УЗИ, МРТ при аутосомно-рецессивном поликистозе почек у плода
Добавил пользователь Дмитрий К. Обновлено: 01.02.2026
Кистозные аномалии почек связаны с продолжительностью жизни, поэтому имеют большое значение. Из-за широкой распространенности, семейного характера и неуклонного прогрессирования заболевания особое место занимает поликистоз. От поликистоза следует отличать медулярную спонгиозную почку с доброкачественным течением и хорошим прогнозом. При других кистозных аномалиях не наблюдается диффузного поражения почки; обычно они односторонние — мультикистозная дисплазия, мультилокулярная киста и солитарная киста. От врожденных кистозных аномалий следует отличать приобретенные ретенционные кисты, имеющие совсем другой патогенез и другое значение.
Наследуемые кисты в почках
Нажимайте на картинки, чтобы увеличить.
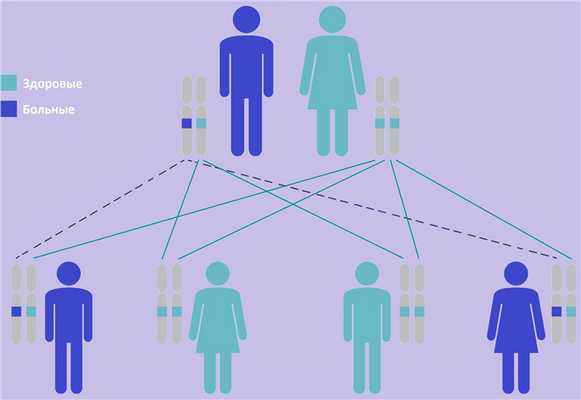
| Фото. А — Как наследуются аутосомно-доминантные признаки: вероятность рождения больного ребенка в семье, где один из родителей болен, составляет 50%. Б — Как наследуются аутосомно-рецессивные признаки: оба родителя здоровы, но являются носителями дефектного гена; вероятность рождения больного ребенка в таком браке составляет 25%. | ||
 |  | |
Поликистоз почек на УЗИ
Почечный поликистоз (поликистозная дегенерация, поликистозная болезнь) — это наследуемая врожденная аномалия, при которой обе почки усеяны множеством кист. Между кистами имеется скудная паренхима без диспластических элементов. Случаи одностороннего поражения являются или мультикистозной дисплазией, или очень рано диагностированным истинным поликистозом, неравномерно развитым в обеих почках. Подобный вид поликистоза следует иметь в виду при обсуждении вопроса о нефрэктомии при «одностороннем» поликистозе. Чем раньше проявляется поликистоз, тем злокачественней он протекает и тем хуже его прогноз.
Наследственный характер почечного поликистоза установил еще в XIX векe Steiner. Злокачественный поликистоз детского возраста передается аутосомно-рецессивно. Взрослый тип поликистоза передается аутосомно-доминантно. Подробнее про поликистоз смотри здесь.
Нефронофтиз и медуллярная кистозная болезнь на УЗИ
Нефронофтиз и медуллярную кистозную болезнь рассматривают вместе, так как они имеют много общих характеристик. Это врожденные заболевания, которые характеризуются хроническим диффузным тубулоинтерстициальным нефритом, который прогрессирует до фиброза и терминальной почечной недостаточности. Небольшие кисты в зоне кортико-медуллярного соустья или в пределах мозгового вещества становятся видимым на поздней стадии болезни. Эти кисты возникают из дистальных извитых канальцев и собирательных трубочек. Базальные мембраны канальцев, как правило, утолщены и прерывисты.
Ухудшение тубулярной концентрационной функции вызывает полиурию и полидипсию. При младенческой форме нефронофтиза терминальная стадия почечной недостаточности развивается в возрасте от 1 года до 3 лет, при ювенильной форме — к 13 годам, при подростковой форме — к 19 годам, а при медуллярной кистозной болезни — к 30-50 годам.
Нефронофтиз встречается у 1-2 на 100000 новорожденных. На него приходится от 10% до 20% случаев почечной недостаточности в детстве. Нефронофтиз — генетически неоднородное заболевание с аутосомно-рецессивным типом наследования. Вероятно мутации в генах, кодирующих тубулярные ресничные протеины, вызывают ресничную дисфункцию, которая приводит к кистозной трансформации. Мутации, вызывающие нефронофтиз, отвечают также и за экстраренальные проявления, такие как окуломоторная апраксия, пигментный ретинит, фиброз печени, конусоформный эпифиз, колобома зрительного нерва с аплазией червя мозжечка.
Медуллярная кистозная болезнь наследуется по аутосомно-доминантному типу. Но около 15% пациентов не имеют семейного анамнеза болезни, что предполагает спорадическую новую мутацию. Гиперурикемия и подагра — единственные экстраренальные проявления при медуллярной кистозной болезни.
При нефронофтизе и медуллярной кистозной болезни на УЗИ контур почки ровный, размеры почек нормальные или уменьшенные, паренхима гиперэхогенная, из-за множества микрокист в зоне между корковым мозговым слоем отсутствует кортикомедуллярная дифференцировка. Кисты почек (медуллярной или кортико-медуллярной локализации) становятся видимыми, когда у пациентов развивается терминальная стадия почечной недостаточности.
Медуллярная губчатая (спонгиозная) почка на УЗИ
Медуллярная губчатая (спонгиозная) почка - это редкая врожденная наследственная аномалия, при которой собирательные трубки в пирамидах расширены кистозно, веретенообразно или диффузно. Между ними находятся нормальные или расширенные канальцы. Кисты малы (от 1 до 4 мм) и наполнены водянистой жидкостью, могут содержать плотные конкременты (обызвествленные сосочки). Изменения локализованы почти исключительно в пирамидах. Корковый слой и колонки Bertini нормальны. Обе почки увеличены, но поверхность их гладкая.
При медуллярной губчатой почке отсутствуют функциональные отклонения, когда не наблюдается осложнений. В 50% неосложненных случаев наблюдается постоянная умереннная протеинурия и микрогематурия или лейкоцитурия, обнаруживаемые случайно. При пальпации можно установить увеличение обеих почек с гладкой поверхностью. Клинические проявления наступают при присоединении нефрокальциноза, калькулеза или инфекции.
