Сопутствующие зрачкам движения глаз. Феномен Вестфаль-Пильца (Westphal-Piltz)
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФазы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Болезнь Вильсона может протекать в брюшной, ригидно-аритмогиперкинетической, дрожательной или экстрапирамидно-корковой форме. Диагностика болезни Вильсона включает офтальмологическое обследование, биохимические анализы мочи и крови, МРТ или КТ головного мозга. Основу патогенетической терапии составляют тиоловые препараты, которые могут приниматься в течении нескольких лет и даже пожизненно.
МКБ-10
Общие сведения
Болезнь Вильсона — наследственное заболевание, передающееся по аутосомно-рецессивному типу. Возникает в условиях мутаций в гене АТР7В, кодирующем белок медьтраснпортирующей АТФ-азы печени. Характерный признак болезни Вильсона — накопление меди в различных органах и тканях, в большей степени в печени и базальных ганглиях. Первооткрыватель заболевания — А.К. Вильсон, описавший заболевание в 1912 году, в отечественной медицине — Н.А. Коновалов. Патогенез болезни Вильсона был выявлен в 1993 году. Понятию «болезнь Вильсона» соответствуют также: болезнь Вильсона-Коновалова, болезнь Вестфаля-Вильсона-Коновалова, дистрофия гепатоцеребральная, дистрофия гепатолентикулярная, дегенерация лентикулярная прогрессирующая.
Причины
Ген АТР7В картирован на длинном плече хромосомы 13 (13q14.3-q21.1). Организм человека содержит около 50-100 мг меди. Суточная потребность меди для человека — 1-2 мг. 95% абсорбированной в кишечнике меди, транспортируется в форме комплекса с церулоплазмином (один из глобулинов сыворотки, синтезируемых печенью) и только 5% в форме комплекса с альбумином. Кроме того, ион меди входит в состав важнейших метаболических ферментов (лизилоксидаза, супероксиддисмутаза, цитохром-С-оксидаза и др.). При болезни Вильсона происходит нарушение двух процессов обмена меди в печени — биосинтез главного медьсвязывающего белка (церулоплазмина) и выведение меди с желчью, следствием чего становится повышение уровня несвязанной меди в крови. Концентрация меди в различных органах (чаще всего в печени, почках, роговице и головном мозге) увеличивается, что приводит к их токсическому поражению.
Классификация
Согласно классификации Н.В. Коновалова различают пять форм болезни Вильсона:
- брюшная
- ригидно-аритмогиперкинетическая
- дрожательно-ригидная
- дрожательная
- экстрапирамидно-корковая
Симптомы
Для болезни Вильсона характерен клинический полиморфизм. Первые проявления заболевания могут появиться в детстве, юношестве, в зрелом возрасте и гораздо реже в зрелом возрасте. В 40-50% случаев Болезнь Вильсона манифестирует с поражения печени, в остальных — с психических и неврологических расстройств. С вовлечением в патологический процесс нервной системы обнаруживается кольцо Кайзера-Флейшера.
Брюшная форма развивается преимущественно до 40 лет. Характерный признак — тяжелое поражение печени по типу цирроза печени, хронического гепатита, фульминантного гепатита.
Ригидно-аритмогиперкинетическая форма манифестирует в детском возрасте. Начальные проявления — мышечная ригидность, амимия, смазанность речи, трудности при выполнении мелких движений, умеренное снижение интеллекта. Для этой формы заболевания характерно прогрессирующее течение, с наличием эпизодов обострения и ремиссии.
Дрожательная форма возникает в возрасте от 10 до 30 лет. Преобладающим симптомом является тремор. Кроме того, могут наблюдаться брадикинезия, брадилалия, тяжелый психоорганический синдром, эпилептические приступы.
Экстрапирамидно-корковая форма встречается весьма редко. Ее начало схоже с началом какой-либо из вышеперечисленных форм. Для нее характерны эпилептические припадки, экстрапирамидные и пирамидные нарушения и выраженный интеллектуальный дефицит.
Диагностика
Офтальмологическое исследование с помощью щелевой лампы выявляет кольцо Кайзера-Флейшера. Биохимические исследования мочи обнаруживают повышенную экскрецию меди в суточной моче, а также снижение концентрации церулоплазмина в крови. С помощью визуализационных методов (КТ и МРТ головного мозга) обнаруживают атрофию полушарий большого мозга и мозжечка, а также базальных ядер.
При диагностике болезни Вильсона неврологу необходимо дифференцировать ее от паркинсонизма, гепатоцеребрального синдрома, болезни Геллервордена-Шпатца. Основным дифференциально-диагностическим признаком этих заболеваний является отсутствие характерных для болезни Вильсона кольца Кайзера-Флейшера и расстройств обмена меди. Для подтверждения болезни Вильсона проводится генодиагностика.
Лечение болезни Вильсона
Основой патогенетического лечения является назначение тиоловых препаратов, в первую очередь — D-пеницилламина либо унитиола. Главное преимущество купренила — низкая токсичность и возможность длительного приема при отсутствии побочных эффектов. Его назначают по 0,15 г (1 капсула) в сутки (только после еды), в дальнейшем, в течение 2,5-3 месяцев дозу увеличивают до 6-10 капсул/сутки (оптимальная доза). Лечение D-пеницилламином проводится годами и даже пожизненно с небольшими перерывами (на 2-3 недели) в случае появления побочных эффектов (тромбоцитопения, лейкопения, обострения язвенной болезни желудка и т. д.).
Унитиол назначают в случае непереносимости (плохой переносимости) D-пеницилламина. Длительность одного курса лечения — 1 месяц, после чего лечение приостанавливают на 2,5-3 месяца. В большинстве случаев наступает улучшение общего состояния пациента, а также регресс неврологических симптомов (скованности, гиперкинезов). В случае доминирования гиперкинезов рекомендовано назначение небольших курсов нейролептиков, при ригидности — леводопы, карбидопы, тригексифенидила.
В случае тяжелого течения болезни Вильсона, при неэффективности консервативного лечения за рубежом прибегают к трансплантации печени. При положительном исходе операции состояние пациента улучшается, восстанавливается обмен меди в организме. В дальнейшем лечение пациента составляет иммуносупрессивная терапия. В России на сегодня постепенно внедряется в клиническую практику метод биогемоперфузии с изолированными живыми клетками селезенки и печени (т. н. аппарат «вспомогательная печень). Немедикаментозное лечение состоит в назначении диеты (стол №5) в целях исключения продуктов богатых медью (кофе, шоколад, бобовые, орехи и т. д.).
