкератоз, иногда акантоз без признаков эпидермолиза. Может наблюдаться паракератоз, были описаны случаи с характерной картиной гипергранулеза. В сосочковом слое дермы - небольшой хронический воспалительный инфильтрат из лимфоцитов и гистиоцитов [11].
Синдром Папийона - Лефевра (ладонно-подошвенный гиперкератоз с периодонтитом) - редкое заболевание, впервые описанное в 1924 г. Считается, что синдром поражает от одного до четырех человек на миллион [12]. Наследуется по аутосомно-рецессивному типу. Синдром Папийона - Лефевра обусловлен мутациями гена CTSC (расположен на хромосоме 11q14.2), кодирующего катепсин C (также известный как дипептидилпептидаза I). CTSC представляет собой олигомерную лизосомальную цистеиновую протеазу, которая играет важную роль в эпидермальной дифференцировке, а также активации серин-протеаз, вырабатываемых клетками иммунной системы. Мутации гена CTSC приводят к практически полной потере активности катепсина C, которая, по всей видимости, является причиной восприимчивости к определенным вирулентным возбудителям. Сообщалось о других клеточных нарушениях (включая окислительный стресс с нарушением антиоксидантной активности), характеризующихся аномально высокими уровнями гидропероксида и измененным содержанием CoQ и витамина E. Кроме того, снижается антимикробная активность нейтрофилов. Периодонтит обусловлен совокупностью факторов, таких как свободные радикалы, активные формы кислорода и митохондриальная дисфункция [9]. Развивается в возрасте от 1 до 4 лет, при этом проявления, как правило, сильнее на коже подошв, чем на ладонях. Начинается с эритемы, далее развивается гиперкератоз, который носит диффузный характер. В дальнейшем очаги гиперкератоза переходят трансгредиентно на тыл кистей и стоп. На локтях и коленях появляются очаги, напоминающие псориазиформные элементы. Наряду с поражением кожи развивается выраженный гингивит, он с большой скоростью прогрессирует, переходит в пародонтит, который сопровождается лизисом костей альвеолярных отростков, приводит к преждевременной потере молочных зубов. На фоне переохлаждения, во время развития острого пародонтита наступает обострение кожного процесса. Отмечено несколько случаев синдрома Папийона - Лефевра с неострым пародонтитом и/или пародонтитом с поздним началом. То же самое происходит и с постоянными зубами, обычно к 14-15 годам пациенты с данным синдромом все беззубые [13].
Большая часть пациентов с синдромом Папийона - Лефевра имеет повышенную восприимчивости к генерализованным инфекциям и инфекциям кожи (пиодермии). Описаны случаи развития абсцесса печени. Первые данные об этом были опубликованы в 1988 г. Патогенные бактерии обычно достигают печени гематогенным путем. Наиболее частым этиологическим агентом является золотистый стафилококк. Следует учитывать риск развития гнойного абсцесса печени у пациентов с синдромом Папийона - Лефевра, ассоциированного с лихорадкой неясного генеза [14]. У пациентов может развиваться гипергидроз со зловонным запахом, фолликулярный гиперкератоз, дистрофия ногтей, обызвествление твердой мозговой оболочки. В редких случаях прослеживается связь синдрома Папийона - Лефевра со злокачественной меланомой или плоскоклеточным раком. Гистологически выявляют утолщение всех слоев эпидермиса, особенно рогового, в дерме - незначительные клеточные скопления лимфоцитов и гистиоцитов.
Дифференциальная диагностика диффузных форм наследственных кератодермий проводится со схожими по клинической картине заболеваниями. Для каждой формы кератодермии характерны свои отличительные особенности. При проведении дифференциальной диагностики учитывают характер наследования, генетический дефект, степень поражения эпидермиса, наличие/отсутствие распространения очагов за пределы кожи ладоней и подошв, сопутствующую патологию. Сравнительная характеристика представлена в табл.
Диагностика кератодермий включает сбор анамнеза (с особым вниманием к семейному), физикальный осмотр, включающий оценку поражения кожных покровов, ногтей, волос и зубов, а также клиническую картину. Диагноз устанавливают при наличии очагового или диффузного гиперкератоза на ладонях и подошвах. Дебют заболевания в детском возрасте, отягощенный семейный анамнез, стойкая клиническая картина с небольшим изменением симптомов, относительная резистентность к терапии - это признаки, как правило, свидетельствующие о наследственной форме кератодермии. Отрицательный семейный анамнез или начальные проявления во взрослом возрасте не исключают ее возможности. При необходимости проводится гистологическое исследование.
