Синдром Ушера (Usher) - синонимы, авторы, клиника
Добавил пользователь Владимир З. Обновлено: 08.01.2026
С.В. АВЕРЬЯНОВА 2 , Т.Н. ЮРЬЕВА 1,3 , Ж.В. ХАГДАЕВА 2
1 Иркутский филиал МНТК «Микрохирургия глаза» имени акад. С.Н. Фёдорова» МЗ РФ, 664033, Иркутск, ул. Лермонтова, д. 337
² Офтальмологический центр «Визус» 670046, г. Улан-Удэ, ул. Ключевская, д. 76а
3 Иркутский государственный медицинский университет МЗ РФ, 664003, г. Иркутск, ул. Красного Восстания, д. 1
Рассмотрены аспекты распространенности синдрома Ушера в «закрытых» сообществах. Уделено внимание пигментному ретиниту, как его видам, так и типам наследования. Представлены результаты определения симптомов нарушения темновой адаптации и тугоухости методом анкетирования у 27 человек, а также путей их наследования путем интервьюирования. Дана сравнительная оценка частоты как изолированных жалоб на тугоухость и нарушение темновой адаптации, так и сочетание данных жалоб. Подчеркнута важность исследования при оказании медико-социальной помощи пациентам с данным синдромом.
Ключевые слова: синдром Ушера, пигментный ретинит, тугоухость, инбридинг, темновая адаптация, староверы.
(Для цитирования: Аверьянова С.В., Юрьева Т.Н., Хагдаева Ж.В. Сравнительная оценка заболеваемости синдромом Ушера в «закрытых» сообществах староверов Республики Бурятия. Практическая медицина. 2018)
S.V. AVERYANOVA 2 , T.N. YURYEVA 1,3 , Zh.V. KHAGDAEVA 2
1 Irkutsk Branch of Federal State Autonomous Institution “Interbranch scientific and technical complex “Eye Microsurgery” named after Acad. S.N. Fedorov” of the Ministry of Health of the Russian Federation, 337 Lermontova Str., Irkutsk, Russian Federation, 664033
²Ophthalmological center “Vizus”, 76a Klyuchevskaya St., Ulan-Ude, Russian Federation, 670046
3 Irkutsk State Medical University of the Ministry of Healthcare of the Russian Federation, 1 KrasnogoVosstaniya Str., Irkutsk, Russian Federation, 664003
Comparative assessment of the incidence of Usher-Hallgren syndrome in “closed” communities of Old Believers of the Republic of Buryatia
The aspects of prevalence of Usher-Hallgren syndrome in “closed” communities are analyzed. Attention is paid to retinitis pigmentosa, both its types and types of inheritance. The results of the detection of symptoms of impaired dark adaptation and diminished hearingby the method of questioning 27 people, as well as the ways of their inheritance by interviewing, are presented. The comparative assessment of the frequency of both isolated complaints of diminished hearingand dark adaptation, as well as a combination of these complaints, is made. The importance of research in providing medical and social care to patients with this syndrome is underlined.
Key words: Usher-Hallgren syndrome, pigmented retinitis, diminished hearing, inbreeding, dark adaptation, Old Believers.
(For citation: Averyanova S.V., Yuryeva T.N., Khagdaeva Zh.V. Comparative assessment of the incidence of Usher-Hallgren syndrome in “closed” communities of Old Believers of the Republic of Buryatia. Practical Medicine. 2018)
Синдром Грефе-Ушера характеризуется врожденной (от умеренной до резко выраженной степени) нейросенсорной потерей слуха с вестибулярной гипофункцией и медленно прогрессирующим пигментным ретинитом [1]. По данным Ушера, частота встречаемости синдрома составляет до 43,5 % случаев пигментного ретинита (ПР), по мнению других авторов — колеблется от 20 до 84 % наблюдений. В семьях, где имеются больные с синдромом Ушера, риск рождения больного ребенка составляет 25 % [2]. Впервые патология, расположенная на стыке офтальмологии и отоларингологии, была описана в 1858 году Альбрехтом фон Грефе. Свое название она получила в честь английского доктора Чарльза Ушера (Ашера), который обосновал ее наследственную природу в 1914 году. Согласно статистике, почти 50 % слепоглухих людей страдают синдромом. В США он встречается у 3-10 человек из 100 000. В Великобритании в 2010 году проживало 132 тысячи слепоглухих — примерно 0,2 % населения. По данным европейского семинара по синдрому Ушера (1997 г.), люди с этим заболеванием составляют до 6 % всех глухих с рождения и до 50 % всех слепоглухих взрослых. Патология наиболее распространена среди евреев ашкенази. Кроме того, синдром Ушера обнаруживается среди франкоканадцев, аргентинцев испанского происхождения, нигерийцев и финнов.
Нейросенсорная потеря слуха у больных отмечается за счет атрофии кортиева органа и эпителия внутреннего и наружного желобка в нижней части базального завитка улитки, дегенеративных изменений в верхнем завитке. Обнаруживаются дефекты вестибулярной системы, которые выражаются в нарушении равновесия при ходьбе. При этом нарушение равновесия обусловлено патологическими изменениями лабиринта, а не мозжечковой патологией.
Потеря зрения (ночная слепота, сужение полей зрения) выявляется в возрасте от 5 до 15 лет и связана с ПР. Пигментный ретинит (синонимы: тапеторетинальная дистрофия, периферическая пигментная абиотрофия сетчатки и др.) представляет собой гетерогенную группу наследственных глазных заболеваний [3, 15], характеризующихся первичным генетически детерминированным поражением фоторецепторного слоя и пигментного эпителия сетчатки [4]. Клиника заболевания включает ночную слепоту, постепенную потерю периферического, а по мере прогрессирования заболевания и центрального зрения, связанную с функциональными изменениями и дегенерацией фоторецепторов. Особенностью процесса является незаметное начало, сопровождающееся постепенным снижением зрительных функций и незначительными изменениями на глазном дне [5].