Прогноз и профилактика
В случае своевременного диагностирования болезни Вильсона и проведения адекватной медьснижающей терапии возможна нормализация общего состояние пациента и обмена меди в организме. Постоянный прием тиоловых препаратов по схеме, назначенной врачом-специалистом, позволяет поддерживать профессиональную и социальную активность пациента.
Для предотвращения рецидивов болезни Вильсона рекомендовано проведение лабораторных исследований крови и мочи пациента несколько раз в год. Необходим контроль следующих показателей: концентрация меди, церулоплазмина и цинка. Кроме того, рекомендовано проведение биохимического анализа крови, общего анализа крови, а также регулярные консультации у терапевта и невролога.
Болезнь Галлервордена-Шпатца ( Пантотенаткиназа-ассоциированная нейродегенерация )
Болезнь Галлервордена-Шпатца — нейродегенеративная наследственная патология, обусловленная отложением железа в базальных ганглиях головного мозга. Проявляется синдромом паркинсонизма, нарушениями интеллектуальной сферы и психики, гиперкинезами, зрительными расстройствами. Основное диагностическое значение имеет обнаружение рисунка «глаз тигра» в зоне бледного шара при проведении МРТ церебральных структур. Лечение симптоматическое: агонисты дофамина, вальпроаты, антиконвульсанты, нейролептики, антидепрессанты. Прогноз неблагоприятный.
Болезнь Галлервордена-Шпатца описана в 1922 г. немецкими морфологами, в честь которых и получила свое название. К наиболее типичным клиническим маркерам данной патологии относят гиперкинезы, синдром паркинсонизма, интеллектуальное снижение, атрофию зрительных нервов, пигментную ретинопатию. Болезнь Галлервордена-Шпатца встречается крайне редко. В зависимости от времени ее манифестации различают детскую, ювенильную (подростковую) и взрослую формы. Ранее заболевание диагностировалось лишь посмертно по данным аутопсии. После внедрения в практическую неврологию МРТ стала возможна прижизненная постановка диагноза. Прорыв в изучении этиологии был сделан в 2001 г., когда было установлено, что в основе заболевания лежит генетический дефект, обуславливающий нарушения в синтезе фермента пантотенаткиназы. После этого болезнь Галлервордена-Шпатца была официально переименована в пантотенаткиназа-ассоциированную нейродегенерацию.
Болезнь Галлервордена-Шпатца является генетической патологией, носящей как семейный, так и спорадический характер, передающейся по наследству аутосомно-рецессивным путем. Генетическим субстратом выступают аберрации в гене пантотенаткиназы (локус 20р12.3-р13 20-й хромосомы). Всего известно более 50 мутаций. Результатом генетического дефекта является уменьшение продукции пантотенаткиназы, что ведет к аккумуляции в базальных структурах цистеина. Последний образует устойчивые химические соединения с ионами железа, которые неблагоприятно воздействуют на белки и запускают процесс перекисного окисления, приводящий к апоптозу нейронов. На месте некротизированных нейронов происходит разрастание глиальной ткани.
Описанный патологический процесс затрагивает преимущественно бледный шар и черное вещество (субстанцию nigra), где морфологически обнаруживаются внеклеточные отложения железа, имеющие коричневую пигментацию. Кроме этого, имеют место сфероидные периаксональные образования, расположенные в белом церебральном веществе, коре мозга, спинном мозге и периферических нервных стволах.
Симптомы болезни Галлервордена-Шпатца
Классическим вариантом болезни Галлервордена-Шпатца считается ранняя детская форма с клинической манифестацией в период от 4 до 10 лет (обычно после 5-летнего возраста). В 90% случаев первым признаком заболевания выступает торсионная дистония, затрагивающая мышцы ног. Ведущей жалобой является затруднение ходьбы. Затем, как правило, происходит генерализация процесса с его распространением на мышцы глотки, лица, туловища. Наряду с генерализованными вариантами могут отмечаться мультифокальный или сегментарный тип дистонии. Наиболее часто наблюдается писчий спазм, блефароспазм, лицевой параспазм, спастическая кривошея. У трети пациентов отмечаются признаки паркинсонизма: мышечная ригидность и гипокинезия. В ряде случаев имеют место эпилептические приступы.
Болезнь Галлервордена-Шпатца характеризуется когнитивными расстройствами в виде снижения внимательности и памяти с постепенным развитием олигофрении; психическими изменениями с преобладанием агрессивности и асоциального поведения. Отмечается дизартрия. У большинства больных имеются нарушения остроты зрения. В 68% случаев они обусловлены атрофией зрительных нервов, в 29% случаев — пигментной ретинопатией. Для детской формы болезни Галлервордена-Шпатца типично быстрое прогрессирование с полной потерей в течение 10-15 лет способности к передвижению.
Подростковый вариант болезни Галлервордена-Шпатца проявляется в возрасте от 10 до 18 лет и характеризуется более замедленным течением. Дебютирует проявлениями фокальной торсионной дистонии, наиболее часто в мышцах конечностей или ортомандибулярной области. Сопровождается психическими, интеллектуальными и поведенческими расстройствами.
Благодаря полиморфизму симптоматики, постановка диагноза болезни Галлервордена-Шпатца представляет трудную задачу для неврологов. Основными критериями заболевания считаются дебют в возрасте до 30 лет, экстрапирамидные расстройства, неуклонное прогрессирование симптомов, наличие типичной МРТ-картины. К дополнительным признакам отнесены наличие пирамидных знаков, прогрессирующее интеллектуальное снижение, эпиприступы, атрофия зрительных нервов, пигментная атрофия сетчатки, аутосомное наследование по рецессивному типу.
В диагностике опираются на данные неврологического статуса и электроэнцефалографии. При нарушении зрения проводят консультацию офтальмолога, визиометрию, офтальмоскопию. Определение типа наследования осуществляет генетик путем составления генеалогического древа. Возможна ДНК-диагностика (поиск мутаций в гене пантотенаткиназы). При проведении ПЭТ головного мозга удается выявить сниженный метаболизм в зоне паллидума. Основанием для исключения болезни Галлервордена-Шпатца является наличие симптомов другой патологии, в рамки которой может укладываться имеющаяся клиническая картина: болезни Вильсона, хореи Гентингтона, нейроакантоцитоза, болезни Мачадо-Джозефа.