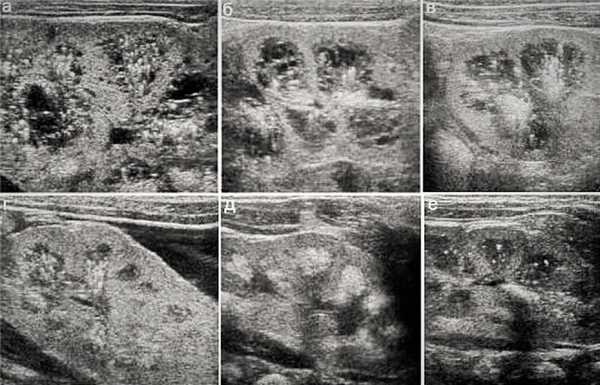
| Фото. Дети с медуллярной губчатой почкой: на УЗИ высокочастотными датчиками кортикальный слой паренхимы почек сохранен, в медуллярном — множественные кисты малых размеров и точечные гиперэхогенные включения. | ||
 | ||
Микрокистозное заболевание почек на УЗИ
Микрокистозное заболевание почек относятся к врожденному нефротическому синдрому финского типа. Распространенность врожденного нефротического синдрома составляет 1 на 10000 живорожденных в Финляндии, и значительно меньше у нефинского населения.
Как аутосомно-рецессивное наследственное заболевание, микрокистозное заболевание почек характеризуется патологическим кистозным расширением проксимальных и дистальных канальцев. При данной патологии ген NPHS1 определяется на длинном плече 19-й хромосомы.
При микрокистозном заболевание на УЗИ почки имеют нормальный размер как внутриутробно, так и при рождении, и со временем заметно увеличиваются с потерей кортико-медуллярной дифференциации.
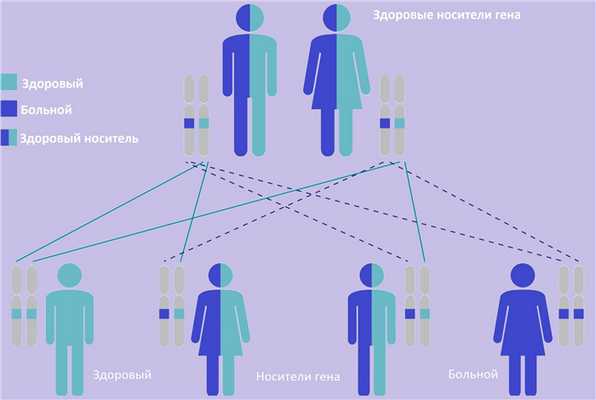
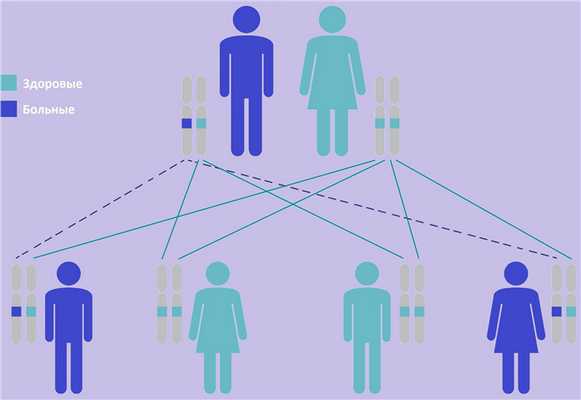
| Фото. А — Как наследуются аутосомно-доминантные признаки: вероятность рождения больного ребенка в семье, где один из родителей болен, составляет 50%. Б — Как наследуются аутосомно-рецессивные признаки: оба родителя здоровы, но являются носителями дефектного гена; вероятность рождения больного ребенка в таком браке составляет 25%. | ||
 | | |
Гломерулокистозные болезни на УЗИ
Гломерулярные кисты не специфичны для одной патологии. Большинство гломерулокистозных болезней (ГКБ) наследуются как аутосомно-доминантный признак. Клинические проявления ГКБ могут развиваться у детей старшего возраста и взрослых, с наличием семейного анамнеза. При гломерулокистозных болезнях пространства капсулы Боумена расширены, а капиллярные петли клубочка сжаты и смещены, корковый слой представлен диффузно расположенными мелкими кистами.
При гломерулокистозных заболеваниях на УЗИ почки могут быть уменьшены, увеличены или нормальных размеров, гиперэхогенные, без кортико-медуллярной дифференциации. Субкапсулярные корковые кисты довольно типичны для ГКБ. Кисты могут развиваться внутриутробно или только после рождения.
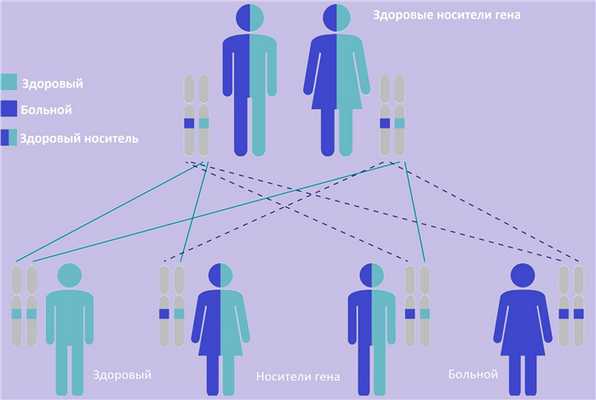
| Фото. А — Как наследуются аутосомно-доминантные признаки: вероятность рождения больного ребенка в семье, где один из родителей болен, составляет 50%. Б — Как наследуются аутосомно-рецессивные признаки: оба родителя здоровы, но являются носителями дефектного гена; вероятность рождения больного ребенка в таком браке составляет 25%. | ||
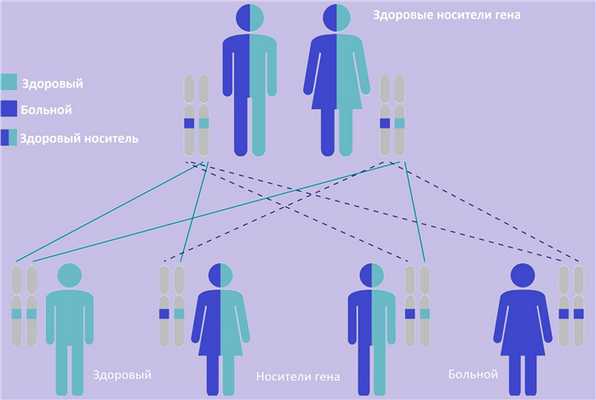 | | |
Гломерулокистозная болезнь нередко является частью многих наследственных синдромов и хромосомной патологии.
Генетические синдромы с кистами в почках смотри здесь
Ненаследуемые кисты в почках
Мультикистозная почка на УЗИ
Мультикистоз — это врожденная, всегда односторонняя аномалия почки. У плода может быть двухсторонняя форма болезни, но при этом он нежизнеспособен. Выживают только дети с односторонним поражением. В основе мультикистоза лежит атрезия лоханочно-мочеточникового соустья в период эмбрионального развития. При этом происходит неполное слияние метанефрогенной ткани (зачатка секреторного аппарата) и мезонефроса (зачатка выводящего аппарата). Развивающаяся метанефрогенная ткань некоторое время продуцирует мочу, которая не выводится, а скапливается в канальцах, приводя к их перерастяжению и постепенному превращению в кисты.