Пигментная дистрофия сетчатки может сочетаться с системными эндокринными и обменными нарушениями. Недостаточность энзимов приводит к мутации генов, что определяет генетическую патологию. К специфическим системным заболеваниям относятся: нарушение метаболизма углеводов, для которого характерно прогрессирующие облаковидные помутнения роговицы, отложение пигмента в сетчатке, атрофия зрительного нерва различной степени выраженности; нарушение метаболизма липидов (синдром Бассена-Корнцвейга, который включает в себя ПР, акантоцитоз, нейромускулярные нарушения с мозжечковой атаксией, нарушением всасывания в кишечнике); нарушение метаболизма липопротеинов, вызывающие церебральные, ретинальные расстройства и задержку умственного развития. Также выделены синдромы, включающие в себя ПР и поражение нервной ткани. Синдром Грефе-Ушера (сочетание глухоты с ПР), Синдром Лауренс-Муна-Барде-Бидле (умственная отсталость с недостаточностью половых желез, ожирением, поли- или синдактилией и пигментной дистрофией [1].
ПР может быть моногенным (вызванным дефектами в одном гене) или дигенными (вызванным дефектами в двух генах). Моногенные формы пигментного ретинита могут иметь аутосомно-доминантный, аутосомно-рецессивный и X — сцепленный рецессивный типы наследования [6, 7, 8].
Синдром Ушера развивается вследствие генетической мутации, наследуемой по аутосомно-рецессивному типу. Для того чтобы симптомы заболевания проявились, ребенок должен унаследовать дефектные гены от обоих родителей. Болезнь Ушера зачастую является следствием инбридинга.
Зарубежный опыт по данной проблеме обширен: как, например, изучение болезни Ушера в такой обособленной группе, как евреи ашкенази. Это социальное сообщество и по сию пору живет на территории Европы. Ашкенази являлись и являются закрытым сообществом, то есть в основном они вступают в браки только внутри своей группы. Внутригрупповые браки — лучший способ избежать ассимиляции, а для евреев это особенно важно, так как многие века у них не было своего государства, и они были рассеяны по всему миру. Во всем есть свои плюсы и минусы. С одной стороны, евреям ашкенази удалось сохранить себя как нацию и не исчезнуть, смешавшись с другими народами. Но, с другой стороны, в таких закрытых группах быстрее накапливаются и чаще встречаются генетические мутации, приводящие к физическим и психофизиологическим патологиям. Сейчас Мировая организация здравоохранения уделяет огромное внимание благополучному всестороннему развитию личности, диагностике психофизиологических заболеваний и их лечению. Между тем в нашей стране эта проблема остается без внимания. Такие пациенты нуждаются в раннем выявлении заболевания и своевременной медико-социальной помощи для эффективной адаптации в стадии клинических проявлений, особенно дети. Официальной статистики по болезни Ушера в Республике Бурятия не ведется, следовательно, не проводятся и профилактические мероприятия. Возникновение этого неизлечимого заболевания можно предотвратить при условии недопущения близкородственных браков, что особенно актуально для малых этноконфессиональных групп. Все это обуславливает актуальность проведения настоящего исследования.
Цель исследования — выявление симптомов тугоухости и нарушения темновой адаптации, а также путей наследования этих изменений у староверов, изолированно проживающих на территории Тарбагатайского района Республики Бурятия.
Материал и методы. Исследуемая группа — староверы, проживающие на территории Тарбагатайского района Республики Бурятия (РБ). Метод исследования — анкетирование, содержащее 17 вопросов о наличии симптомов снижения зрения и слуха, кровных родственников с данной патологией. Опрошено 1237 человек, из них 397 мужчин и 840 женщин в возрасте от 18 до 76 лет.
Результаты исследования. По итогам опроса было выявлено 27 человек с симптомами, свидетельствующими о наличии тугоухости и снижения темновой адаптации. Распределение по полу мужчин к женщинам составило 16:11, возраст пациентов варьировал от 35 до 76 лет (48,7±2,4). Все пациенты распределены на три группы в зависимости от наличия или отсутствия жалоб на патологическое снижение зрения и слуха. Изолированные жалобы на тугоухость выявлены в 15 % случаев, возраст пациентов варьировал от 42 до 76 лет (53,4±2,6); изолированные жалобы на снижение темновой адаптации в 8 % случаев, возраст больных от 37 до 74 лет (55,5±2,3). В подавляющем большинстве, в 77 % случаев отмечалось сочетание жалоб на тугоухость и снижение темновой адаптации, практически в равном соотношении у мужчин и женщин в возрасте от 35 до 75 лет (54,2±2,1).
Синдром Ашера Симптомы, причины, лечение
Синдром ашера он состоит из группы нарушений врожденного наследственного происхождения, характеризующихся нейросенсорными изменениями (Nàjera, Baneyto and Millán, 2005).
На клиническом уровне эта патология определяется наличием двусторонней глухоты, пигментного ретинита и различных вестибулярных изменений (Nàjera, Baneyto and Millán, 2005).
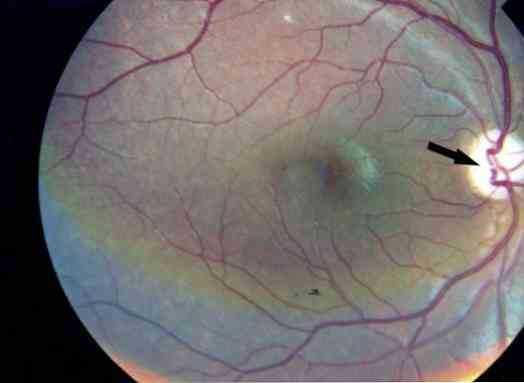
В зависимости от тяжести и пораженных областей синдром Ашера обычно делится на три клинические формы: Синдром Ашера I (USH1), Синдром Ашера II (USH2) и Синдром Ашера III (USH3) (Jaijo, Aller, Beneyto, Nájera and Millán, 2005).