Основополагающим методом диагностики болезни Галлервордена-Шпатца выступает МРТ. Во всех типичных вариантах патологии в режиме Т2 на МРТ головного мозга определяется расположенная в области бледного шара гиперинтенсивная зона овальной формы, окруженная еще большей гипоинтенсивной зоной. Подобная МРТ-картина является патогномоничной и получила название «глаз тигра». Гипоинтенсивная зона представляет собой «глаз», а гиперинтенсивная — его «зрачок». Время появления этого признака на томограммах пока дискутируется. По мнению одних авторов «глаз тигра» может появляться еще до клинической манифестации болезни, по мнению других — через несколько лет от дебюта клинических симптомов.
Лечение болезни Галлервордена-Шпатца
В настоящее время болезнь Галлервордена-Шпатца не имеет эффективных методов лечения. Попытки терапии препятствующими накоплению железа хелатными соединениями (дефероксамином) и антиоксидантами не имели успеха. В связи с этим применяется симптоматическое лечение. Синдром паркинсонизма служит показанием к назначению дофаминовых агонистов (пирибедила, прамипексола) или производных амантадина. Однако при данном заболевании он, как правило, резистентен к проводимому лечению.
При гиперкинезах применяют вальпроаты, бензодиазепины (диазепам, клоназепам). При спастике рекомендованы миорелаксанты (баклофен, толперизона гидрохлорид), при эпиприступах — топирамат или вальпроаты, при когнитивных расстройствах — ипидакрин и холина альфосцерат, при психических отклонениях — нейролептики (рисперидон, кветиапин, клоназепам), антидепрессанты 3-го поколения (венлафаксин, циталопрам, дапоксетин).
Симптоматическая терапия болезни Галлервордена-Шпатца позволяет уменьшить проявленность клинических симптомов, продлить способность пациентов к самообслуживанию. Вместе с тем продолжается разработка новых способов лечения. Исследуется эффективность применения пантотеновой кислоты. Получены данные о положительном влиянии на течение заболевания магнитной стимуляции бледного шара.
Прогноз
Прогноз зависит от формы заболевания. Наиболее неблагоприятное течение имеет ранняя форма, при которой полная инвалидизация наступает в промежутке от 10 до 15 лет с момента дебюта симптомов. Более благоприятен взрослый вариант, особенно в случаях, когда деменция слабо выражена. Его средняя продолжительность составляет более 20 лет.
Пароксизмальное позиционное головокружение
Пароксизмальное позиционное головокружение — повторные преходящие краткосрочные приступы системного головокружения, провоцируемые изменением положения головы. Связаны с наличием плавающих в эндолимфе или фиксированных на купуле отолитов. Кроме тошноты и иногда рвоты, приступы пароксизмального головокружения не сопровождаются какой-либо другой симптоматикой. Диагноз базируется на жалобах пациента, положительной пробе Дикса-Холлпайка, результатах вращательного теста. Лечение состоит в проведении специальных лечебных методик Эпли или Семонта, выполнении вестибулярной гимнастики.
Пароксизмальное позиционное головокружение (ППГ) представляет собой доброкачественное приступообразное системное головокружение, продолжительностью от нескольких секунд до 0,5 мин, возникающее при движениях головы, чаще в горизонтальном положении тела. Описано в 1921 г. Робертом Барани. В 1952 г. Дикс и Холлпайк предположили связь заболевания в нарушениями в органе равновесия и предложили к клиническому использованию провокационную диагностическую пробу, которой до сих пор пользуются специалисты в области неврологии и вестибулологии. Поскольку пароксизмальное позиционное головокружение не связано с органическим поражением внутреннего уха, а обусловлено лишь механическим фактором, к его названию зачастую прибавляют «доброкачественное». ППГ чаще встречается у женщин. Заболеваемость составляет около 0,6% населения в год. Люди старше 60-летнего возраста заболевают в 7 раз чаще, чем более молодые. Наиболее подверженный ППГ возрастной период — от 70 до 78 лет.
Причины пароксизмального позиционного головокружения
Вестибулярный аппарат образован 3-мя полукружными каналами и 2-мя мешочками. Каналы наполнены эндолимфой и высланы волосковыми клетками — вестибулярными рецепторами, воспринимающими угловые ускорения. Сверху волосковые клетки покрывает отолитова мембрана, на поверхности которой образуются отолиты (отоконии) — кристаллы бикарбоната кальция. В процессе жизнедеятельности организма отработавшие отолиты разрушаются и утилизируются.
При нарушении метаболизма (гиперпродукции или ослабленной утилизации) отоконий, их части свободно плавают в эндолимфе полукружных каналов, наиболее часто скапливаясь в заднем канале. В других случаях отолиты попадают в ампулы (расширения) каналов и прилипают там к купуле, покрывающей рецепторные клетки. Во время движений головы, отоконии перемещаются в эндолимфе каналов или смещают купулу, тем самым раздражающим образом воздействуя на волосковые клетки и вызывая головокружение. После окончания движения отолиты оседают на дно канала (или перестают смещать купулу) и головокружение прекращается. Если отоконии располагаются в просвете каналов, то говорят о каналолитиазе, если они осаждаются на купуле — то о купулолитиазе.
Не смотря на подробно изученный механизм возникновения ППГ, причины образования свободных отоконий в большинстве случаев остаются не ясны. Известно, что у ряда пациентов отолиты образуются вследствие травматического повреждения отолитовой мембраны при черепно-мозговой травме. К этиофакторам, обуславливающим пароксизмальное позиционное головокружение, относят также перенесенный ранее лабиринтит вирусной этиологии, болезнь Меньера, спазм кровоснабжающей лабиринт артерии (при мигрени), хирургические манипуляции на внутреннем ухе, прием ототоксических фармпрепаратов (в первую очередь, антибиотиков гентамицинового ряда). Кроме того, ППГ может выступать в качестве сопутствующей патологии при других заболеваниях.
Симптомы пароксизмального позиционного головокружения
Основу клинической картины составляет транзиторное системное головокружение — ощущение движения предметов в горизонтальной или вертикальной плоскости, как бы вращающихся вокруг тела пациента. Подобный пароксизм головокружения провоцируется движениями головой (поворотами, запрокидыванием). Наиболее часто возникает в положении лежа, при переворачивании в кровати. Поэтому большинство атак ППГ приходится на утреннее время, когда пациенты лежат в кровати после просыпания. Иногда пароксизмы головокружения возникают во сне и приводят к пробуждению больного.