Почечной паренхимы при мультикистозе нет. На месте почки определяется «гроздь винограда» — конгломерат кист (полостных образований с тонкой капсулой) различных размеров и количества. Между кистами располагается рыхлая соединительная ткань, а также очаги хрящевой и костной ткани. У взрослых нередко наблюдается обызвествление стенок кист. При ЦДК часто визуализируется сосудистая ножка. Увидеть атрезированную нижнюю треть мочеточника не всегда представляется возможным.
Здоровая почка, которая работает «за себя и за того парня», часто увеличена в размерах. Это компенсаторная (викарная) гипертрофия, которая начинается с внутриутробной жизни. Отсутствие выраженной гипертрофии контралатеральной почки чаще свидетельствует о нарушении ее функции.
Клинически мультикистозная почка может ничем себя не проявлять и обнаруживается только при вскрытии. Часто мультикистозная почка выявляется случайно при скрининговом ультразвуковом обследовании.
Почки могут быть большими — их неточно называют поликистозными (правильнее — мультикистоз); они также бывают маленькими (гипопластичные, апластичные или дисгенетичные). Мультикистозная дисплазия охватывает целиком паренхиму одной почки, которая напоминает кисть винограда, придерживающуюся к почечным воротам одной ножкой. Эта почка не функционирует. Мочеточник отсутствует, или рудиментарен, лоханки и чашечек нет. Иногда к гроздевидной опухолевой массе присоединяется придаток яичка или яичко на соответствующей стороне.
Мультикистозная почка может протекать бессимптомно. Односторонняя мультикистозная почка может представлять собой доброкачественное непрогрессирующее поражение, при котором имеется хороший прогноз, если противоположная почка не изменена. Иногда ее можно установить при пальпации в виде неравномерной опухолевой массы в области одной из почек. Придавливая соседние ткани или органы, мультикистозная почка вызывает соответствующие симптомы. В таких случаях ее удаляют оперативным путем. У новорожденных прогноз зависит от возможного сочетания с другими врожденными аномалиями.
У 33% пациентов с поликистозной дисплазией почек происходит полная атрофия в возрасте 2-х лет, у 47% — в 5 лет и у 59% — в 10 лет. Инволюция поликистозной дисплазии почек может имитировать почечную агенезию. Существуют эпизодические данные о развитии опухоли Вильмса и почечно-клеточной карциномы на фоне поликистозной дисплазии почек.
Мультилокулярная и простая киста почки на УЗИ
Мультилокулярная киста является многокамерным кистозным образованием в части почки, остальная паренхима которой нормально и функционирует. Вокруг кисты наблюдаются дисплазические элементы (эмбриональные канальцы, скопление лимфоцитов, хрящевая ткань и пр.), указывающие на раннее эмбриональное происхождение аномалии и ее родство с мультикистозной дисплазией.
Врожденная солитарная киста также является односторонней, но иногда бывает и двусторонней. Киста может быть различной величины — от 1 до 10 см в диаметре — и расположена обычно в нижнем полюсе почки. Разрастаясь, киста сдавливает почечную паренхиму и лоханку. Солитарная киста является редкой аномалией, но встречается чаще в почке с другими аномалиями — подковообразной, эктопической почке, почке с дополнительными сосудами.
Клинически солитарная и многокамерная кисты проявляются редко. Когда кистозное образование разрастается до значительных размеров, его можно прощупать в виде опухоли в полости живота. Совсем редко наблюдаются боли.
Кистозные новообразования почек на УЗИ
Кистозная нефрома и кистозная частично дифференцированная нефробластома на УЗИ выглядят как многокамерное кистозное образование с тонкими перегородками, без каких-либо твердых элементов. Они неотличимы в дооперационном периоде и лечатся путем нефректомии. При кистозной нефроме, перегородки не содержат бластных клеток, тогда как кистозная частично дифференцированная нефробластома имеет признаки септальной бластемы. Кистозная нефрома в 65% случаев возникает в возрасте до 4-х лет, 5% — в возрасте от 5 до 30 лет, и 30% — в возрастестарше 30 лет. Кистозная частично дифференцированная нефробластома главным образом поражает детей до 2-х лет.
Опухоль Вильмса с образованием кисты вследствие кровоизлияния и некроза, светлоклеточная саркома (ранее называлась «анапластический подтип Вильмса»), кистозная аденомиосаркома (клеточный подтип) и кистозный рак почки — это новообразования детского возраста, которые могут также проявляться, как многокамерное кистозные образования с твердым узловым компонентом.
Приобретенные кисты почки на УЗИ
Приобретенный поликистоз почек характеризуется двусторонними кистозными изменениями, которые распределены по всей почечной коре и мозговому веществу у пациентов с терминальной стадией почечной патологии и не связанные с наследственными кистозными заболеваниями почек. Кисты, как правило, небольшие по размеру. Считается, что такие кисты образуются из-за закупорки просвета канальцев вследствие фиброза и кальцификации. Размер почки вариабельный, обычно уменьшенный, но иногда нормальный или даже увеличенный.
Распространенность и тяжесть приобретенного поликистоза почек увеличивается пропорционально длительности азотемии. Приобретенный поликистоз почек определяется у 22% пациентов с терминальной стадией болезни почек до диализа, у 58% пациентов на диализе от 2 до 4 лет, у 75% — на диализе от 4 до 8 лет, и у 92% — на диализе более 8 лет. Кисты могут регрессировать после успешной пересадки почек. Кровотечение и неопластическая трансформация (почечная карцинома) являются основным осложнением приобретенного поликистоза почек.
Кисты почек нетубулярного происхождения на УЗИ
Кисты синусов почек (киста ворот в пределах липоматоза синуса, окололоханочная киста лимфатического происхождения) встречается у взрослых и редко у детей. Паранефральные лимфангиомы достаточно редкая патология и встречаются у пациентов с туберозным склерозом.
Субкапсулярная и паранефральная уриномы (псевдокисты мочевыводящих путей) являются обычно вторичными обструктивными уропатиями, либо в следствие аномалий развития (задний клапан уретры, обструкция лоханочно-мочеточникового соустья, обструкция уретро-везикального соустья), либо приобретенные (конкременты мочеточника, травма). Чашечно-лоханочные кисты и дивертикулы развиваются вероятнее всего вследствие аномалий развития, и подразделяются на два типа. Тип I, более распространенный, связан обычно с малой чашечкой и расположен обычно в полюсе почке (особенно верхнем). Тип II представляет собой дивертикул большой чашечки или лоханки.
Чашечно-лоханочные кисты возникают спорадически, встречаются во всех возрастных группах, и, как правило, являются односторонними. При внутривенной урографии, они обнаруживаются в 0,5% случаев. Они имеют клинические проявления, когда осложняются почечнокаменной болезнью, инфекцией или воспалением. В последнем случае, киста заметно увеличивается по отношению к блокированной шейке дивертикула. Частота формирования конкрементов в дивертикулах почечных чашечек находится между 10% и 40%.