Этиологическая причина этого синдрома связана с аутосомно-рецессивным паттерном, определяемым широкой генетической гетерогенностью (Dyce Gordon, Mapolón Arcedor, Santana Álvarez, 2011).
Было выявлено более 8 различных генов, связанных с появлением синдрома Ашера. Они ответственны за каждый из клинических подтипов (López, Gelvez and Tamayo, 2011).
Диагноз этого заболевания требует использования различных офтальмологических и аудиологических анализов. Кроме того, генетическое исследование обычно проводится для анализа специфических мутаций (Sabaté Cintas, 2009).
Не существует лечебного терапевтического подхода к этому расстройству. Наиболее распространенным является использование методов физической адаптации, реабилитации, обучения ориентации / мобильности и специального образования (Sabaté Cintas, 2009).
Кроме того, медицинский прогноз у пострадавших, как правило, характеризуется прогрессирующим развитием психиатрических и / или неврологических психологических изменений, которые значительно ухудшат их качество жизни (Dyce Gordon, Mapolón Arcedor, Santana Álvarez, 2011 ).
Характеристика синдрома Ушера
Синдром Ушера (SU) является одной из наиболее распространенных причин слепоты и глухоты генетического происхождения (Американская академия офтальмологии, 2013).
Это заболевание характеризуется клинической картиной слухового ухудшения сенсоневрального характера, потери остроты зрения и вестибулярных аномалий (Американская академия офтальмологии, 2013).
Клинический курс связан с (Наера, Банейто и Миллан, 2005):
- Травмы и нарушения во внутреннем ухе (нарушение слуха и равновесия).
- Пигментный ретинит (нарушение зрения)
Это расстройство особенно определяется его клинической и генетической изменчивостью. Клинические исследования, как правило, используют термин синдром Ушера в качестве группы расстройств (USH1, USH2 и USH3) (Genetics Home Reference, 2016).
Это заболевание, представляющее большой медицинский и психологический интерес из-за той степени сенсорной и социальной изоляции, которую имеют затронутые люди (Jaijo et al., 2005)..
Первые клинические описания этого расстройства относятся к фону Грефе и Либрайху, которые выявили значительную медицинскую связь между глухотой и пигментным ретинитом (Braga Norte, Cortez Juares, Nardi, Dell'Aringa и Kobari, 2007)..
Его наследственный характер выявлен в 1914 году благодаря исследованиям британского офтальмолога Ашера, от которого он получил свое имя (Cleveland Clinic, 2016)..
Однако Белл (1933) был одним из пионеров в выявлении большой клинической неоднородности, которая определяет этот синдром (Dyce Gordon, Mapolón Arcedor, Santana Álvarez, 2011).
статистика
Большинство клинических, эпидемиологических и / или экспериментальных исследований считают, что синдром Ашера является частью редких или редких заболеваний (Wallber, 2009).
Тем не менее, синдром Ашера является наиболее распространенной причиной слепоты среди людей (Wallber, 2009)..
Происхождение клинических характеристик у 6% врожденных глухих и 18% людей, страдающих пигментным ретинитом, обусловлено состоянием синдрома Ашера (López, Gelvez and Tamayo, 2011).
Общая распространенность этого синдрома оценивается в 3-4 случая на 100 000 человек в общей популяции, если есть конкретная связь с полом, расой или географическим происхождением (Sabaté Cintas, 2009).
Однако другие авторы, такие как López, Gelvez и Tamayo (2011), считают, что распространенность составляет 3,5-6,2 случая на 100 000 человек..
В случае Испании показатели распространенности могут достигать 4,2 случая на 100 000 жителей, предполагая, что около 1600 человек пострадали на всей территории (Jaijo, Aller, Beneyto, Nájera and Millán, 2005).
В Соединенных Штатах он был обнаружен примерно в 5 случаях на 100 000 жителей; в скандинавских регионах - 3 на 100 000 человек, а в Колумбии - около 3,2 случая на 100 000 человек (López, Gelvez and Tamayo, 2011).
Наконец, что касается распределения случаев по подтипам, мы можем указать на следующие данные (Genetics Home Reference, 2016):
- Тип I и II как наиболее частые формы синдрома Ашера.
- Тип III, наименее распространенный, представляющий 2% от общего числа случаев.
Признаки и симптомы
Клинические характеристики синдрома Ашера в основном связаны с нейросенсорной глухотой, потерей остроты зрения и изменением вестибулярной системы..
Нейросенсорная глухота
Уровень остроты слуха может значительно варьироваться среди пострадавших и в зависимости от подтипа синдрома Ашера, который страдает (Sabaté Cintas, 2009).
Люди могут страдать от общей врожденной глухоты, умеренных проблем со слухом или нормальной или эффективной остроты зрения (Sabaté Cintas, 2009).
Все проблемы, связанные со слуховой зоной, берут свое начало в присутствии нейросенсорных изменений. Таким образом, наиболее распространенным является наблюдение типа глухоты или нейросенсорной тугоухости (Genetics Home Reference, 2016).
Эта патология относится к наличию врожденных поражений во внутреннем ухе и изменчивому изменению волокон и нервных окончаний, связанных со слуховым нервом (Cochlear, 2016).
Потеря остроты зрения
Нарушения зрения обычно составляют основное клиническое изменение синдрома Ашера (Американская академия офтальмологии, 2016).
Пострадавшие люди представляют курс, характеризующийся постепенным снижением остроты зрения, определяемой по следующей схеме (Genetics Home References, 2016):
- Потеря ночного видения.
- Потеря бокового зрения.
- Появление слепых пятен.
- Развитие непрозрачности в хрусталике (катаракты).
Все эти офтальмологические аномалии берут свое начало в проявлении пигментного ретинита (RP)..
Пигментный ретинит представляет собой заболевание, которое относится к прогрессирующему развитию поражений в клетках глаза, чувствительных к свету (Американская академия охталомологии, 2016)..