В среднем атака ППГ длится не более 0,5 мин, хотя пациентам этот период представляется более длительным, в своих жалобах они зачастую указывают, что головокружение продолжается несколько минут. Характерно, что приступ не сопровождается шумом в ушах, головной болью, падением слуха (тугоухостью). Возможна тошнота, в отдельных случаях — рвота. В течении нескольких часов после приступа или периодически в промежутках между ними некоторые пациенты отмечают наличие несистемного головокружения — чувства покачивания, неустойчивости, «дурноты». Иногда атаки ППГ имеют единичный характер, но в большинстве случаев в период обострения они возникают несколько раз в неделю или в сутки. Затем следует период ремиссии, при котором пароксизмы головокружения отсутствуют. Он может продолжаться до нескольких лет.
Приступы позиционного головокружения не представляют собой опасности для жизни или здоровья пациента. Исключение составляют случаи, когда пароксизм случается при нахождении человека на большой высоте, подводном погружении или вождении транспортного средства. Кроме того, повторяющиеся атаки могут негативно влиять на психо-эмоциональное состоянии пациента, провоцируя развитие ипохондрии, депрессивного невроза, неврастении.
Диагностика пароксизмального позиционного головокружения
Диагноз ППГ базируется преимущественно на клинических данных. С целью его подтверждения невролог или вестибулолог проводят пробу Дикса-Холлпайка. Изначально пациент сидит, повернув голову на 45 градусов в пораженную сторону и фиксируя взгляд на переносице врача. Затем пациента резко переводят в положение лежа, запрокидывая при этом его голову на 30 градусов. По прошествии латентного периода (1-5 секунд) возникает системное головокружение, сопровождающееся ротаторным нистагмом. Для регистрации последнего необходима видеоокулография или электронистагмография, поскольку периферический нистагм подавляется при фиксации взора и визуально может быть не зафиксирован. После исчезновения нистагма пациента возвращают в положение сидя, что сопровождается легким головокружением и ротаторным нистагмом, направленным в обратную сторону по отношению к ранее вызванному.
Провокационную пробу выполняют с 2-х сторон. Двусторонняя положительная проба Дикса-Холлпайка, как правило, встречается при ППГ травматического генеза. Если в ходе пробы отсутствовало как головокружение, так и нистагм, она считается отрицательной. Если отмечалось головокружение без нистагма, то проба считается положительной, диагностируется т. н. «субъективное ППГ». После неоднократного повторения пробы нистагм истощается, головокружение не возникает, поскольку в результате повторных движений отолиты рассеиваются по полукружному каналу и не формируют скопление, способное воздействовать на рецепторный аппарат.
Дополнительной диагностической пробой выступает вращательный тест, который проводится в положении лежа с запрокинутой на 30 градусов головой. При положительной пробе после резкого поворота головы спустя латентный интервал возникает горизонтальный нистагм, который хорошо регистрируется при визуальном наблюдении. По направлению нистагма можно отличить каналолитиаз от купулолитиаза и диагностировать какой именно полукружный канал поражен.
Дифференциальную диагностику ППГ необходимо проводить с позиционным головокружением при артериальной гипотонии, синдроме позвоночной артерии, синдроме Барре-Льеу, болезни Меньера, вестибулярном нейроните, фистуле лабиринта, заболеваниях ЦНС (рассеянном склерозе, новообразованиях задней черепной ямки). Основу дифдиагноза составляет отсутствие наряду с позиционным головокружением других, характерных для этих заболеваний, симптомов (тугоухости, «потемнения» в глазах, болей в шее, головных болей, ушного шума, неврологических нарушений и т. п.).
Лечение пароксизмального позиционного головокружения
Большинству пациентов рекомендована консервативная терапия, которая зависит от вида ППГ. Так, при купулолитиазе применяют вестибулярную гимнастику Семонта, а при каналолитиазе — специальные лечебные методики, направленные на изменение расположения отоконий. При остаточной и легкой симптоматике рекомендованы упражнения для тренировки вестибулярного аппарата. Фармакотерапия может иметь смысл в периоды обострения. Ее основу составляют такие препараты как циннаризин, гинкго билоба, бетагистин, флунаризин. Однако медикаментозная терапия может служить лишь дополнением к лечению специальными методиками. Следует сказать, что некоторые авторы высказывают большие сомнения в отношении ее целесообразности.
К наиболее распространенным лечебным методикам относится прием Эпли, заключающийся в последовательной фиксации головы в 5-ти различных положениях. Прием позволяет переместить отолиты из канала в овальный мешочек лабиринта, что приводит к купированию симптомов ППГ у 85-95% пациентов. При приеме Семонта пациента из положения сидя с повернутой в здоровую сторону головой переводят в положения лежа на пораженной стороне, а затем, не изменяя поворота головы, через положение сидя в положение лежа на здоровой стороне. Такая быстрая смена положения головы позволяет освободить купулу от осевших на нее отолитов.
В тяжелых случаях с частыми атаками позиционного головокружения, не купирующегося применением методик Эпли и Семонта, рассматривается вопрос хирургического лечения. Операционное вмешательство может состоять в пломбировании пораженного полукружного канала, избирательном пересечении отдельных вестибулярных волокон, лазеродеструкции лабиринта.
Синдром запертого человека

Данное состояние описывалось под разными названиями, например: синдром изоляции, синдром деэфферентации, псевдокома. Термин «синдром запертого человека» (locked-in syndrome / LIS) был введен в 1966 году в публикации Plum and Posner [1]. Однако впервые довольно точно данное состояние было описано в художественной литературе еще в XIX веке. В романе 1846 года «Граф Монте-Кристо» Александр Дюма (отец) описывает своего героя, господина Нуартье де Вильфор, как «трупа с живыми глазами»: «зрение и слух были единственными чувствами, которые, подобно двум искрам, еще тлели в этом теле, … да и то из этих двух чувств только одно могло свидетельствовать о внутренней жизни, еще теплившейся в этом истукане — взгляд. В черных глазах старого Нуартье … сосредоточилась вся энергия, вся воля, вся сила, весь разум, некогда оживлявшие его тело и дух. Конечно, недоставало жеста руки, звука голоса, движений тела, но этот властный взор заменял все. Глаза отдавали приказания, глаза благодарили».
В 1868 году Эмиля Золя в романе «Тереза Ракен» описал прогрессирующую форму синдрома запертого человека: у госпожи Ракен «только глаза и были в движении; они стремительно вращались в глазницах, зато щеки, губы как бы окаменели, их неподвижность наводила ужас. Несомненно, она все видела, слышала и рассуждала трезво и ясно, но она не могла пошевелить ни одним членом: лишенная голоса, она не могла передать вовне мысли, рождавшиеся в ее сознании. Тереза … довольно ловко общалась с замурованным сознанием старухи, еще живым, но погребенным в недрах мертвого тела».