Поликистоз почек у детей
Поликистоз почек у детей - врожденная аномалия почек, характеризующаяся наличием в почечной ткани множественных мелкокистозных изменений, нарушающих функционирование органа. При манифестации поликистоза почек в детском возрасте отмечается стойкая высокая артериальная гипертензия, боли в пояснице, рецидивирующий пиелонефрит, почечная недостаточность. Диагноз поликистоза почек у детей подтверждается данными УЗИ почек, экскреторной урографии, ангиографии, сцинтиграфии и КТ почек. Лечение поликистоза почек у детей направлено на борьбу с ХПН; иногда проводится чрескожная пункция или лапароскопическое иссечение кист, трансплантация почки.
Общие сведения
Поликистоз почек у детей (поликистозная болезнь, поликистозная дегенерация почек) - тяжелый структурный порок развития почек, при котором нормальная почечная ткань замещается множественными кистами различной величины. Поликистоз почек у детей встречается с частотой 1 случай на 250-1000 новорожденных, однако в связи с латентным течением в детском возрасте диагностируется редко. В зависимости от клинической формы, поликистоз почек может обнаружиться или проявиться в различные возрастные периоды: у новорожденных, детей раннего возраста, подростков или взрослых преимущественно в возрасте 30-40 лет. Кистозные изменения почек у детей нередко сочетаются с поликистозом печени, поджелудочной железы, селезенки, легких, поликистозными яичниками, мегауретером, добавочной почкой.
В связи с тем, что поликистоз почек у детей является наследственным заболеванием, его изучение является задачей урологии и генетики.

Классификация поликистоза почек
С учетом генетических аспектов выделят:
- поликистоз почек взрослых, тип I (мутация гена PKD1 в коротком плече 16-ой хромосомы)
- поликистоз почек взрослых, тип II (мутация гена РKD2 или РKD4 в 4-ой хромосоме)
- аутосомно-рецессивную поликистозную болезнь почек (мутация генов PKHD, ARPKD в 6-ой хромосоме). Данная форма поликистоза почек у детей сочетается с множественными пороками развития (расщелинами лица, врожденными пороками сердца и пр.).
- тяжелый инфантильный поликистоз почек с туберозным склерозом (мутация гена PKDTS в 16-ой хромосоме)
- врожденный микрокистоз почек финского типа (встречают у населения Финляндии и севера России)
- прочие разновидности поликистоза почек: поликистоз почек у детей в сочетании с катарактой и врожденной слепотой; поликистоз в сочетании микроцефалией, брахицефалией, гипертелоризмом и непропорционально короткими конечностями и др.
Поликистоз почек новорожденных наследуется по аутосомно-рецессивному типу. Поликистоз почек, наследуемый по аутосомно-доминантному типу, встречается у детей старшего возраста, подростков и взрослых.
В зависимости от степени кистозного поражения почек различают солитарные кисты, очаговую и тотальную форму поликистоза.
Причины поликистоза почек у детей
Формирование поликистоза почек у детей происходит уже в первые недели эмбриогенеза вследствие несрастания канальцев метанефроса и собирательных канальцев зачатка мочеточника. Согласно одной из гипотез, это может происходить вследствие иммунологической несовместимости метанефрогенной бластомы и мочеточникового ростка. Подтверждением этого предположения служит уменьшение в сыворотке пациентов с поликистозом концентрации С3-комплемента.
Кистозные полости, образующиеся из почечных канальцев, не соединенных с выводящей системой, могут быть гломерулярными, тубулярными и экскреторными. Кисты гломерулярного типа не связаны с канальцевой системой, поэтому не склонны к увеличению размеров. Данный тип характерен для поликистоза почек новорожденных и способствует раннему развитию хронической почечной недостаточности и смерти ребенка. Кисты тубулярного типа образуются из канальцев, а экскреторного типа - из собирательных трубочек. Увеличение кист происходит неравномерно, но постоянно из-за трудностей с опорожнением. Растущие кисты вызывают компрессию паренхимы и гибель значительной части нефронов.
Возникновение мутации генов, обусловливающих развитие поликистоза у детей, может быть вызвано воздействием химических и лекарственных веществ (консервантов продуктов, инсектицидов, препаратов лития, цитостатиков и пр.), вирусов (цитомегаловируса и т. д.) и другими неблагоприятными факторами.
Симптомы поликистоза почек у детей
Поликистоз почек новорожденных имеет злокачественное течение: нередко отмечается мертворождение, в остальных случаях летальный исход наступает первые месяцы или первый года жизни. Кистозное перерождение охватывает до 90% паренхимы почек. У новорожденных детей отмечается рвота, резкое увеличение почек и объемов живота; быстро нарастают признаки почечной недостаточности (увеличение в крови уровня мочевины и остаточного азота, олигурия и анурия), отеки, повышение АД. При этом гибель детей нередко наступает от сопутствующего респираторного дистресс-синдрома, обусловленного гипоплазией легких и пневмотораксом.
При поликистозе почек у детей старшего возраста большая часть почечных кист является открытыми; в почках сохраняется от 40 до 75% неповрежденной паренхимы. В связи с этим клиническая манифестация поликистоза почек у детей происходит позднее, во многом напоминая течение заболевания у взрослых. У детей обнаруживается увеличение размеров почек, гепатоспленомегалия, стойкая высокая артериальная гипертензия. Растяжение почечной ткани растущими кистами вызывает болевые ощущения в пояснице или в боку. Дети существенно отстают в росте, страдают анемией. Характерными осложнениями поликистоза почек у детей раннего и старшего возраста служат гематурия, мочекаменная болезнь, пиелонефриты, хроническая почечная недостаточность. Развитие печеночного фиброза приводит к портальной гипертензии, пищеводным и желудочно-кишечным кровотечениям. Гибель детей наступает от почечной или печеночной недостаточности через 2-15 лет от начала развития заболевания.
Диагностика поликистоза почек детей
Для постановки точного диагноза необходимо проведение полного клинико-лабораторного и инструментального обследования ребенка, анализ родословной. Дети с подозрением на поликистоз почек должны быть проконсультированы детским нефрологом (детским урологом) и генетиком.
Исследование общего анализа мочи при поликистозе почек у детей выявляет протеинурию, микро- или макрогематурию, лейкоцитурию (при пиелонефрите), гипо- или изостенурию. Для оценки функционального состояния почек проводится биохимическое исследование крови, исследование мочи по Зимницкому, проба Реберга.
Окончательное подтверждение и верификация диагноза осуществляется с помощью экскреторной урографии, почечной ангиографии, УЗИ, динамической сцинтиграфии, КТ почек и МР-урографии. Важнейшими признаками поликистоза почек у детей служат изменение размеров, контуров и расположения почек, деформация чашечек и лоханки, изменение сосудистой системы и др. При поликистозе почек детям может потребоваться УЗИ поджелудочной железы, печени, селезенки, яичников, поскольку нередко наблюдается кистозная трансформация этих органов.