Эти клетки, называемые колбочками и палочками, расположены в сетчатке и способны преобразовывать световые стимулы в электрические сигналы, интерпретируемые на уровне мозга (Американская академия охталомологии, 2016)..
Частота возникновения различных факторов, таких как генетические аномалии, может привести к гибели этих клеток (Американская академия охталомологии, 2016).
Первоначально поражает трости, в основном отвечающие за ночное и периферическое зрение. Впоследствии происходит ухудшение конусов, отвечающих за центральное зрение и восприятие цветов (Американская академия офтальмологии, 2016).
Изменение вестибулярной системы
Врожденные аномалии, присутствующие во внутреннем ухе, могут также вызывать некоторые значительные изменения в вестибулярной системе (Nàjera, Baneyto and Millán, 2005).
Вестибулярная система сформирована различными структурами, которые играют фундаментальную роль в балансе и эффективном поддержании положения тела.
Эта система объединяет несколько периферических компонентов (вестибулярные нервные окончания и внутреннее ухо) и другие центральные компоненты на уровне мозга и позвоночника..
При синдроме Ашера участие любого из этих компонентов вызовет различные симптомы, в основном связанные с балансом (Genetics Home Reference, 2016).
Как следствие, часто встречаются проблемы с ориентацией, частой потерей равновесия, приобретением сидячего состояния и опозданием, среди прочего (Genetics Home Reference, 2016).
Какие бывают подтипы?
Синдром Ашера можно разделить на несколько подтипов в зависимости от возраста появления первых симптомов, клинических характеристик и тяжести состояния здоровья (Jaijo, Aller, Beneyto, Nájera and Millán, 2005).
Синдром Ашера Тип I
Первый подтип синдрома Ашера можно идентифицировать с рождения, хотя некоторые специфические признаки являются прогрессивными (Sabaté Cintas, 2009):
Слуховые аномалии характеризуются наличием глубокой глухоты врожденного характера, то есть от рождения. Кроме того, невозможно использовать специальные приспособления, такие как слуховые аппараты, для улучшения этой способности..
Визуальные изменения появляются коварно. Первые проблемы со зрением появляются около 10 лет и могут прогрессировать до слепоты с возрастом.
Также возможно выявить нарушения, связанные с вестибулярной системой. Это в основном через серьезные проблемы баланса.
Тип II синдром Ашера
Подтип II синдрома Ашера представляет более поздний дебют. Типичные возрасты появления первых симптомов обычно находятся в подростковом возрасте (Sabaté Cintas, 2009):
Слуховые изменения обычно представляют менее серьезный характер. Хотя возможно развитие умеренных нарушений слуха, можно использовать слуховые аппараты для повышения их эффективности..
Кроме того, наличие остаточного слуха позволяет им использовать устный язык в качестве основного средства общения.
Нарушения зрения, как правило, связаны с прогрессирующим развитием пигментного ретинита, в то время как баланс существенно не нарушается..
Синдром Ашера, тип III
Третий и последний подтип синдрома Ашера имеет типичное проявление в зрелом возрасте. Хотя некоторые клинические признаки могут появляться раньше (Sabaté Cintas, 2009):
Острота слуха характеризуется нормальным или нормальным началом, которое должно быть снижено в зрелом возрасте, что приводит к глухоте.
Нарушения зрения определяются проявлением пигментного ретинита у подростков и развитием слепоты на промежуточных стадиях взрослой стадии..
Наконец, также затрагивается вестибулярная система, что приводит к развитию важных проблем координации и баланса..
причины
Как мы указали в первоначальном описании, синдром Ашера имеет аутосомно-рецессивное наследственное происхождение (López, Gelvez and Tamayo, 2011).
Генетические изменения в основном определяются гетерогенностью, поскольку разные аномалии соответствуют каждому из разных подтипов (López, Gelvez and Tamayo, 2011).
Удалось идентифицировать более 12 мест различных генетических изменений, сопровождаемых более чем 8 специфическими мутациями: MYO7A, USH3, USH1C, VLGR1, CDH23, SANS, CLRN1, OCDH15 (Nàjera, Baneyto and Millán, 2005).
Большинство случаев типа I связаны с мутациями гена MYO7A и CDH12. Хотя тип II больше связан со специфическими мутациями в гене USH2A. Наконец, тип III обусловлен мутациями в гене CLRN1 (Genetics Home Reference, 2016).
диагностика
Клинические характеристики синдрома Ашера ставят диагноз для исследования слуховой, офтальмологической и вестибулярной системы (Американская академия офтальмологии, 2016).
Поэтому крайне важно оценить слуховые способности, остроту зрения и наличие возможных изменений баланса и координации тела (Американская академия офтальмологии, 2016).
- Аудиторская экспертиза: аудиометрия, отоакустическая эмиссия, вызванные улитки потенциалы и отоскопия (Sabaté Cintas, 2009).
- Офтальмологическое обследование: глазное дно, кампиметрия, электроретинограмма, электроокулограмма и электронистагмограмма.
- Вестибулярный осмотр: хотя некоторые из предыдущих тестов могут выявить некоторые изменения вестибулярной системы, наиболее обычным является проведение теста баланса.
В дополнение к подходам, описанным выше, жизненно важно провести генетическое исследование из-за наследственной природы этой патологии..
Фундаментальная цель этого типа тестов состоит в том, чтобы идентифицировать специфическую генетическую мутацию, которая приводит к клиническому подтипу, от которого страдает пациент, и определить характер наследования..
лечение
Там нет ни лечения, ни терапевтического подхода, разработанного специально для синдрома Ашера (Sabaté Cintas, 2009).
Различные специалисты и учреждения, такие как Американская академия офтальмологии (2016), отмечают, что лучшим санитарным подходом является идентификация и ранняя диагностика.
Классическая терапия включает в себя:
- Слуховые компенсационные устройства, такие как кохлеарный имплантат.
- Устройства визуальной компенсации, такие как линзы или приспособления.
- Витаминотерапия на основе введения витамина А для контроля пигментного ретинита.