В 1875 году французский врач M. Darolles дал одно из первых точных описаний клиники и патологии случая «запертого человека» [2]. У его пациента — женщины старше 30 лет, — развилось состояние, при котором она не могла говорить, но сознание оставалось сохранным. У нее наблюдалась прогрессирующая надъядерная квадриплегия с децеребрационной ригидностью, неспособностью говорить и глотать. Контакт с пациенткой был установлен благодаря сохранным вертикальным движениям глаз, при горизонтальном парезе взора. Состояние пациентки развилось за несколько часов перед смертью. Патологоанатомическое исследование выявило односторонний инфаркт моста мозга, размером с «лесной орех», из-за полной окклюзии базилярной артерии тромбом.
Что такое синдром запертого человека?
Синдром запертого человека — редкое неврологическое расстройство, которое характеризуется полным параличом всех мышц, за исключением мышц, контролирующих движения глаз. Пациенты с синдромом запертого человека не могут произвольно жевать, глотать, дышать, говорить или совершать любые движения мышцами. Под контролем пациента могут оставаться только мышцы, контролирующие движения глаз и век, при этом возможны только вертикальные движения глазами (вверх-вниз), но не горизонтальные (из стороны в сторону). Сохраняется сознание и ориентировка в месте, времени и собственной личности, нормальные циклы сна и бодрствования также сохранны. Таким образом, пациенты с синдромом запертого человека находятся в сознании, но не имеют возможности двигаться (кроме глаз) и говорить.
Когнитивные функции относительно сохранны, у некоторых наблюдаются нарушения памяти и внимания. Пациенты понимают обращенную к ним речь, однако коммуникация возможна только с помощью движений глаз или мигания. В острой/инициальной фазе синдрома коммуникация с пациентом при помощи движений глаз и оценка его когнитивного статуса затруднены, так как уровень бодрствования/сознания колеблется, пациент легко истощаем, движения глаз могут быть плохо контролируемыми и непоследовательными.
Люди с такими поражениями часто находятся в коме в течение нескольких дней или недель, нуждаются в искусственной вентиляции легких, а затем постепенно приходят в сознание, но остаются парализованными и неспособными говорить, внешне напоминающими пациентов в вегетативном состоянии.
Синдром запертого человека вызван повреждением моста — частью ствола мозга, который содержит нервные волокна, которые передают информацию в другие области мозга.
Симптомы / клиническая картина
- Тетраплегия (квадриплегия).
- Анартрия.
- Сохранность сознания и большинства (или всех) когнитивных функций: восприятие, мышление, внимание, память и др. (уровень сохранности функций зависит от размеров очага поражения).
- Сознательное управление движениями век и вертикальными движениями глазных яблок.
- Возможность контакта с пациентом при помощи движений глаз.
Причины
Наиболее частая причина синдрома запертого человека — двусторонние обширные повреждения моста головного мозга (части ствола мозга). Мост содержит важные двигательные тракты: нисходящие кортикоспинальные и кортиконуклеарные пути. При синдроме запертого человека происходит повреждение моторных волокон и двигательных центров в стволе мозга, обеспечивающих контроль мышц лица и речь.

Таблица 1. Причины и механизмы развития синдрома запертого человека.
Диагностика
Синдром запертого человека — сложное для диагностики состояние. Исследование 44 случаев синдрома выявило, что в среднем медикам необходимо до 3 месяцев для постановки точного диагноза. Основная сложность при диагностике заключается в том, что большинство пациентов находятся в коме продолжительное время, потом постепенно приходят в сознание, оставаясь парализованными. Поэтому часто первыми, кто замечает реакцию пациента на внешние стимулы, являются его близкие.
Если у пациента в коме, не отвечающего на внешние раздражители, на МРТ выявлено поражение вентральной части моста, необходимо дополнительная диагностика возможности совершать вертикальные движения глаз.
ЭЭГ при синдроме запертого человека выявляет нормальную активность коры и сохранные циклы сна и бодрствования.
Для исключения повреждений мышц и периферических нервов можно использовать электронейромиографию.
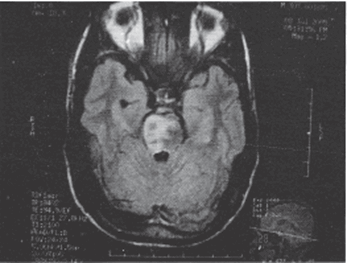
Рисунок 1. T2 Flair МРТ снимок, демонстрирующий билатеральный инфаркт моста (затронувший правую сторону больше, чем левую) (Paliwal et al., 2007)
Похожие заболевания / дифференциальная диагностика
Симптомы некоторых других заболеваний могут быть схожи с проявлениями синдрома запертого человека. Дифференциальный диагноз данных состояний может представлять определенные трудности.
Акинетический мутизм — редкое неврологическое состояние, при котором пациент не двигается (акинезия) и не говорит (мутизм), находясь при этом в сознании. У пациентов с акинетическим мутизмом сохраняются нормальные циклы сна и бодрствования. Акинетический мутизм — это разновидность «бодрствующей комы» (апаллический синдром), которая развивается из-за повреждений коры лобной доли, таламуса или поясной коры.
Вегетативное состояние — неврологическое состояние, характеризующееся нарушением функции полушарий при нормальном функционировании диэнцефальной области и ствола. В отличие от синдрома запертого человека, у пациента в вегетативном состоянии при сохранности вегетативных функций (обеспечиваемых диэнцефальным отделом и стволом) отсутствуют нормальная корковая активность, регистрируемая на ЭЭГ, и любые осознанные реакции на внешние раздражители.
Некоторые другие заболевания могут иметь клиническую картину, схожую с синдромом запертого человека, например: сидром Гиена-Барре, миастения гравис, полиомиелит, полиневрит.
Классификация
Согласно классификации Bauer et al. выделяют три клинические формы синдрома запертого человека на основе сохранности произвольных движений [3]:
- Неполная форма (incomplete form), при которой некоторые произвольные движения, помимо движений глаз, сохранны.
- Чистая форма (pure form) при которой у пациента отсутствует возможность совершать произвольные движения, за исключением моргания и вертикальных движений глаз.
- Полная/тотальная форма (total form), при которой пациент полностью теряет способность двигаться. Данное состояние наиболее драматично сказывается на психологическом состоянии пациента и его качестве жизни, так как он совершенно не способен взаимодействовать с людьми, сообщать о своих потребностях, выражать свои мысли.