Помимо неспецифических проявлений, таких как гиперкератоз, гранулез и акантоз, можно выявить характерные признаки, такие как эпидермолитический гиперкератоз, нарушение адгезии кератиноцитов (свидетельствует о десмосомных дефектах) или паракератоз (характерный для лорикриновой кератодермии). Разграничение эпидермолитических и неэпидермолитических форм ладонно-подошвенных кератодермий имеет терапевтическое значение, учитывая, что эпидермолитические формы на фоне приема системных ретиноидов имеют тенденцию к ухудшению [15]. Генетическое тестирование позволяет установить точный диагноз. В случае пародонтоза при кератодермии Папийона - Лефевра пациент направляется на консультацию к стоматологу, где стоматологическая рентгенография позволяет выявить атрофию альвеолярной кости.
Современные методы лечения наследственных форм кератодермий представляют собой в основном симптоматическую терапию. Основными принципами терапии являются увлажнение, восстановление гидролипидной мантии и отшелушивание роговых наслоений. Даются общие рекомендации пациентам: регулярные ванны, очищение и увлажнение области ороговения; избегать длительной механической нагрузки на кожу ладоней и подошв; носить ортопедическую обувь, использовать стельки. В качестве местной терапии применяются кератолитические средства (мочевина, салициловая кислота, молочная кислота). Возможно применение данных лекарственных препаратов под окклюзионную повязку на ночь [16]. При системной терапии используют препараты ретинола (витамина А). Чаще всего это ацитритин (0,3-1,0 мг/кг в зависимости от тяжести процесса). Ретиноиды регулируют процессы роста и трансформации клеток, оказывают терапевтический эффект путем модуляции дифференцировки кератиноцитов, подавления гиперпролиферации и уменьшения инфильтрации воспалительными клетками. Для этого используются синтетические ретиноиды, фитоэстрогены. Клинический эффект наблюдается не сразу, а по истечении 5-6 месяцев после начала приема препаратов и прекращается после их отмены [17]. Ретиноиды обычно применяют для лечения тяжелых, приводящих к инвалидности ладонно-подошвенных кератодермий. Ограниченность применения обусловлена большим количеством побочных эффектов (тератогенность, сухость слизистых оболочек, головная боль, повышение уровня в сыворотке крови холестерина, триглицеридов, трансаминаз и др.). У пациентов с эпидермолитической кератодермией лечение ретиноидами может привести к усилению образования пузырей [16]. Дополнительно используется профессиональная гигиена полости рта, проводится системная антибактериальная терапия периодонтита у пациентов с кератодермией Папийона - Лефевра. При присоединении бактериальной и грибковой флоры необходима наружная терапия антибактериальными и противогрибковыми средствами.
Таким образом, детальное изучение механизмов развития патогенетических, клинических, диагностических особенностей кератодермий открывает новые возможности для разработки методов лечения, повышая интерес к данной патологии.
КОНФЛИКТ ИНТЕРЕСОВ. Авторы статьи подтвердили отсутствие конфликта интересов, о котором необходимо сообщить.
CONFLICT OF INTERESTS. Not declared.
Литература/References
КГМА - филиал ФГБОУ ДПО РМАНПО Минздрава России; 420012, Россия, Казань, ул. Толстого, 4, корп. 2
Сведения об авторах:
Information about the authors:
Синдром Унны-Тоста: клинический случай.
Пугнер А.С.
Научный руководитель: к.м.н., ассистент Ерёмина М.Г.
Кафедра кожных и венерических болезней
Ладонно-подошвенные кератодермии - большая группа наследственных заболеваний, отличающихся по интенсивности, распространенности кератоза и выраженности различных симптомов. Их объединяет усиление ороговения кожи ладоней и подошв за счет интенсивного образования кератиноцитов при замедленной их десквамации. Синдром Унны-Тоста проявляется в раннем детском возрасте, наследуется по аутосомно-доминантному типу и связан с мутацией в генах, кодирующих кератин I типа. Он представляет собой диффузную кератодермию ладоней и подошв без перехода на другие участки кожи, возможно сочетание с различными патологиями внутренних органов и опорно-двигательного аппарата.
Цель: показать основные клинические проявления синдрома Унны-Тоста, являющимся редким генодерматозом из группы ладонно-подошвенных кератодермий.