- Физическая реабилитация для улучшения баланса проблем и координации тела.
- Коммуникативная терапия для генерации альтернативных форм общения.
Кроме того, проводятся исследования альтернативных методов лечения последнего поколения, связанных с генетической заменой..
Синдром Ушера ( Синдром Ашера )
Синдром Ушера - это редко встречающееся генетическое заболевание, протекающее с врожденной сенсоневральной тугоухостью, прогрессирующим пигментным ретинитом и вестибулярной атаксией. В зависимости от типа синдрома у пациентов присутствуют следующие признаки: значительная потеря слуха либо глухота, снижение зрения, нарушение равновесия, когнитивные расстройства. Диагностика включает офтальмологическое (визометрия, офтальмоскопия, электроретинография), отоневрологическое (аудиометрия, вестибулярные пробы), генетическое обследование. Лечение направлено на коррекцию слуха (слухопротезирование, КИ), поддержание зрения (вит. А, Е, омега-3).
МКБ-10
Общие сведения
Синдром Ушера (синдром Ашера) - самая частая причина наследственной слепоглухоты. Его распространенность в популяции оценивается в 3,2-6,2 случая на 100 тыс. населения (по другим данным - 1:6000). Наибольшая заболеваемость отмечается среди евреев-ашкенази, франкоканадцев, испанских аргентинцев, финнов и др. «Первооткрывателем» заболевания считается германский офтальмолог А. фон Грефе. Однако свое «имя» синдром получил в честь британского окулиста Ч. Ушера, который в 1914 г. указал на наследственный характер болезни. Молекулярно-генетические механизмы синдрома были расшифрованы в 1995 г., что открыло широкие возможности для его изучения.
В настоящее время известно более десятка генов, дефекты которых могут привести к развитию синдрома Ушера. Все эти гены, несмотря на разную локализацию и функцию, входят в состав трансмембранного белкового комплекса, участвующего в перемещении миозина, функционировании фоторецепторов сетчатки, а также волосковых клеток улитки. Наиболее часто мутации обнаруживаются в следующих генах:
- CDH23 - «ген глухоты», кодирует белок кадгерин-23;
- MYO7A - ген миозина VIIA;
- PCDH15 - ген протокадгерина-15;
- USH1С - ген гармонина;
- USH1G - ген анкириноподобного белка;
- USH2A - ген ушерина;
- ADGRV1 - ген адгезии G-белка;
- CLRN1 - ген кларина-1 и др.
Синдром Ушера наследуется аутосомно-рецессивным путем от обоих родителей, являющихся носителями дефектных генов. Чаще болеют представители закрытых этнических групп, среди которых достаточно часты близкородственные браки.
Патогенез
Белковые продукты, кодируемые названными генами, принимают непосредственное участие в развитии и функционировании рецепторного аппарата сетчатки глаза и внутреннего уха. Генетические мутации приводят к нарушению формирования воспринимающего аппарата - фоторецепторов и волосковых клеток, что сопровождается врожденными или рано дебютирующими нарушениями зрительной, слуховой и вестибулярной функции.
При синдроме Ушера на глазном дне накапливаются гранулы пигмента, которые распространяются от центра к периферии. Со временем происходит сужение полей и снижение остроты зрения. Патология также затрагивает структуры внутреннего уха: отмечается атрофия спирального узла, нервных волокон кортиева органа, сосудистой полоски улитки.
Классификация
Синдром Ушера генетически неоднороден. Выделяют 4 клинических подтипа, различающихся молекулярно-генетическими механизмами, возрастом манифестации и выраженностью симптоматики:
- Тип 1. Сопряжен с мутациями в генах MYO7A, CDH23, PCDH15, USH1G, USH1C. Протекает наиболее тяжело. Характерна врожденная глухота или глубокая тугоухость. Вестибулярные нарушения и признаки пигментного ретинита развиваются до 5 лет. Составляет около 30% всех случаев.
- Тип 2. Вызывается мутациями USH2A, DFNB31, ADGRV1. Сопровождается непрогрессирующей тугоухостью, ухудшением зрения после 10 лет. Вестибулярная функция не нарушена. Диагностируется примерно у 60% пациентов.
- Тип 3. Связан с мутациями гена CLRN1. Протекает с постепенным ухудшением слуха и зрения, часто - с вестибулярной дисфункцией. Изменения сетчатки развиваются после 20 лет. Выявляется у 3% больных.
- Тип 4. Атипичный вариант синдрома Ушера. Обусловлен дефектами генов HARS, PDZD7, CEP250, C2orf71, может наследоваться Х-сцеплено.
Симптомы
Классическая клиническая картина развивается при 1-м типе синдрома Ушера. Уже в раннем детстве у ребенка диагностируется тяжелая сенсоневральная тугоухость или полная глухота. Наблюдается задержка психомоторного развития, дети поздно начинают ходить. В дошкольном возрасте обнаруживается зрительная дисфункция: в результате усугубления пигментного ретинита быстро ухудшается периферическое зрение, развивается ночная слепота (гемералопия). Это проявляется затруднением ориентации в темноте, частыми спотыканиями, столкновениями с препятствием (другой человек, дверной проем, мебель).
Возникают вестибулярные расстройства: головокружение, нарушение равновесия, атактическая походка. В некоторых случаях может отмечаться когнитивный дефицит, психозы. При других типах болезни Ушера зрительные и слуховые нарушения развиваются позднее и выражены в меньшей степени; вестибулярный синдром не отмечается.
Осложнения
Нейросенсорная тугоухость и постепенная утрата зрения приводят к глубокой инвалидизации. Дети с синдромом Ушера нуждаются в специальном обучении, психолого-педагогическом сопровождении, создании безопасной окружающей среды. Вестибулярные нарушения могут провоцировать падения, повышать риск травматизма. Вторично страдает речь и интеллектуальная сфера. Одним из частых осложнений является катаракта. Прогрессивное снижение зрения приводит к тому, что к 40-50 годам больные синдромом Ушера могут полностью ослепнуть. В тяжелых случаях возникает слепоглухота.