Для выявления мутантного гена поликистоза почек у детей или родителей используются методы молекулярной гибридизации. На современном этапе поликистоз почек у ребенка может быть выявлен еще внутриутробно при скриннинговом акушерском УЗИ после 30-ой недели беременности.
Дифференциальную диагностику поликистоза почек у детей необходимо проводить с губчатой почкой, нефробластомой (опухолью Вильмса).
Лечение поликистоза почек детей
Терапия поликистоза почек у детей является пожизненной и направлена на предупреждение или борьбу с осложнениями, сохранение и улучшение функции почек. Дети с поликистозом почек нуждаются в повторных курсах медикаментозной терапии, постоянном соблюдении диетического и питьевого режима.
При сопутствующем воспалительном процессе в почках проводится курс лечения пиелонефрита антибиотиками и уросептиками. В случае присоединения артериальной гипертензии назначаются гипотензивные средства. При развитии ХПН на фоне поликистоза почек у детей проводится гемодиализ и ставится вопрос о трансплантации почки (иногда вместе с трансплантацией печени).
В случае быстрого увеличения кист в объеме может потребоваться хирургическое лечение: чрескожная пункционная аспирация кист под контролем УЗИ, лапароскопическое иссечение кист. При развитии портальной гипертензии может быть проведено портокавальное или спленоренальное шунтирование.
Прогноз при поликистозе почек детей
Прогноз при поликистозе почек у детей всегда серьезный; при этом, чем раньше заболевание начало прогрессировать, тем хуже. Младенческая форма поликистоза почек протекает крайне неблагоприятно и рано заканчивается гибелью детей. Практически у всех выживших детей к 20 годам формируется тяжелая почечная недостаточность. В этом случае единственным шансом на продление жизни больных является пересадка донорской почки.
Поликистозная болезнь почек с аутосомно-рецессивным типом наследования

Поликистозная болезнь почек с аутосомно-рецессивным типом наследования (ПБП-АР) принадлежит к группе врожденных гепаторенальных фибрознокистозных заболеваний. Первично всегда поражаются почки и печень, но с развитием заболевания в патологический процесс могут включаться и другие органы. Заболевание характеризуется двусторонним увеличением почек из-за наличия небольших множественных кист и поражением печени.
Эпидемиология
Младенческая форма. Встречается редко (1:10000-1:40000), частота может расти в изолированных популяциях. На данный момент не выявлено существующих гендерных и расовых предрасположенностей.
Этиология
Несмотря на клиническую вариабельность и множественное поражение органов, только единичная мутация гена PKHD1 в 6 хромосоме (6p12) отвечает за заболевание. Ген PKHD1 отвечает за экспрессию белка фиброцистина преимущественно в печени, почках и поджелудочной железе. Белок фиброцистин входит в состав первичных ресничек, располагающихся на апикальной мембране клеток эпителия почек и желчных путей.
Классификация
В зависимости от времени начала первых проявлений и степени поражения печени выделяют следующие типы заболевания:
1. Перинатальный тип — наиболее распространенный:
- сопровождается маловодием и гипоплазией легкого;
- 75 % умирают в течение 24 часов после родов;
- минимальная степень фиброза печени;
2. Неонатальный тип — минимальная степень перипортального фиброза печени;
3. Младенческий тип — умеренный перипортальный фиброз;
4. Ювенильный тип — выраженный перипортальный фиброз в сочетании с портальной гипертензией, спленомегалией и портосистемным варикозом.
Патологическая анатомия
Макроскопическое строение
Плод имеет характерный внешний вид (лицо Поттера): узкие щели век, характерная борозда под линией века, микрогнатия, приплюснутый нос, мягкие большие уши аномальной формы. Почки увеличены в размерах, при этом сохраняют свою бобовидную форму, но не могут выполнять свою функцию. Плохо функционирующие почки не производят достаточное количество фетальной мочи, что приводит к маловодию и гипоплазии легких. Гидростатическое давление амниотической жидкости обеспечивает нормальное развитие дыхательной системы.
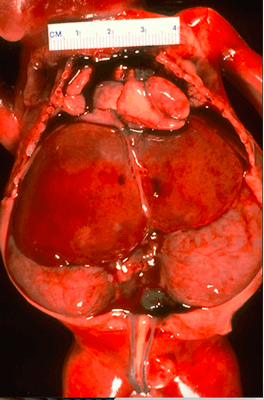
Рисунок 1 | Плод, 23 недели. Смерть плода после преждевременных родов наступила из-за гипоплазии легких, причиной которой является маловодие вследствие ПБП.
Увеличение обеих почек со смещением органов брюшной полости в сочетании с гипоплазией легкого.
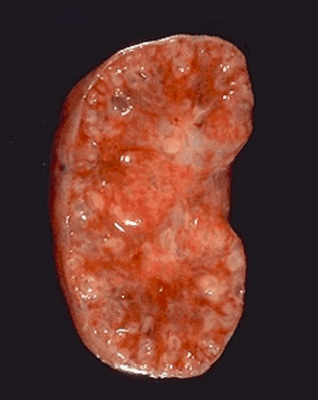
Рисунок 2 | Продольный срез почки с ПКБ.
Микроскопия
Почки
Радиально ориентированные расширенные собирательные канальцы формируют почечные кисты диаметром 1-2 мм, между которыми видны нормальные клубочки и канальцы. Размеры кист могут варьировать в зависимости от возраста. На ранних стадиях болезнь дебютирует с микрокист, которые затем растут и превращаются в макрокисты. Кистозное поражение почек сопровождается незначительным интерстициальным фиброзом паренхимы почек.
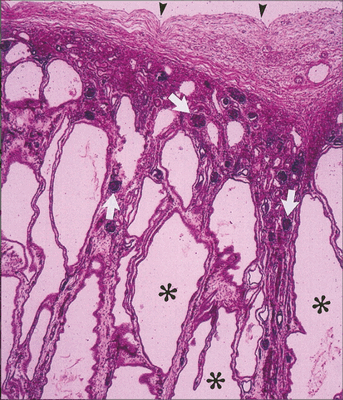
Рисунок 3 | Микропрепарат почки пациента с ПБК-АР. Двадцатикратное увеличение, окраска гематоксилин-эозин.
✱ — радиальные почечные кисты;
▼— почечная капсула.
Стрелками обозначены нормальные клубочки между расширенными собирательными канальцами.
Печень
Гистологические изменения печени включают следующие пороки развития дуктальной пластинки: гиперплазию желчевыводящих путей, билиарную эктазию и перипортальный фиброз. Нарушения морфогенеза развития желчных путей приводит к их дилатации. С последующим прогрессированием заболевания расширенные протоки превращаются в макрокисты, связанные нормальными протоками, что позволяет достаточно хорошо их верифицировать с помощью МРХПГ.