Пациентка Е. 67 лет находится под наблюдением Клиники кожных болезней Саратовского Государственного Медицинского Университета им. В.И. Разумовского в течении 10 лет с диагнозом кератодермия Унны-Тоста. Из анамнеза известно - изменения кожи возникли в 7 летнем возрасте. У родственников по женской линии (у бабушки и мамы) наблюдалось подобное заболевание. Неоднократно использовала различные физио - и бальнеометоды лечения, кератолитики с временным положительным эффектом. Объективно: на ладонях и подошвах на фоне участков гиперкератоза коричневатого цвета с четко очерченным эритематозным венчиком в 1 мм, отмечаются болезненные трещины и пластинчатое шелушение. Ногтевые пластинки изменены незначительно по типу онихогрифоза. На задней поверхности шеи, спускаясь в межлопаточное пространство, локализуется сосудистый невус розовато-синюшной окраски размером 12 x 17 см.
Общие клинические анализы в пределах возрастной нормы. При рентгенологическом исследовании выявляется деформирующий артроз тазобедренных суставов II степени. Последние 9 лет наблюдается у офтальмолога с диагнозом хронический конъюнктивит, макулодистрофия.
Заключение: кератоз ладоней и подошв может быть симптомом ряда наследственных синдромов - эктодермальной дисплазии, кератодермии Меледа, Папийона-Лефевра и других, что затрудняет клиническую диагностику в данной группе заболеваний, занимающих второе место после ихтиозоформных поражений.
Унны — Тоста синдром
1. Малая медицинская энциклопедия. — М.: Медицинская энциклопедия. 1991—96 гг. 2. Первая медицинская помощь. — М.: Большая Российская Энциклопедия. 1994 г. 3. Энциклопедический словарь медицинских терминов. — М.: Советская энциклопедия. — 1982—1984 гг .
Смотреть что такое "Унны — Тоста синдром" в других словарях:
Унны-Тоста синдром — (P. G. Unna; Thost) см. Унны Тоста наследственная кератодермия … Большой медицинский словарь
Унны-Тоста наследственная кератодермия — (P. G. Unna; Thost; син.: Унны Тоста кератоз, Унны Тоста синдром) наследственный дерматоз, характеризующийся возникновением массивных роговых наслоений на ладонях и подошвах, сочетающихся с локальным гипергидрозом, гиперонихией, иногда с… … Большой медицинский словарь
У́нны — То́ста насле́дственная кератодерми́я — (P.G. Unna; Thost; син.: Унны Тоста кератоз, Унны Тоста синдром) наследственный дерматоз, характеризующийся возникновением массивных роговых наслоений на ладонях и подошвах, сочетающихся с локальным гипергидрозом, гиперонихией, иногда с… … Медицинская энциклопедия
Керато́з — (keratosis; Керат + оз) общее название дерматозов, характеризующихся утолщением рогового слоя эпидермиса. Кератоз бородавчатый семейный наследственный (k. verrucosa familiaris hereditaria; син.: бородавки семейные наследственные, гелодермия)… … Медицинская энциклопедия
Кератодерми́я — (keratodermia; Керато + греч. derma кожа; син.: акрокератома, кератоз ладонно подошвенный, тилез) кератоз, при котором участки ороговения расположены преимущественно на ладонях и подошвах. Кератодермия веррукозная узловатая (k. verrucosa nodosa)… … Медицинская энциклопедия
Унны-Тоста синдром
Кератодермия Унны-Тоста: особенности заболевания и его лечение
Кератодермии (мутилирующая, Меледа, Папийона-Лефевра) представлены преимущественно наследственно передающимися поражениями дермы. Лишь в единичных случаях эта патология является приобретенной. Основной чертой болезни считается ороговение дермы, которое обычно происходит в районе ладоней, подошв. Кератодермия Унны-Тоста, которую мы рассмотрим более детально, является одной из таких болезней.
Особенности болезни
Болезнь Унны-Тоста также известна в кругу специалистов, как ихтиоз ладоней, подошв (врожденный), синдром Унны-Тоста, врождённая ладонно-подошвенная кератома. Эта болезнь характеризуется аутосомно-доминантным типом наследования.
Чаще всего поражение охватывает ладони, стопы. Лишь в некоторых случаях ороговение дермы отмечается исключительно на стопах человека.
Развитие кератодермии Унны-Тоста
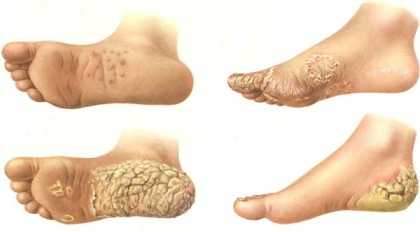
Причины возникновения
Врожденный ихтиоз является результатом нарушения процесса нормального ороговения дермы. Патологии характерен аутосомно-доминантный тип наследования.
Болезнь передается детям в том случае, когда оба родителя имеют мутированный ген. Данная патология может протекать в разных формах:
Симптомы
Обычно кератодермия Уины-Тоста фиксируется у очень недоношенных детей. У таких малышей врачи обычно диагностируют очень много пороков в развитии.
- Дерма малыша выглядит подобно панцирю, поверх которого наблюдаются множественные трещины (весьма глубокие). Образовавшиеся трещины обычно кровоточат.
- У детей синдромом Уины-Тоста может проявляться в деформации лица. Слизистые оболочки весьма активно растут, они могут даже выворачиваться наружу. Практически сразу после рождения малыш с такой патологией умирает.
Иногда специалистам удается спасти ребенка с указанной болезнью. Со временем врожденная болезнь переходит в эритродермию. Редко зафиксированы случаи, когда болезнь исчезла полностью.
Иногда наблюдается легкая форма ихтиоза. Ей характерны весьма незначительные изменения дермы, патология внутренних органов практически не наблюдается. На коже видны множественные очаги покраснения, к которому присоединяется отечность. Трещины, характерные этой патологии, могут возникать в таких участках:
- локти;
- колени;
- лицо (в редких случаях);
- пах.
У больного рассматриваемой патологией наблюдается гипергидроз. Кроме поражения дермы на вышеуказанных частях тела, может наблюдаться также поражения таких структур:
Болезнь могут сопровождать аномалии строения скелета, патологии некоторых внутренних органов (эндокринной, нервной систем).
Кератодермия Унны-Тоста и другие формы недуга рассмотрены специалистом в этом видео:
Диагностика
Особенностью ихтиоза является то, что патология дермы появляется в материнской утробе. При рождении у малыша уже есть видимые изменения кожных покровов. Очень редко патология проявляется на протяжении первого года развития ребенка. Основываясь на таких особенностях, специалисты обычно ставят точный диагноз.
- акантоз (увеличенное количество клеток росткового слоя);
- гранулез (утолщение зернистого слоя); (шелушение рогового слоя).
Может понадобиться дифдиагностика с другими диффузными видами кератодермии.
Лечение кератодермии Унны-Тоста
Терапевтическим способом
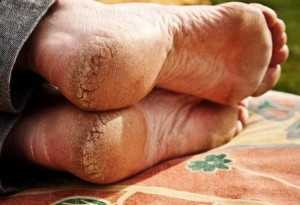
Вылечить полностью эту своеобразную врожденную патологию практически невозможно. Единственное, чего может добиться профессиональный врач, это стойкая ремиссия. Чтобы достичь такого эффекта специалист обычно назначает прием на протяжении месяца нижеуказанные витамины:
После перерыва в месяц, витаминотерапию повторяют снова на протяжении месяца. Более удобно применять масляный раствор. Его назначают по 3 - 5 капель дважды в день.
Медикаментозным способом
Если болезнь протекает в очень тяжелой форме, вышеуказанные витамины вводят внутримышечно. Кроме того, врач может назначать такие препараты:
- Калий.
- Анаболики.
- Ароматический ретиноид.
- Кальций.
- Витамины В.
- Метандростенолон. .
- Нерабол.
- Железа глицерофосфат.
- Аскорбиновая кислота.
Специалист может назначить глюкокортикостероиды, если случай весьма тяжелый.
Профилактика заболевания
Профилактических мероприятий нет, ведь патология является врожденной.
Осложнения
Из-за значительного снижения иммунитета, его полного отсутствия, к основной болезни могут присоединяться различные инфекции.
Прогноз

- Очень часто патология приводит к летальному исходу новорожденного. Иногда болезнь может проходить самостоятельно (единичные случаи).
- Даже комплексная терапия не дает длительного эффекта. Несмотря на то, что современная медицина использует лишь новейшие медикаменты, полного излечения патологии достичь невозможно.
Благодаря своевременно начатой терапии, профессиональному подходу к лечению болезни, врачи могут в значительной степени ослабить симптоматику поражения дермы, снизить дискомфорт.
Читайте также:
- Двигательные рефлексы глаз. Взаимосвязь мозжечка и вестибулярного аппарата
- Инфекция вызванная Mycoplasma pneumoniae: клиника, диагностика
- Хиломикроны. ЛПОНП. ЛПНП. ЛПВП.
- Внежелудочковая обструктивная гидроцефалия на КТ, МРТ головного мозга
- Рецепторная теория действия лекарств. Комплекс агонист-рецептор