Поскольку нарушения при синдроме Ушера затрагивают несколько анатомо-функциональных систем, диагностика должна носить мультидисциплинарный подход. Больным необходимы консультации отоларинголога, офтальмолога, генетика, проведение комплексного инструментально-лабораторного обследования:
- Офтальмологический осмотр.Визометрия и осмотр глазного дна не всегда позволяют обнаружить начальные признаки пигментной дегенерации сетчатки. В этом отношении наиболее чувствительным тестом является электроретинография, выявляющая изменения еще на доклиническом уровне. По результатам периметрии отмечается концентрическое сужение полей зрения.
- Исследование слуха. Степень снижения слуха устанавливается с помощью аудиометрии. Дополнительно регистрируется отоакустическая эмиссия, стволовые ВП, проводится электрокохлеарография. Больного осматривает врач-сурдолог.
- Исследование вестибулярного анализатора. Для выявления вестибулярных нарушений выполняется видеонистагмография, вестибулометрические пробы.
- Генодиагностика. Проводится секвенирование образцов генетического материала пациента. На основе обнаруженных изменений определяется генетический вариант синдрома Ушера. Также необходимо медико-генетическое консультирование членов семьи.
Дифференциальная диагностика
Синдром Ушера необходимо отличать от других синдромальных и несиндромальных форм глухоты и слепоты, развивающихся при:
- врожденной краснухе - врожденная тугоухость, катаракта, ВПС;
- синдроме Альстрема - дегенерация сетчатки, сенсоневральная тугоухость, ожирение, СД, кардиомиопатия, нефропатия;
- синдроме Халлгрена (акустико-ретино-церебеллярной дегенерации) - пигментный ретинит, катаракта, тугоухость, мозжечковый синдром.
Лечение синдрома Ушера
Консервативная терапия
На сегодняшний день методов полного излечения заболевания не разработано. Все предпринимаемые меры направлены на компенсацию нарушенных функций и замедление течения патологии. Больным синдромом Ушера рекомендовано проведение ежегодных поддерживающих курсов медикаментозной терапии, включающей ноотропы, сосудорасширяющие препараты, антиоксиданты. С целью торможения прогрессирования пигментного ретинита рекомендуется прием больших доз ретинола пальмитата, соблюдение диеты, богатой содержанием витаминов А, Е, ПНЖК.
Способ коррекции слуховой функции подбирается индивидуально. Выбор может быть сделан в пользу слухового протезирования или установки кохлеарного импланта. Для адаптации детей с синдром Ушера к жизни в социуме важна помощь психологов, сурдопедагогов.
Экспериментальное лечение
Сообщается о разработке генной терапии для больных с синдромом Ушера 1 типа. Клинические испытания проходит препарат UshStat, который представляет собой лентивирусный вектор для доставки неповрежденного гена MYO7A. Предполагается субретинальное введение UshStat с целью восстановления функции зрения.
Прогноз и профилактика
Качество жизни пациентов с синдромом Ушера значительно страдает из-за проблем со зрением и слухом. Около 50% лиц с 1-м и 70% со 2-м типом заболевания сохраняют остроту зрения от 20 до 40% на обоих или одном глазу. Больным необходимо пожизненное соблюдение диеты, защита глаз от ультрафиолета. Профилактика заключается в проведении генетических консультаций и лабораторного тестирования в семьях, где имеются случаи данного заболевания. Возможна пренатальная диагностика синдрома Ушера.
1. Изучение интерактома при синдроме Ашера в российской популяции для выбора приоритетных патогенетически ориентированных терапевтических подходов/ Иванова М.Е , Атарщиков Д.С. , Демчинский А.М. , Стрельников В.В. , Бар Д. , Порядин Г.В. , Балашова Л.М. , Салмаси Ж.М.// РМЖ «Клиническая Офтальмология». - 2019. - №4.
3. Этиологические аспекты врожденной тугоухости/ Коноплев О.И. и др./ Медицинский вестник Северного Кавказа. - 2019.
Синдром Ушера
Синдром Ушера
Описание
Синдром Ашера. Это редкое генетическое заболевание, связанное с врожденной нейросенсорной тугоухостью, прогрессирующим пигментным ретинитом и вестибулярной атаксией. В зависимости от типа синдрома у пациентов наблюдаются следующие симптомы: значительная потеря слуха или глухота, снижение зрения, дисбаланс и когнитивные нарушения. Диагностика включает офтальмологическое обследование (визометрия, офтальмоскопия, электроретинография), отоневрологическое обследование (аудиометрия, вестибулярные тесты), генетическое обследование. Лечение направлено на коррекцию слуха (слуховые аппараты, IC), поддержание зрения (вит. А, Е, омега-3).
Дополнительные факты
Синдром Ушера (синдром Ушера) - наиболее частая причина наследственной слепоглухоты. Распространенность его среди населения оценивается в 3,2-6,2 случая на 100 тыс. Жителей (по другим данным - 1: 6000). Наибольшая заболеваемость отмечается среди евреев-ашкенази, франко-канадцев, испанских аргентинцев, финнов и «Первооткрывателем» заболевания является немецкий офтальмолог А. Фон Грефе. Однако свое «название» синдром получил в честь британского офтальмолога Ашера, который в 1914 году указал на наследственную природу болезни. Молекулярно-генетические механизмы синдрома были расшифрованы в 1995 г., что открыло широкие возможности для его изучения.
В настоящее время известно более десятка генов, дефекты которых могут привести к развитию синдрома Ашера. Все эти гены, несмотря на их различную локализацию и функции, являются частью трансмембранного белкового комплекса, участвующего в движении миозина, функционировании фоторецепторов сетчатки, а также волосковых клеток улитки. Наиболее частые мутации обнаруживаются в следующих генах:
• CDH23 - «ген глухоты», кодирующий белок кадгерин-23.