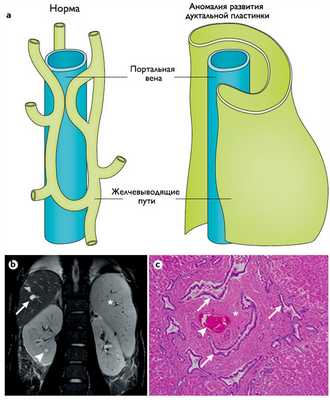
Рисунок 4 | Патологические изменения печени.
a) Строение нормально разветвленной портовенозной и решетчатой систем желчных путей (слева) нарушается вследствие дефекта развития дуктальной пластинки и ошибок в терминальной дифференцировке холангиоцитов (справа);
b) Корональное Т2-взвешенное изображение брюшной полости:
— стрелкой показано кистозное веретенообразное расширение желчных протоков;
— стрелкой по типу наконечника стрелы — нефромегалия с маленькими цистами;
✱ — спленомегалия.
c) Препарат фрагмента печени, окраска гематоксилин-эозин, увеличение в 40 раз:
✱ — обширный фиброз портальной области;
стрелка — расширенные извилистые желчные протоки;
стрелка по типу наконечника стрелы — гипоплазия притоков портальной вены.
Формирование кист
При ПБП-АР кисты формируются преимущественно из дистальных канальцев и собирательных трубочек, детальные молекулярные механизмы этого до сих остаются покрыты мраком. В результате неправильного восприятия сигналов из-за нарушенной работы первичных ресничек в клетках почечного эпителия происходит нарушение внутренних сигнальных механизмов пролиферации и дифференцировки, что приводит к образованию пузырьков, наполненных жидкостью.
Клиническая характеристика
Первые симптомы у большинства пациентов проявляются в неонатальном периоде.
Почки
При осмотре: увеличение правых и левых боковых областей живота, где пальпируются объемные образования. Объем выделяемой мочи обычно уменьшен, хотя при нарушении концентрационной функции почек могут иметь место явления полиурии и полидипсии. Тяжелые формы с развитием острой почечной недостаточности (ОПН) будут проявляться олигурией, гипонатриемией, увеличением креатинина и азота мочевины в крови. Но с возрастом благодаря развитию нормальной почечной ткани происходит улучшение почечной функции и уменьшение проявлений ОПН. Несмотря на это, примерно у 50 % пораженных в первую декаду жизни болезнь прогрессирует в терминальную почечную недостаточность. Симптомы тяжелой артериальной гипертензии, которая дебютирует с первых дней, также становятся менее тяжелыми с течением времени.
Печень
С развитием методов заместительной терапии функций почек и трансплантологии улучшается долгосрочная выживаемость пациентов. Благодаря этому гепатобилиарные клинические проявления становятся основной проблемой старших возрастных групп. Врожденный фиброз печени приводит к прогрессирующей портальной гипертензии, что проявляется варикозно расширенными венами пищевода, желудка, геморроидальных вен, гепатоспленомегалией, энтеропатией с потерей белка и желудочно-кишечными кровотечениями.
Синдром Кароли— необструктивная дилатация внутрипеченочных желчных протоков и общего желчного протока, встречающаяся более чем в 60 % случаев ПКБ-АР. Недостаточность дренажной функции желчной системы предрасполагает к развитию бактериального восходящего холангита и сепсиса. Мальабсорбция из-за холестаза приводит к дефициту жирорастворимых витаминов (A, D, E и K). У взрослых пациентов гиперплазия желчевыводящих путей предрасполагает к возникновению добро- и злокачественных опухолей.
Диагностика
Диагноз ПБП-АР должен быть заподозрен во всех случаях диффузного эхогенного увеличения обеих почек при УЗИ. Для постановки точного диагноза обычно бывает достаточно клинических признаков и наличия радиографических изменений. Но для точной верификации заболевания разработаны специальные диагностические критерии: сочетание патогномоничных признаков изменения почки — одного или нескольких пунктов из следующих:
- эктазия желчных протоков (см. ультрасонографию);
- клинические и лабораторные признаки врожденного фиброза печени (ВФП);
- гепатобилиарная патология, характерная для аномалий развития дуктальной пластины и сопутствующая ВФП;
- наличие морфологических (биопсия или аутопсия) и генетических признаков.
Ультразвуковое исследование
УЗИ высокого разрешения значительно улучшило диагностику легких форм заболевания, когда клинические проявления невыраженные, обеспечивая неинвазивную детальную визуализацию изменений в почках без использования радиации и контрастных веществ.
Ультрасонография является методом выбора при исследовании плода и детских форм ПКБ-АР. УЗИ диагностика обладает большим перечнем преимуществ: низкая стоимость, безболезненность, широкая распространенность и отсутствие необходимости в излучении и седации. Но несмотря на все перечисленные достоинства УЗИ, с целью верификации диагностика дополняется МРТ.
Патогномоничные изменения почек, диагностируемые при помощи УЗИ:
- увеличение размера почек (с учетом возрастных особенностей);
- повышенная эхогенность;
- снижение кортико-медуллярной дифференциации.
- обнаруживаются увеличенные бобовидные почки со сглаженной кортико-медуллярной дифференциацией, маловодие и пустой мочевой пузырь в некоторых случаях;
- сильно выраженное маловодие приводит к легочной гипоплазии и высокой смертности вследствие легочной недостаточности. Помимо этого, олигоамнион может сочетаться с множественными аномалиями лица, конечностей и других органов по типу синдрома Поттера.
Младенчество (до года):
- наличие пальпируемых объемных образований в обеих боковых областях живота с идиопатической хронической легочной патологией, маловодие в анамнезе матери или спонтанный пневмоторакс у новорожденного и артериальная гипертония позволяют предположить ПБП-АР;
- сочетание аномалий билиарной системы вместе с признаками портальной гипертензии, такими как гепатоспленомегалия, добавляет уверенности для постановки диагноза ПБП-АР.
Детство и юношеский возраст:
- с возрастом размер и степень фиброза почек будут прогрессировать вместе с аномалиями гепатобилиарного тракта, что приводит к прогрессирующей портальной гипертензии;
Рисунок 5 | Обе почки увеличены в продольном размере (правая 10 см и левая 9 см), с повышенной эхогенностью (за счет акустического усиления крошечных кист) и измененной кортикомедуллярной дифференцировкой.