Лечение
Лечение в первую очередь должно быть направлено на причину. Например, при удалении тромба из базилярной артерии и восстановлении нормального церебрального кровотока в течение 6 часов после закупорки возможно полное восстановление моторных и когнитивных функций.
Пациенты с синдромом запертого человека часто нуждаются в искусственной вентиляции легких. Также нормальный прием пищи через рот невозможен, так как попадание частиц пищи в легкие может вызвать респираторную инфекцию, поэтому данным пациентам может быть показана гастростомия.
Важно установить канал коммуникации с пациентом при помощи движений глаз. Медицинские работники и близкие пациента должны определить наиболее простой и удобный для пациента «код» — движение глазами, — и впоследствии все люди, контактирующие с пациентом, должны придерживаться единой системы кодировки. Например, «посмотри вверх» — «да» и «посмотри вниз» — «нет». В таком случае общение ограничено ответами на закрытые вопросы «да-нет», поэтому впоследствии система должна быть модифицирована для кодирования отдельных букв. Например, медицинский работник медленно называет буквы алфавита, пациент должен посмотреть вниз или закрыть глаза, чтобы выбрать букву. Для упрощения можно предъявлять буквы соответственно частоте их встречаемости в языке или использовать таблицы с буквами, организованные в колонки согласно их частотности в языке.
Рисунок 2. AEIOU-алфавит. Ассистент называет цвета, а пациент выбирает нужный ряд движением или миганием. Затем ассистент зачитывает буквы, пациент выбирает нужную букву, которая записывается на доске. Так последовательно формируются слова и предложения. Пример для английского алфавита.
В настоящее время разрабатываются устройства для коммуникации с пациентами с синдромом запертого человека на основе ай-трекинга [Отслеживания движений глаз. — прим. ред.] и компьютерных технологий. Например, сочетание технологий ай-трекинга и генерации компьютерного голоса позволяет пациентам обрести возможность говорить и взаимодействовать со средой.
Важное направление реабилитации — раннее восстановление произвольных движений (чаще движение одним пальцем или ступней, глотание). Исследование показало, что электромиостимуляция может быть эффективна в восстановлении мышечных функций у данной группы пациентов.
Качество жизни
Несмотря на то, что синдром запертого человека — наиболее драматичная форма расстройства моторики, некоторые исследователи отмечают, что качество их жизни несколько выше, чем можно ожидать в данном состоянии. В недавнем исследовании самооценки качества жизни пациентов с хроническим синдромом запертого человека было выявлено, что многие пациенты оценивают свою жизнь как счастливую и наполненную смыслом, особенно когда пациентам доступны адекватные средства для коммуникации с окружающими людьми.
Прогноз
В таблице представлены результаты двух исследований, оценивающих продолжительность жизни пациентов с синдромом запертого человека, и процент пациентов, восстановивших некоторые моторные функции [4, 5].
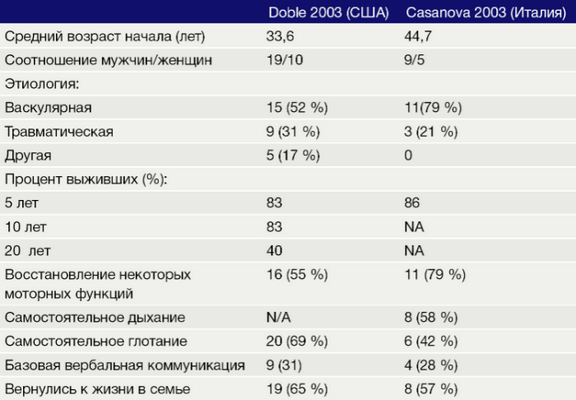
Таблица 2. Прогнозы жизни пациентов с синдромом запертого человека.
Синдром запертого человека в литературе, фильмах
Jean-Dominique Bauby «The Diving Bell and the Butterfly: A Memoir of Life in Death». И фильм по этой книге: «Скафандр и бабочка» («Le scaphandre et le papillon»).
Нарушение движений глазных яблок
Движения глаз, глазодвигательный, блоковый и отводящий нерв
Движения глазных яблок осуществляются глазодвигательными мышцами. Глазодвигательные мышцы получают иннервацию от III, IV, VI пары черепных нервов.
Глазодвигательный нерв (n. oculomotorius, III пара ЧМН)
Мотонейроны глазодвигательных нервов (n. oculomotorius, III пара ЧМН) располагается по обе стороны от средней линии в ростральной части среднего мозга. Эти ядра глазодвигательного нерва иннервирует пять наружных мышц глазного яблока, включая мышцу, поднимающую верхнее веко. Ядра глазодвигательного нерва также содержат парасимпатические нейроны (ядро Эдингера-Вестфаля), участвующие в процессах сужения зрачка и аккомодации.
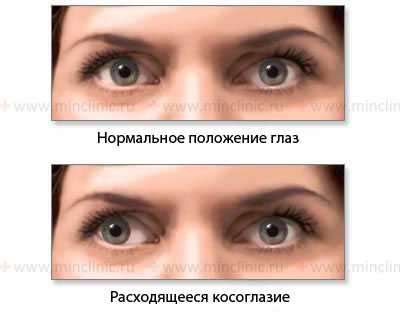
Показано нормальное положение глазных яблок и расходящееся косоглазие при слабости медиальной (внутренней) прямой мышцы глаза справа (n. oculomotorius, III пара ЧМН).
Существует разделение надъядерных групп двигательных нейронов для каждой отдельной мышцы глаза. Волокна глазодвигательного нерва, иннервирующие медиальную прямую, нижнюю косую и нижнюю прямую мышцы глаза, находятся на одноименной стороне. Подъядро глазодвигательного нерва для верхней прямой мышцы располагается на контралатеральной стороне. Мышца поднимающая верхнее веко иннервируется центральной группой клеток глазодвигательного нерва.
Блоковый нерв (n. trochlearis, IV пара ЧМН)
Мотонейроны блокового нерва (n. trochlearis, IV пара ЧМН) тесно прилегают к основной части комплекса ядер глазодвигательного нерва. Левое ядро блокового нерва иннервирует правую верхнюю косую мышцу глаза, правое ядро — левую верхнюю косую мышцу глаза.
Отводящий нерв (n. abducens, VI пара ЧМН)
Мотонейроны отводящего нерва (n. abducens, VI пара ЧМН), иннервирующего латеральную (наружную) прямую мышцу глаза на одноименной стороне, располагаются в ядре отводящего нерва в каудальной части моста. Все три глазодвигательных нерва, выйдя из ствола головного мозга, проходят через кавернозный синус и входят в орбиту через верхнюю глазничную щель.
Чёткое бинокулярное зрение обеспечивается именно совместной деятельностью отдельных мышц глаза (глазодвигательные мышцы). Содружественные движения глазных яблок контролируются надъядерными центрами взора и их связями. В функциональном отношении существуют пять различных надъядерных систем. Эти системы обеспечивают различные виды движений глазных яблок. Среди них есть центры, контролирующие:
- саккадические (быстрые) движения глаз
- целенаправленные движения глаз
- сходящиеся движения глаз
- удерживание взора в определённом положении
- вестибулярные центры
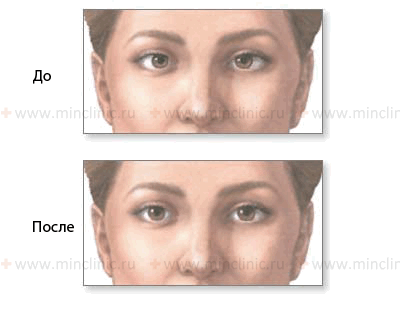
Показана слабость латеральной (наружной) прямой мышцы правого глаза (n. abducens, VI пара ЧМН) до и после лечения.
Саккадические (быстрые) движения глаз
Саккадические (быстрые) движения глазного яблока возникают в виде команды в противоположном зрительном поле коры лобной области головного мозга (поле 8). Исключением являются быстрые (саккадические) движения, возникающие при раздражении центральной ямки сетчатки, которые исходят из затылочно-теменной области головного мозга. Эти лобные и затылочные контролирующие центры в головном мозге имеют с обеих сторон проекции в надъядерных стволовых центрах. Деятельность этих надъядерных стволовых центров зрения подвергается также воздействиям и со стороны мозжечка и комплекса вестибулярных ядер. Парацентральные отделы ретикулярной формации моста являются стволовым центром, обеспечивающим содружественные быстрые (саккадические) движения глазных яблок. Одновременная иннервация внутренней (медиальной) прямой и противоположной наружной (латеральной) прямой мышц при передвижении глазных яблок по горизонтали обеспечивается медиальным продольным пучком. Этот медиальный продольный пучок связывает ядро отводящего нерва с подъядром комплекса глазодвигательных ядер, которые отвечают за иннервацию противоположной внутренней (медиальной) прямой мышцы глаза. Для начала вертикальных быстрых (саккадических) движений глаз требуется двустороннее раздражение парацентральных отделов ретикулярной формации моста со стороны корковых структур головного мозга. Парацентральные отделы ретикулярной формации моста передают сигналы от ствола головного мозга к надъядерным центрам, контролирующим движения глазных яблок по вертикали. К такому надъядерному центру движения глаз относится ростральное межуточное ядро медиального продольного пучка, расположенное в среднем мозге.
Целенаправленные движения глаз
Корковый центр для плавных целенаправленных или следящих движений глазных яблок располагается в затылочно-теменной области головного мозга. Контроль осуществляется с одноименной стороны, т. е. правая затылочно-теменная область головного мозга контролирует плавные целенаправленные движения глаз вправо.
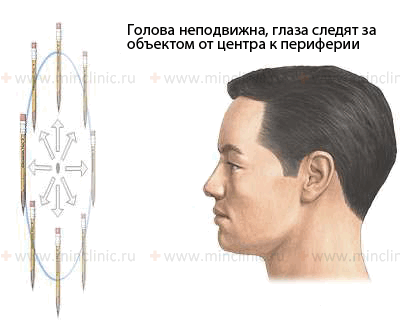
Проверка целенаправленного движения глаз осуществляется путём слежения за объектом от центра к периферии при неподвижной голове пациента.
Сходящиеся движения глаз
Механизмы контроля сходящихся движений менее понятны, однако, как известно, нейроны, отвечающие за сходящиеся движения глаз, расположены в ретикулярной формации среднего мозга, окружающей комплекс ядер глазодвигательного нерва. Они дают проекции в двигательные нейроны внутренней (медиальной) прямой мышцы глаза.
Удерживание взора в определённом положении
Стволовые центры движения глаз, называемые нейрональными интеграторами. Они отвечают за удерживание взора в определённом положении. Эти центры меняют поступающие сигналы о скорости передвижений глазных яблок в информацию о их положении. Нейроны, обладающие этим свойством, располагаются в мосту ниже (каудальнее) ядра отводящего нерва.
Движение глаз при изменение силы тяжести и ускорения
Координация движений глазных яблок в ответ на изменения силы тяжести и ускорения осуществляются вестибулярной системой (вестибулоокулярный рефлекс). При нарушении согласованности движений обоих глаз развивается двоение, поскольку изображения проецируются на диспаратные (несоответствующие) участки сетчатки. При врождённом страбизме, или косоглазии, нарушение сбалансированности мышц, приводящее к неправильному расположению глазных яблок (непаралитический страбизм), может способствовать тому, что головной мозг будет подавлять одно из изображений. Такое снижение остроты зрения в нефиксирующем глазе называют амблиопией без анопсии. При паралитическом страбизме двоение возникает в результате паралича мышц глазного яблока, обычно вследствие поражения глазодвигательного (III), блокового (IV) или отводящего (VI) черепных нервов.
Мышцы глазного яблока и параличи взора
Различают три вида параличей наружных мышц глазного яблока:
Паралич отдельных мышц глаза
Характерные клинические проявления возникают при изолированных повреждениях глазодвигательного (III), блокового (IV) или отводящего (VI) нерва.
Полное повреждение глазодвигательного (III) нерва приводит к возникновению птоза. Птоз проявляется в виде ослабления (пареза) мышцы, поднимающей верхнее веко и нарушения произвольных движений глазного яблока кверху, книзу и кнутри, а также к расходящимся косоглазием вследствие сохранности функций боковой (латеральной) прямой мышцы. При повреждении глазодвигательного (III) нерва возникают также расширение зрачка и отсутствие его реакции на свет (иридоплегия) и паралич аккомодации (циклоплегия). Изолированный паралич мышц радужки и цилиарного тела называют внутренней офтальмоплегией.
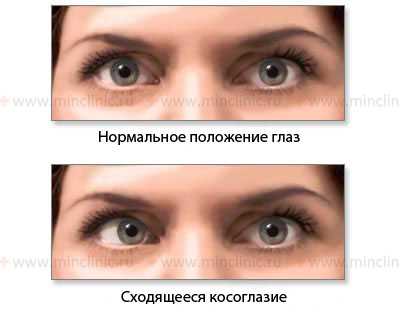
Показано нормальное положение глазных яблок и сходящееся косоглазие при слабости латеральной (наружной) прямой мышцы глаза справа (n. abducens, VI пара ЧМН).
Повреждения блокового (IV) нерва вызывают паралич верхней косой мышцы глаза. Подобные повреждения блокового (IV) нерва приводят к отклонению глазного яблока кнаружи и затруднении движения (парез) взора вниз. Парез взора вниз наиболее отчётливо проявляется при повороте глаз кнутри. Диплопия (двоение) исчезает при наклоне головы к противоположному плечу, при котором происходит компенсаторное отклонение интактного глазного яблока кнутри.
Повреждение отводящего (VI) нерва приводит к параличу мышц, отводящих глазное яблоко в сторону. При повреждении отводящего (VI) нерва развивается сходящееся косоглазие за счёт преобладания воздействия тонуса нормально работающей внутренней (медиальной) прямой мышцы глаза. При неполном параличе отводящего (VI) нерва больной может повернуть голову в сторону поражённой отводящей мышцы глаза, чтобы устранить имеющееся у него двоение с помощью компенсаторного воздействия на ослабленную боковую (латеральную) прямую мышцу глаза.
Выраженность приведённых выше симптомов при повреждениях глазодвигательного (III), блокового (IV) или отводящего (VI) нерва будет зависеть от степени тяжести поражения и места его локализации у пациента.
Паралич содружественного взора
Содружественным взором называют одновременное движение обоих глаз в одном направлении. Острое поражение одной из лобных долей, например, при инфаркте мозга (ишемическом инсульте), может привести к преходящему параличу произвольных содружественных движений глазных яблок в горизонтальном направлении. При этом самостоятельные движения глаз во всех направлениях буду полностью сохранены. Паралич произвольных содружественных движений глазных яблок в горизонтальном направлении выявляется при помощи глазного феномена куклы при пассивном повороте головы горизонтально лежащего человека или с помощью калорической стимуляции (вливание в наружный слуховой проход холодной воды).
Одностороннее повреждение расположенного книзу парацентрального отдела ретикулярной формации моста на уровне ядра отводящего нерва вызывает стойкий паралич взора в сторону поражения и выпадение окулоцефалического рефлекса. Окулоцефалический рефлекса — это двигательная реакция глаз на раздражение вестибулярного аппарата, как при феномене головы и глаз куклы или калорической стимуляции стенок наружного слухового прохода холодной водой.
Поражение рострального межуточного ядра медиального продольного пучка в передней части среднего мозга и/или повреждение задней спайки вызывают надъядерный паралич взора вверх. К этому очаговому неврологическому симптому добавляется и диссоциированная реакция зрачков пациента на свет:
- вялая реакция зрачков на свет
- быстрая реакция зрачков на аккомодацию (изменение фокусного расстояния глаза) и взгляд на близко расположенные предметы
В некоторых случаях у больного развивается также паралич конвергенции (движение глаз друг к другу, при котором взор будет фокусироваться на переносице). Данный симптомокомплекс называется синдромом Парино. Синдромом Парино встречается при опухолях в области шишковидной железы, в некоторых случаях при инфаркте головного мозга (ишемическом инсульте), рассеянном склерозе и гидроцефалии.
Изолированный паралич взора вниз встречается у пациентов редко. Когда это происходит, причиной чаще всего являются закупорка просвета (окклюзии) проникающих артерий срединной линии и двусторонние инфаркты (ишемические инсульты) среднего мозга. Некоторые наследственные экстрапирамидные заболевания (хорея Гентингтона, прогрессирующий надъядерный паралич) могут вызывать ограничения движений глазных яблок во всех направлениях, особенно вверх.
Смешанный паралич взора и отдельных мышц глазного яблока
Одновременное сочетание у пациента паралича взора и паралича отдельных мышц, двигающих глазное яблоко, обычно является признаком поражения среднего мозга или моста головного мозга. Поражение нижних отделов моста головного мозга с разрушением расположенных там ядра отводящего нерва может привести к параличу быстрых (саккадических) движений глазных яблок по горизонтали и параличу латеральной (наружной) прямой мышцы глаза (отводящий нерв, VI) на стороне поражения.
При поражениях медиального продольного пучка возникают различные расстройства взора в горизонтальном направлении (межъядерная офтальмоплегия).
Односторонние повреждения медиального продольного пучка, обусловленные инфарктом (ишемическим инсультом) или демиелинизацией, приводят к нарушению приведения глазного яблока кнутри (к переносице). Это может клинически проявляться в виде полного паралича с невозможностью отведения глазного яблока кнутри от средней линии, или же в виде умеренного пареза, который проявится в виде снижения скорости приводящих быстрых (саккадических) движений глаза к переносице (приводящая (аддукционная) задержка). На стороне, противоположной поражению медиального продольного пучка, как правило, наблюдают отводящий (абдукционный) нистагм: нистагм, возникающий при отведении глазных яблок кнаружи с медленной фазой, направленной к средней линии, и быстрыми горизонтальными саккадическими движениями. Асимметричное расположение глазных яблок относительно вертикальной линии часто развивается при односторонней межъядерной офтальмоплегии. На стороне поражения глаз будет расположен более высоко (гипертропия).
Двусторонняя межъядерная офтальмоплегия возникает при демиелинизирующих процессах, опухолях, инфарктах или артериовенозных мальформациях. Двусторонняя межъядерная офтальмоплегия приводит к более полному синдрому расстройства движений глазных яблок, которые проявляются двусторонним парезом мышц, приводящих глазное яблоко к переносице, нарушением движений по вертикали, следящих целенаправленных движений и движений, обусловленных влиянием вестибулярной системы. Отмечают нарушение взора по вертикальной линии, нистагм вверх при взгляде вверх и нистагм вниз при взгляде вниз. Поражения медиального продольного пучка в вышележащих (ростральных) отделах среднего мозга сопровождаются нарушением конвергенции (сходящемся движении глаз друг к другу, в сторону переносицы).
Читайте также:
- Профилактика профессиональных отравлений и методы борьбы с ними
- Менингиома крыла клиновидной кости прорастающая в глазницу: признаки, гистология, лечение, прогноз
- Рекомендуемые таблетки для лечения воспаления яичников. Выбор эффективной
- Разновидности скрабов для пилинга. Правила нанесения скрабов
- Генетика хронического миелолейкоза. Механизмы развития лейкоза