• MYO7A - ген миозина VIIA.
• PCDH15 - ген протокадгерина-15.
• USH1C - ген гармонина.
• USH1G - ген анкириноподобного белка.
• USH2A - ген ушерина.
• ADGRV1 - ген адгезии G-белков.
• CLRN1 - ген кларина-1 и.
Синдром Ушера наследуется по аутосомно-рецессивному типу от обоих родителей, несущих дефектные гены. Чаще болеют представители закрытых этносов, среди которых довольно часты близкородственные браки.
Белковые продукты, кодируемые этими генами, непосредственно участвуют в развитии и функционировании рецепторного аппарата сетчатки и внутреннего уха. Генетические мутации приводят к нарушению формирования аппарата восприятия - фоторецепторов и волосковых клеток, что сопровождается врожденными или ранними нарушениями функций зрения, слуха и равновесия.
При синдроме Ашера пигментные гранулы собираются на глазном дне и распространяются от центра к периферии. Со временем происходит сужение полей и снижение остроты зрения. Патология поражает и структуры внутреннего уха: наблюдается атрофия спирального узла, нервных волокон кортиевого органа и сосудистой полоски улитки.
Синдром Ушера генетически неоднороден. Выделяют 4 клинических подтипа, которые различаются молекулярно-генетическими механизмами, возрастом проявления и тяжестью симптомов:
• Тип 1. Связан с мутациями в генах MYO7A, CDH23, PCDH15, USH1G, USH1C. Курс посложнее. Характерны врожденная глухота или глубокая потеря слуха. Вестибулярные нарушения и признаки пигментного ретинита развиваются до 5 лет. На его долю приходится около 30% всех случаев.
• Тип 2. Вызван мутациями USH2A, DFNB31, ADGRV1. Сопровождается непрогрессирующей тугоухостью, нарушением зрения через 10 лет. Вестибулярная функция не нарушена. Выявляется примерно у 60% пациентов.
• Тип 3. Связан с мутациями в гене CLRN1. Он протекает с постепенным ухудшением слуха и зрения, часто с вестибулярной дисфункцией. Изменения сетчатки развиваются через 20 лет. Выявляется у 3% пациентов.
• Тип 4. Атипичный вариант синдрома Ашера. Это вызвано дефектами генов HARS, PDZD7, CEP250, C2orf71, он может быть унаследован с X-сцеплением.
Клиническая картина
Классическая клиническая картина развивается при первом типе синдрома Ушера. В раннем детстве у ребенка диагностируют тяжелую нейросенсорную тугоухость или полную глухоту. Отмечается задержка психомоторного развития, дети поздно начинают ходить. В дошкольном возрасте выявляется нарушение функции зрения: в результате обострения пигментного ретинита быстро ухудшается периферическое зрение и развивается куриная слепота (гемералопия). Это проявляется затруднением ориентации в темноте, частыми поездками, столкновениями с препятствием (другим человеком, дверью, мебелью).
Возникают вестибулярные нарушения: головокружение, нарушение равновесия, атактическая походка. В некоторых случаях могут наблюдаться когнитивные нарушения, психозы. При других типах болезни Ашера зрительные и слуховые нарушения развиваются позже и менее выражены; вестибулярный синдром не наблюдается.
Ассоциированные симптомы: Шаткая походка.
Возможные осложнения
Нейросенсорная тугоухость и постепенная потеря зрения приводят к тяжелой инвалидности. Дети с синдромом Ашера нуждаются в специальном образовании, психологической и педагогической поддержке и создании безопасной среды. Вестибулярные расстройства могут стать причиной падений и повысить риск травм. Речевая и интеллектуальная сфера страдает вторично. Одно из самых частых осложнений - катаракта. Прогрессирующее снижение зрения приводит к тому, что к 40-50 годам пациенты с синдромом Ашера могут полностью ослепнуть. В тяжелых случаях возникает слепоглухота.
Поскольку нарушения при синдроме Ашера затрагивают разные анатомические и функциональные системы, диагноз должен быть поставлен с применением мультидисциплинарного подхода. Пациентам необходимы консультации отоларинголога, офтальмолога, генетика и комплексное инструментальное и лабораторное обследование:
• Офтальмологический визит. Визометрия и осмотр глазного дна не всегда выявляют первые признаки дегенерации пигмента сетчатки. В этом отношении наиболее чувствительным тестом является электроретинография, выявляющая изменения даже на доклиническом уровне. По результатам периметрии отмечается концентрическое сужение полей зрения.
• Исследование слуха. Степень потери слуха определяется с помощью аудиометрии. Кроме того, записывается отоакустическая эмиссия, ВП ствола, проводится электрокхлеарение. Больной осмотрен сурдологом.
• Исследование вестибулярного анализатора. Для выявления вестибулярных нарушений проводится видеонистагмография, вестибулометрические пробы.
• Генодиагностика. Выполняется секвенирование образцов генетического материала пациента. На основании обнаруженных изменений определяется генетический вариант синдрома Ашера. Также требуется медико-генетическое консультирование членов семьи.
Диф. диагностика
Синдром Ушера необходимо отличать от других синдромных и несиндромальных форм глухоты и слепоты, которые развиваются при:
• Врожденная краснуха - врожденная потеря слуха, катаракта, врожденные пороки сердца.
• Синдром Альстрома - дегенерация сетчатки, нейросенсорная тугоухость, ожирение, сахарный диабет, кардиомиопатия, нефропатия.
• Синдром Халльгрена (акусто-ретино-мозжечковая дегенерация) - пигментный ретинит, катаракта, потеря слуха, мозжечковый синдром.
На сегодняшний день не разработаны методы полного излечения от болезни. Все принимаемые меры направлены на компенсацию нарушенных функций и замедление течения болезни. Пациентам с синдромом Ашера рекомендуется ежегодно проходить поддерживающие курсы медикаментозной терапии, включая ноотропы, вазодилататоры и антиоксиданты. Чтобы подавить прогрессирование пигментного ретинита, рекомендуется принимать высокие дозы пальмитата ретинола и поддерживать диету, богатую витаминами А, Е и ПНЖК.
Способ коррекции функции слуха подбирается индивидуально. Выбор может быть сделан в пользу слуховых аппаратов или кохлеарных имплантатов. Для адаптации детей с синдромом Ашера к жизни в обществе важна помощь психологов и глухих учителей.
Сообщается о разработке генной терапии для пациентов с синдромом Ушера 1 типа. UshStat, лентивирусный вектор для доставки интактного гена MYO7A, в настоящее время проходит клинические испытания. Для восстановления зрительной функции предлагается субретинальное введение УшСтата.
Прогноз
На качество жизни пациентов с синдромом Ашера в значительной степени влияют проблемы со зрением и слухом. Около 50% людей с типом 1 и 70% с заболеванием типа 2 поддерживают от 20 до 40% остроты зрения на оба или один глаз. Пациентам необходима пожизненная диета, защита глаз от ультрафиолета. Профилактика заключается в генетическом консультировании и лабораторных исследованиях в семьях с заболевшими. Возможный пренатальный диагноз синдрома Ушера.
Список литературы
1. Изучение интерактома при синдроме Ашера в российской популяции для выбора приоритетных патогенетически ориентированных терапевтических подходов/ Иванова М.Е , Атарщиков Д.С. , Демчинский А.М. , Стрельников В.В. , Бар Д. , Порядин Г.В. , Балашова Л.М. , Салмаси Ж.М.// РМЖ «Клиническая Офтальмология». - 2019. - №4.
2. Сравнительная оценка заболеваемости синдромом Ушера в.
3. Этиологические аспекты врожденной тугоухости/ Коноплев О.И. И / Медицинский вестник Северного Кавказа. - 2019.
Синдром Ушера, типы 1C, 1D, 1B, 2A, 3A определение 30 мутаций в генах USH2A, CLRN1, MYO7A, CDH23, USH1C в Барнауле
Исследование позволяет выявить 30 мутаций в генах USH2A, CLRN1, MYO7A, CDH23, USH1C, которые приводят к развитию синдрома Ушера — наиболее распространённой причины слепоглухоты. Анализ рекомендуется пациентам с подозрением на синдром Ушера для подтверждения диагноза.
Приём и исследование биоматериала
Когда нужно сдавать анализ Синдром Ушера, типы 1C, 1D, 1B, 2A, 3A определение 30 мутаций в генах USH2A, CLRN1, MYO7A, CDH23, USH1C?
Основные показания к проведению исследования:
- подозрение на синдром Ушера,
- диагностика причин нарушения слуха и зрения.
Подробное описание исследования
Синдром Ушера
Синдром Ушера — генетическая патология, наиболее распространённая причина слепоты и глухоты. Встречается у 1 из 6 000 человек.
Синдром передаётся по аутосомно-рецессивному типу наследования. Это значит, что он возникает, когда оба родителя имеют патологическую мутацию в гене и передают её ребёнку. При этом сами родители здоровы и выступают лишь бессимптомными носителями. Вероятность рождения ребёнка с синдромом Ушера в такой паре — 25%.
При мутации некоторых генов, в том числе USH2A, CLRN1, MYO7A, CDH23, USH1C, особый комплекс белков, который отвечает за работу зрительных и слуховых рецепторов, перестаёт нормально функционировать. У больного развивается тугоухость и прогрессирует пигментный ретинит — патология, при которой фоторецепторы глаза постепенно перестают различать свет.
Формы синдрома Ушера
В зависимости от вида мутации и тяжести патологии выделяют три основные формы синдрома Ушера.
Первая форма — наиболее тяжёлая, проявляется врождённой глухотой или очень плохим слухом у ребёнка (глубокая тугоухость). Слепота и вестибулярные нарушения проявляются рано — в возрасте 5-10 лет.
Вторая форма — самая распространённая, встречается в 60% случаев. Больные с этой формой патологии, как правило, слабослышащие. Глухота не прогрессирует, но зрение начинает ухудшаться после 10 лет. Нарушения координации отсутствуют.
Третья форма — самая редкая. Изначально у детей нормальные слух и зрение, но в течение жизни постепенно развивается глухота, появляются проблемы с координацией и равновесием. После 20 лет начинает прогрессировать пигментный ретинит.
В некоторых случаях встречается атипичное течение болезни.
Каждая форма синдрома Ушера разделяется на несколько типов, которые отличаются друг от друга по нарушениям в отдельных генах.
Исследование позволяет выявить:
- три типа первой формы заболевания — USH1B (1B), USH1C (1C), USH1D (1D), которые связаны с мутацией генов MYO7A, CDH23, USH1C;
- тип заболевания 2A второй формы, связанный с мутацией гена USH2A;
- тип заболевания 3A третьей формы, который связан с мутацией гена CLRN1.
Диагностика и лечение синдрома Ушера
Заподозрить синдром Ушера может отоларинголог при проверке слуха или офтальмолог при осмотре сетчатки или проведении электроретинограммы. Предварительный диагноз подтверждают генетическим тестом с определением формы и типа заболевания.
Эффективного способа вылечить или замедлить развитие глухоты, вызванной синдромом Ушера, в современной медицине не существует. Терапия направлена только на поддержание состояния больного и улучшение качества его жизни. В некоторых случаях тугоухость можно компенсировать слуховыми (кохлеарными) имплантами.
Прогрессирующая слепота также не лечится, но в большинстве случаев пациенты с первой и второй формой заболевания сохраняют слабое зрение.
Читайте также:
- Диагностика склероза артериол головного мозга на КТ, МРТ
- Дифференциальный диагноз при нарушениях памяти. Диагностика при нарушениях памяти.
- Влияние внешних факторов на бронхо-легочную систему.
- Рентгенограмма, КТ при обструктивной непроходимости тонкого кишечника
- Дифференциальная диагностика гистиоцитоза Х