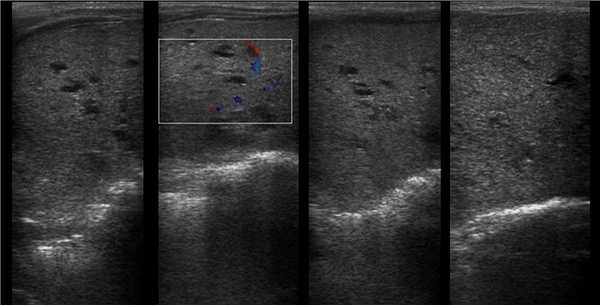
Рисунок 6 | Печень с врожденным фиброзом и кистозными расширениями в правой доле.
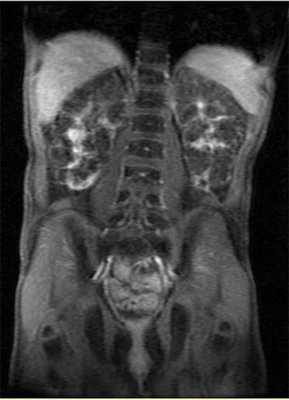
Рисунок 7 | МРТ, корональная Т2-проекция, пациент с ПБП-АР.
Магнитно-резонансный томограф не имеет никаких преимуществ перед ультразвуковыми сканерами высокого класса и генетическим тестированием в диагностике ПКП-АД.
Магнитно-резонансная холангиопанкреатография (МРХПГ)
Изображения, получаемые с помощью МРХПГ, обеспечивают хорошее качество визуализации гепатобилиарной системы у пациентов с ПБП-АР. МРХПГ является чувствительным методом диагностики билиарной эктазии, который в сочетании с визуализацией почек почти полностью заменил такие инвазивные методы диагностики, как биопсия почек и печени.
NB! Биопсия почек и печени не должны использоваться для диагностики ПБП.
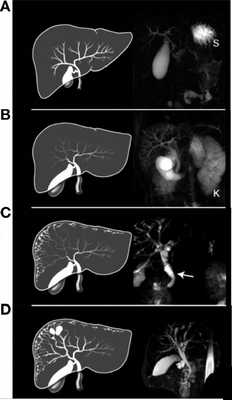
Рисунок 8 | МРХПГ и последующий рендеринг билиарной системы с аномалиями, свойственными для пациентов с ПБП-АР:
A. Нормальная печень с увеличенным желчным пузырем, (S) желудок.
B. Увеличенный общий желчный проток с желчным пузырем. Расширенные внутрипеченочные протоки.
C. Веретенообразные кисты периферических и центральных желчных протоков небольшого размера с расширенным общим желчным протоком (стрелка) и желчным пузырем.
D. Веретенообразные макроцисты с расширенными печеночными протоками.
У всех пациентов была печень аномальной формы с непропорционально увеличенной левой долей.
Дифференциальная диагностика
ПБП-АР следует дифференцировать со следующими состояниями:
- аутосомно-доминантная поликистозная болезнь почек;
- почечная дисплазия, связанная с трисомией 13 хромосомы;
- синдром Ме́ккеля — Гру́бера;
- синдром Лоренса — Муна — Барде — Бидля;
- синдром Беквита — Видемана;
- гломерулярная кистозная (гломерулокистозная) болезнь почек.
Подтверждение диагноза
Если имеются серьезные подозрения на ПБП-АР, необходим генетический анализ и консультирование. Молекулярно-генетическое тестирование показано пробанду с наличием у него кистозного увеличения почек и ВФП для выявления обеих патологических аллелей PKHD1. Должны быть проанализированы как минимум 3 поколения, т. к. заболевание передается рецессивно. Молекулярно-генетическое подходы к тестированию включают в себя панели для диагностики единичных точечных мутаций, мультигенные панели или полное геномное исследование.
Лечение
Аппарат искусственного кровообращения необходим всем пациентам с гипоплазией легкого, у которых оксигенация 100 % кислородом неэффективна из-за гипервентиляции вследствие давления на диафрагму. Новорожденным с врожденной легочной недостаточностью может потребоваться ИВЛ с первых минут жизни до определения причины. Когда увеличенные почки сдавливают ЖКТ, ряд авторов рекомендуют двух- или одностороннюю нефрэктомию, но при односторонней нефрэктомии может наблюдаться компенсаторное еще большее увеличение оставшейся почки. После двусторонней нефрэктомии пациенты нуждаются в гемодиализе.
Хронический перитонеальный диализ, с короткими периодами на гемодиализ для восстановления брюшины — метод выбора. Продолжительность этих процедур, также как и превентивная трансплантация почки, будут зависеть от ряда таких факторов как возраст, вес, клинический статус пациента и наличие здорового подходящего донора. Лечение гипонатриемии должно проводиться в зависимости от степени обезвоживания — раннее распознавание и лечение дегидратации имеют решающее значение. Для введения дополнительного питания и жидкостей может потребоваться установка назогастрального зонда. Артериальная гипертония (АГ) обычно хорошо поддается лечению ингибиторами АПФ и блокаторами рецепторов ангиотензина, которые являются препаратами выбора. Порой АГ может потребовать сочетания нескольких препаратов. Для лечения анемии у детей с хронической почечной недостаточностью необходимо добавление препаратов железа или стимуляторов выработки эритропоэтина. Лечение билиарной дисфункции должно быть сфокусировано на мальабсорбции питательных веществ и жирорастворимых витаминов и снижении риска восходящего холангита. Варикозно расширенные вены пищевода можно клипировать или использовать склеротерапию. Прогрессирующая портальная гипертензия требует портосистемного шунтирования. Благодаря возможностям современной медицины, в тяжелых случаях портальной гипертензии, почечной и печеночной недостаточности стала возможна двойная почечно-печеночная трансплантация.
УЗИ кистозных дисплазий почек у детей на клинических примерах. Лекция для врачей
Для понимания происхождения различных видов кортикальных дисплазий необходимо изучение и понимание процессов эмбриогенеза почек.
Развитие кистозных аномалий, таких как мультикистозная дисплазия, мультилокулярная киста, солитарная киста, чашечковый дивертикул связывают с отклонениями в эмбриогенезе в первые недели внутриутробной жизни. Постоянная почка формируется из метанефрогенной бластемы и выроста протока мезонефроса, дающего начало мочеточнику. Краниальный конец эмбрионального мочеточника вначале мешкообразно расширяется, а затем на нем появляются выросты, соответствующие лоханке и чашечкам первого и второго порядка. На этом этапе возможны различные отклонения от нормального эмбрионального развития. При очень быстром росте одна из первичных чашечек может отшнуроваться от общего протока метанефроса и превратиться в изолированную полость.
Так возникают парапельвикальные, и околомочеточниковые кисты, а при сохранении соединения первичной чашечки и протока метанефроса - чашечковые дивертикулы.
Поликистоз почек ( Поликистозная болезнь почек )
Поликистоз почек - это врожденная двусторонняя кистозная трансформация почечной паренхимы, приводящая к прогрессирующему снижению функции почек. Патология может проявляться артериальной гипертензией, болевым синдромом в поясничной области и животе, гематурией, дизурией, развитием инфекции и камней в почках, почечной недостаточностью. Диагностика заболевания включает изучение семейного анамнеза, лабораторные исследования, УЗИ почек, урографию, ангиографию, МРТ и КТ. Лечение при поликистозе почек консервативное, симптоматическое.
МКБ-10



Поликистозная болезнь (поликистоз почек) - порок эмбрионального развития почечных канальцев, характеризующийся образованием множественных мелких кист в паренхиме почек. Поликистоз почек всегда двусторонний. Кисты могут иметь размер от спичечной головки до крупной вишни и больше, содержать светлый либо шоколадного цвета желеобразный секрет.
Увеличение кист со временем приводит к сокращению объема функционирующей паренхимы и развитию почечной недостаточности. Как правило, поликистоз не ограничивается только поражением почек; к внепочечным формам поликистоза относится формирование кист в поджелудочной железе, селезенке, печени, семенных пузырьках, легких и других органах.

Причины
Поликистоз почек в большинстве случаев является патологией, наследуемой по аутосомно-доминантному типу. Развитие аутосомно-доминантного поликистоза может произойти, если аналогичное заболевание имеется у одного из родителей; эта форма наследования определяет 85-90% случаев болезни. Признаки заболевания в этом случае чаще развиваются к 30-40 годам, реже в детском возрасте.
Для развития аутосомно-рецессивного поликистоза почек необходима передача мутантных генов, обусловливающих дефект, от обоих родителей. Это менее распространенная форма, которая обнаруживается у новорожденных. При отсутствии семейной истории предполагается возникновение новой мутации в половой клетке одного из родителей.
Патогенез
В патогенезе поликистоза почек ведущая роль принадлежит нарушению слияния секреторных и экскреторных структур нефрона на стадии развития вторичной почки. Это затрудняет выделение первичной мочи, увеличивает давление в почечных канальцах, что приводит к деформации их просвета и образованию кистозно расширенных полостей. Отмечается значительное увеличение размеров и массы почек, которые макроскопически имеют бугристую неровную поверхность из-за множественных выступающих кист.
Стенки кист представлены соединительной тканью, полость выстлана плоским или кубическим эпителием, внутри содержится жидкость желтоватого или коричневого цвета, близкая по составу к моче. Между отдельными кистами имеются участки паренхимы, которые из-за давления кист могут подвергаться дистрофическим изменениям, атрофии или ишемии. Чашечки и лоханки значительно деформированы и увеличены.
Классификация
Симптомы поликистоза почек
У новорожденных патология обычно протекает крайне неблагоприятно и достаточно рано заканчивается гибелью ребенка от уремии. У взрослых поликистоз почек развивается медленно, проходя компенсированную, субкомпенсированную и декомпенсированную стадии.
На стадии компенсации проявления длительно отсутствуют. Со временем появляется чувство давления в пояснице, неопределенные боли в животе, дизурия, обусловленные растяжением почек. Отмечается утомляемость, головная боль, иногда - гематурия неясного генеза. Функция почек в компенсированной стадии остается не нарушенной.
В стадии субкомпенсации нарастают признаки почечной недостаточности, проявляющиеся тошнотой, сухостью во рту, жаждой, приступами мигрени, стойкой и высокой артериальной гипертензией. Нарушения функции почек характеризуются полиурией с изостенурией, эритроцитурией, цилинрурией, при возникновении пиелонефрита - лейкоцитурией. В случае нагноения кист присоединяется лихорадка, интоксикация, ознобы; при камнях в почках развиваются приступы почечной колики.
В декомпенсированной стадии болезни возникает хроническая уремия. Прогрессированию поликистоза почек может способствовать артериальная гипертензия, травмы, хирургические вмешательства, беременность, кровотечения. Присоединение вторичной инфекции (гриппа, ОРВИ, пневмонии и др.) может вызвать резкое ухудшение состояния вплоть до гибели пациента; при нагноении кист нередко развивается уросепсис.
Осложнения
Стойкое повышение кровяного давления со временем может осложняться гипертрофией левого желудочка, пролапсом митрального клапана и сердечной недостаточностью, аневризмой сосудов мозга и геморрагическим инсультом. Поликистоз почек может вызывать развитие поздних токсикозов беременности - преэклампсии и эклампсии. В группе особого риска - женщины, страдавшие гипертонией до наступления беременности. У пациентов с этой почечной патологией повышена вероятность образования кист печени, дивертикулярной болезни толстого кишечника.
Диагностика
Данные анамнеза в ряде случаев позволяют выявить семейные случаи поликистоза почек у родственников одной линии. Пальпировать увеличенные и кистозно измененные почки удается не всегда, в связи с чем решающее значение в диагностике отводится инструментальным методикам:
Посредством УЗИ в увеличенных почках определяются множественные кисты. В неясных случаях прибегают к ретроградной пиелографии, почечной ангиографии, которые также позволяют обнаружить кистозное перерождение почек. Для выяснения степени компенсации функции почек проводят исследования мочи (общий анализ, пробу Зимницкого и Реберга), биохимическое исследование крови. При развитии пиелонефрита моча подвергается бактериологическому посеву. С целью установления семейных форм поликистоза почек показано генетическое исследование.
Поликистоз почек необходимо дифференцировать от хронического гломерулонефрита и хронического пиелонефрита, опухолей почки. При подозрении на аневризму сосудов головного мозга выполняется ангиография сосудов головного мозга, УЗДГ, магнитно-резонансная ангиография.
Лечение поликистоза почек
Проводится симптоматическая терапия. К общим рекомендациям относится исключение чрезмерных и длительных нагрузок, профилактика хронических инфекций (кариеса, ОРВИ, тонзиллита, синуситов и др.), соблюдение высококалорийной, богатой витаминами диеты с ограничением белка и поваренной соли. При развитии пиелонефрита назначается курсовое лечение антибиотиками и уроантисептиками. В случае макрогематурии осуществляется гемостатическая терапия; при снижении диуреза показан прием диуретиков; при артериальной гипертонии - гипотензивных средств.
В компенсированной стадии поликистоза почек может выполняться вскрытие и опорожнение крупных кист. Это приводит к уменьшению размеров почек, улучшению их кровообращения и функций. В терминальных стадиях, при почечной недостаточности ставится вопрос о хроническом гемодиализе и трансплантации почки.
Прогноз и профилактика
Своевременная коррекция артериальной гипертензии и устранение инфекций мочевых путей позволяют замедлить прогрессирование поликистоза почек. Тем не менее, у большинства больных с поликистозом почек в различные сроки от выявления болезни развивается почечная недостаточность. При семейных формах поликистоза почек необходима консультация врача-генетика для определения рисков рождения ребенка с подобной почечной аномалией. При установленном диагнозе требуется постоянное наблюдение пациента врачом-нефрологом и урологом.
Читайте также: