УЗИ, рентгенограмма при кампомелической дисплазии у плода
Добавил пользователь Валентин П. Обновлено: 22.01.2026
Кампомелическая дисплазия. Летальное генетическое заболевание из группы остеохондродисплазий, характеризующееся тяжелыми скелетными аномалиями и другими нарушениями, которые нередко приводят к внутриутробной гибели плода или смерти новорожденного в первые недели жизни. Симптомами этого состояния являются сильное искривление трубчатых костей конечностей, наличие 11-ти пар ребер и колоколообразная деформация грудной клетки. В ряде случаев происходит инверсия пола. Диагностика кампомелической дисплазии чаще всего производится на основании данных пренатальных ультразвуковых исследований, кариотипирования и молекулярно-генетических исследований, после рождения ребенка для подтверждения могут использоваться данные рентгенографии. Лечения этого генетического заболевания не существует.
Кампомелическая дисплазия
Дополнительные факты
Причины
На заре изучения кампомелической дисплазии многих врачей-генетиков ставило в тупик необычное сочетание нарушений - скелетных аномалий конечностей, лопаток, ребер и патологии черепа (расщелина твердого нёба, микроцефалия) с гермафродитизмом и половой инверсией. Причина столь необычного сочетания крылась в гене, мутации которого становятся причиной заболевания - гене SOX-9, расположенном на 17-й хромосоме. Ген кодирует одноименный белок, который является одним из важнейших транскрипционных факторов в организме человека. Например, он контролирует работу гена Col2A1, который локализуется на 12-й хромосоме и кодирует последовательность основной разновидности коллагена 2-го типа, принимающего участие в процессах формирования скелета и оссификации. Дефекты в структуре протеина SOX-9 ведут к резкому снижению экспрессии гена Col2A1, дефициту коллагена и как следствие - к кампомелической дисплазии.
Другой важной функцией SOX-9 является активация гена AMH, располагающегося на 19-й хромосоме. В основном этот ген экспрессируется в клетках Сертоли у мужчин в процессе эмбрионального развития. AMH кодирует так называемый мюллеров ингибирующий фактор, который приводит к разрушению Мюллерова протока, играющего важную роль в половой дифференцировке плода. При мутациях гена SOX-9 эта функция нарушается, в результате примерно 75% больных кампомелической дисплазией при кариотипе XY имеют фенотипически женские половые признаки. В прошлом из-за несовпадения гентотипа и фенотипа считалось, что данное заболевание намного чаще поражает девочек, нежели мальчиков. Многие исследователи предполагают активное участие белка SOX-9 и в контроле других генов, на что косвенно указывает наличие многих других аномалий развития при кампомелической дисплазии.
Диагностика
Основную роль в определении кампомелической дисплазии играют методики пренатальной диагностики: ультразвуковое исследование, кариотипирование и молекулярно-генетические анализы. Ультразвуковыми методами данное заболевание можно выявить уже со 2-го триместра вынашивания ребенка, ведущими проявлениями будет искривление костей конечностей, наличие 11-ти пар ребер и колоколообразная форма грудной клетки. Также могут определяться пороки развития черепа, лица, сердца и почек, гипоплазия лопаток и тел позвонков, увеличение размеров тазового отверстия. На основании перечисленных данных пренатального ультразвукового исследования можно обоснованно предположить наличие кампомелической дисплазии и поставить вопрос о прерывании беременности по медицинским показаниям.
Кариотипирование производят с целью исключения анеуплоидий или хромосомных аномалий, которые могут вызывать схожие проявления и пороки развития плода. При этом в случае кампомелической дисплазии определение кариотипа может указать на мужской пол плода, тогда как по результатам ультразвуковых исследований будет определяться девочка. Это служит дополнительным подтверждением данного заболевания, поскольку именно для него характерны гермафродитизм и половые инверсии. Для наиболее точной диагностики могут использоваться молекулярно-генетические анализы методом секвенирования последовательности гена SOX-9. Обнаружение мутаций в данном гене является окончательным подтверждением наличия кампомелической дисплазии.
Лечение
В некоторых случаях диагностику кампомелической дисплазии производят у живого новорожденного. Для этого, помимо вышеуказанных методик, применяют рентгенологическое исследование скелета. На рентгенограммах определяются многочисленные деформации трубчатых костей и другие признаки, которые были выявлены еще на УЗИ. Врачи-неонатологи также могут определить у новорожденного с кампомелической дисплазией порок сердца, аномалии развития почек и респираторные нарушения. Нередко окончательное подтверждение диагноза производится на основании данных патологоанатомического исследования. Практические любые лечебные мероприятия, даже паллиативного характера, при данном заболевании неэффективны и неспособны продлить жизнь больного.
Кампомелическая дисплазия
Кампомелическая дисплазия - летальное генетическое заболевание из группы остеохондродисплазий, характеризующееся тяжелыми скелетными аномалиями и другими нарушениями, которые нередко приводят к внутриутробной гибели плода или смерти новорожденного в первые недели жизни. Симптомами этого состояния являются сильное искривление трубчатых костей конечностей, наличие 11-ти пар ребер и колоколообразная деформация грудной клетки. В ряде случаев происходит инверсия пола. Диагностика кампомелической дисплазии чаще всего производится на основании данных пренатальных ультразвуковых исследований, кариотипирования и молекулярно-генетических исследований, после рождения ребенка для подтверждения могут использоваться данные рентгенографии. Лечения этого генетического заболевания не существует.
Общие сведения
Причины кампомелической дисплазии
На заре изучения кампомелической дисплазии многих врачей-генетиков ставило в тупик необычное сочетание нарушений - скелетных аномалий конечностей, лопаток, ребер и патологии черепа (расщелина твердого нёба, микроцефалия) с гермафродитизмом и половой инверсией. Причина столь необычного сочетания крылась в гене, мутации которого становятся причиной заболевания - гене SOX-9, расположенном на 17-й хромосоме. Ген кодирует одноименный белок, который является одним из важнейших транскрипционных факторов в организме человека. Например, он контролирует работу гена Col2A1, который локализуется на 12-й хромосоме и кодирует последовательность основной разновидности коллагена 2-го типа, принимающего участие в процессах формирования скелета и оссификации. Дефекты в структуре протеина SOX-9 ведут к резкому снижению экспрессии гена Col2A1, дефициту коллагена и как следствие - к кампомелической дисплазии.
Другой важной функцией SOX-9 является активация гена AMH, располагающегося на 19-й хромосоме. В основном этот ген экспрессируется в клетках Сертоли у мужчин в процессе эмбрионального развития. AMH кодирует так называемый мюллеров ингибирующий фактор, который приводит к разрушению Мюллерова протока, играющего важную роль в половой дифференцировке плода. При мутациях гена SOX-9 эта функция нарушается, в результате примерно 75% больных кампомелической дисплазией при кариотипе XY имеют фенотипически женские половые признаки. В прошлом из-за несовпадения гентотипа и фенотипа считалось, что данное заболевание намного чаще поражает девочек, нежели мальчиков. Многие исследователи предполагают активное участие белка SOX-9 и в контроле других генов, на что косвенно указывает наличие многих других аномалий развития при кампомелической дисплазии.
Симптомы кампомелической дисплазии
При кампомелической дисплазии достаточно часто возникает пре- или интранатальная смерть, обусловленная резкой дыхательной недостаточностью у младенца. В тех случаях, когда больной рождается живым, обнаруживаются выраженная деформация длинных трубчатых костей рук и ног, удлинение мозговой части черепа со значительным уменьшением лицевого отдела, микрогнатия и уменьшенные размеры тела. У больных кампомелической дисплазией выявляется плоское лицо, нередко наблюдаются расщелина твердого нёба и врожденный вывих бедра. При обследовании также определяются гипоплазия лопаток, деформации стоп, гидронефроз и различные врожденные пороки сердца. Кампомелическая дисплазия приводит к смерти больного на протяжении первых дней или недель жизни. Как правило, летальный исход наступает из-за выраженного респираторного дистресс-синдрома, свой вклад вносят пороки развития черепа, мочевыделительной и сердечно-сосудистой систем.
Диагностика и лечение кампомелической дисплазии
Основную роль в определении кампомелической дисплазии играют методики пренатальной диагностики: ультразвуковое исследование, кариотипирование и молекулярно-генетические анализы. Ультразвуковыми методами данное заболевание можно выявить уже со 2-го триместра вынашивания ребенка, ведущими проявлениями будет искривление костей конечностей, наличие 11-ти пар ребер и колоколообразная форма грудной клетки. Также могут определяться пороки развития черепа, лица, сердца и почек, гипоплазия лопаток и тел позвонков, увеличение размеров тазового отверстия. На основании перечисленных данных пренатального ультразвукового исследования можно обоснованно предположить наличие кампомелической дисплазии и поставить вопрос о прерывании беременности по медицинским показаниям.
Кариотипирование производят с целью исключения анеуплоидий или хромосомных аномалий, которые могут вызывать схожие проявления и пороки развития плода. При этом в случае кампомелической дисплазии определение кариотипа может указать на мужской пол плода, тогда как по результатам ультразвуковых исследований будет определяться девочка. Это служит дополнительным подтверждением данного заболевания, поскольку именно для него характерны гермафродитизм и половые инверсии. Для наиболее точной диагностики могут использоваться молекулярно-генетические анализы методом секвенирования последовательности гена SOX-9. Обнаружение мутаций в данном гене является окончательным подтверждением наличия кампомелической дисплазии.
Прогноз и профилактика кампомелической дисплазии
Диастрофическая дисплазия
Диастрофическая дисплазия - одна из разновидностей скелетных дисплазий, которая характеризуется нарушением формирования некоторых типов хрящевой ткани и связанным с этим затрудненным образованием эндохондральной кости. Симптомы заболевания выявляются сразу при рождении или в рамках пренатальной диагностики и заключаются в уменьшенной длине тела новорожденного и низкорослости в дальнейшем, контрактуре суставов, сколиозе и других пороках развития. Диагностика диастрофической дисплазии производится на основании данных осмотра больного, рентгенологических и молекулярно-генетических исследований. Специфического лечения патологии не существует, используют симптоматическую терапию. При выявлении характерных нарушений на ранних сроках вынашивания ребенка осуществляют прерывание беременности по медицинским показаниям.
Диастрофическая дисплазия - наследственное заболевание из группы костно-хрящевых дисплазий, характеризующееся многочисленными пороками развития скелета. Впервые данная патология была описана в 1960-м году французским врачом-генетиком М. Лами совместно с его учеником, педиатром П. Марото. Исследователи смогли определить особенности скелетных аномалий при этом состоянии и установить их наследственный характер. Диастрофическая дисплазия является заболеванием с аутосомно-рецессивным механизмом наследования. Встречается очень редко, что несколько затрудняет достоверное определение его распространенности. При этом удалось выяснить, что такое состояние чаще встречается в странах балтийского региона, особенно в Финляндии. Из-за аутосомно-рецессивной передачи диастрофической дисплазии половое распределение заболевания не имеет каких-либо особенностей - от него в равной степени страдают как мальчики, так и девочки.
Причины диастрофической дисплазии
Основной причиной развития диастрофической дисплазии является мутация в гене SLC26A2, который располагается на 5-й хромосоме. Этот ген широко известен в медицинских кругах, так как его дефекты обуславливают большое количество наследственных и врожденных аномалий развития скелета, в том числе - некоторых типов ахондрогенеза и ателостеогенеза, множественной эпифизарной дисплазии и синдрома Де ля Шапеля. Причина заключается в том, что SLC26A2 кодирует особый белок-переносчик сульфат-ионов, принимающий активное участие в образовании протеогликанов хрящей и других соединительных тканей. Различные по своему типу мутации гена ведут к неодинаковым структурным изменениям данного протеина, что, в свою очередь, по-разному меняет его функциональную активность и обуславливает разнообразие пороков развития.
Согласно данным современной генетики, причиной развития диастрофической дисплазии (особенно финляндского типа) является мутация IVS1+2T>C. При этом сульфирование протеогликанов хрящей становится недостаточным, что приводит к накоплению «необработанных» продуктов в матриксе хрящевой ткани. Нарушается плотность хряща и его функциональная активность, что ведет к проблемам при формировании костей с эндохондральным окостенением (костей туловища, конечностей и основания черепа). Именно этими процессами обусловлены практически все симптомы диастрофической дисплазии, которые наблюдаются у больных и обнаруживаются в ходе пренатальной диагностики. Все мутации гена SLC26A2 делятся на летальные и нелетальные. Диастрофическая дисплазия относится к последней группе, больные в ряде случаев способны доживать до преклонного возраста.
Симптомы диастрофической дисплазии
Первые симптомы диастрофической дисплазии можно обнаружить сразу при рождении ребенка. Врачи-неонатологи регистрируют уменьшенную длину тела (не более 42 сантиметров) и массу тела (до 2800 грамм) при нормальных сроках вынашивания. Это свидетельствует о внутриутробной задержке развития плода, что нередко может быть выявлено и при профилактических ультразвуковых исследованиях. Из других ранних постнатальных проявлений диастрофической дисплазии можно отметить микроцефалию и воспаление хрящей ушных раковин, которое развивается в течение 1-5 месяцев жизни ребенка. После затухания воспаления происходит деформация хрящевой основы органа.
В дальнейшем у больного диастрофической дисплазией развивается целый ряд других патологий: деформации кисти с тугоподвижностью в межфаланговых суставах, короткие пальцы, выраженное проксимальное расположение большого пальца. Возникают контрактуры тазобедренных и коленных суставов. Длинные трубчатые кости конечностей укорочены относительно пропорций тела. Уменьшение длины костей конечностей ведет к низкорослости больных диастрофической дисплазией, средний рост мужчин с данной патологией составляет 132 сантиметра, женщин - 126 сантиметров. Выявляются прогрессирующие искривления позвоночника (сколиоз, кифоз). Других нарушений (в частности - расстройств интеллекта и эндокринной системы) при диастрофической дисплазии, как правило, не наблюдается.
Диагностика диастрофической дисплазии
Диагностика диастрофической дисплазии производится на основании данных физикального осмотра, рентгенологического исследования скелета и молекулярно-генетического анализа. При осмотре новорожденного отмечаются признаки пренатального отставания в физическом развитии (уменьшенная длина и масса тела, микроцефалия), в дальнейшем эти показатели остаются более низкими, чем у здоровых сверстников. В старшем возрасте при диастрофической дисплазии выявляются короткие конечности, деформации кистей и пальцев, контрактуры коленных и тазобедренных суставов, низкий рост. Почти у 80% больных наблюдаются утолщение и деформация хрящей ушных раковин как следствие перенесенного в раннем детстве воспаления.
Рентгенологически у больных диастрофической дисплазией определяется уменьшение относительной длины трубчатых костей конечностей, часто сочетающееся с их дугообразной деформацией. Выявляются расширение метафизов, деформация головок бедренных костей, подвывихи и вывихи крупных суставов (коленных, локтевых, тазобедренных). Пястные кости и фаланги пальцев нередко укорочены, аналогичные изменения просматриваются и на костях плюсны. Практически всегда при диастрофической дисплазии обнаруживаются искривления позвоночника - сколиоз и кифоз различной степени выраженности. Молекулярно-генетическая диагностика заболевания сводится к прямому секвенированию гена SLC26A2 с целью подтверждения характерных генетических дефектов. Этот метод позволяет наиболее точно дифференцировать диастрофическую дисплазию от других скелетных аномалий, обусловленных мутациями SLC26A2.
Лечение и прогноз диастрофической дисплазии
Специфического лечения диастрофической дисплазии не существует, осуществляют симптоматическую коррекцию нарушений, в том числе - хирургическими методами. В число возможных операций входят вмешательства по устранению искривлений и фиксации позвоночного столба, показанные при тяжелом повреждении спинномозговых корешков. При умеренном радикулите используют противовоспалительные средства, физиопроцедуры, лечебную гимнастику и другие методики. Прогноз диастрофической дисплазии относительно выживаемости больных неопределенный, даже при благоприятном исходе состояние становится причиной инвалидизации. В ряде случаев больные с такой патологией доживают до взрослого и даже преклонного возраста.
Профилактика диастрофической дисплазии
Профилактические мероприятия при диастрофической дисплазии сводятся к своевременной пренатальной диагностике заболевания и определению носительства патологической формы гена SLC26A2. Посредством ультразвукового исследования патологию можно выявить у плода со второго триместра гестации. При обнаружении дисплазии ставится вопрос о прерывании беременности по медицинским показаниям, но окончательное решение по этому поводу принимают родители. Молекулярно-генетическими техниками пренатальной диагностики подтвердить диастрофическую дисплазию у плода можно еще до начала второго триместра, материал для исследования получают посредством биопсии ворсин хориона или аминоцентеза. Использование таких техник особенно актуально, когда родители предположительно входят в число носителей патологической формы гена SLC26A2 (заболевание проявлялось у кровных родственников) или когда генетическими методами было доказано, что оба родителя являются гетерозиготами по мутантной форме SLC26A2 - в подобных случаях вероятность рождения ребенка с диастрофической дисплазией составляет 25%.
Современные подходы к изучению плодового фенотипа в эру генетического ультразвука
Медико-генетическое отделение Московского областного НИИ акушерства и гинекологии, Москва.
Курс пренатальной диагностики, ФГБОУ ДПО РМАПО Минздрава России, Москва.

УЗИ аппарат RS85
Революционные изменения в экспертной диагностике. Безупречное качество изображения, молниеносная скорость работы, новое поколение технологий визуализации и количественного анализа данных УЗ-сканирования.
Настоящая работа посвящена изучению плодового фенотипа при ультразвуковом исследовании в различные сроки беременности при помощи новейших технологий Samsung, оптимизирующих диагностику многих врожденных пороков развития, аномалий, нарушений строения, особенностей развития с оценкой значимости отдельных ультразвуковых патологических маркеров в качестве потенциальных признаков генетических синдромов и ассоциаций.
Одной из ведущих причин детской и младенческой смертности являются врожденные пороки развития (ВПР), среди которых в 20% случаев встречаются множественные ВПР (МВПР) с установленной частотой 1:250 новорожденных [1]. Особое значение в профилактике МВПР имеет пренатальная диагностика - современный и высокоэффективный метод диспансеризации плода, направленный на своевременное выявление патологии, впоследствии определяющий выбор адекватной акушерской тактики при данной беременности и специфических мер профилактики болезни в данной семье в дальнейшем [2]. Современные ее возможности позволяют из комплекса патологических нарушений у плода выделить достоверно значимые ультразвуковые маркеры для постановки диагноза генетических синдромов различной этиологии. Сегодня пренатальная синдромология активно развивается, как в плане расширения спектра возможных лабораторных исследований, так и описания значимых, ключевых, таргетных ультразвуковых признаков многих наследственных синдромов различного происхождения.
Группа МВПР включает сотни нозологических форм, различных по этиологии, фенотипическим проявлениям и прогнозу. В силу огромной гетерогенности МВПР представляют собой сложную проблему для постановки окончательного диагноза. Основными этиологическими факторами, приводящими к возникновению синдромов с множественными пороками развития, являются генные, хромосомные мутации и действие на плод неблагоприятных факторов внешней среды [3, 4].
Любые наследственные или врожденные генетические болезни являются результатом повреждения генетической информации на геномном, хромосомном или генном уровне. В 60% всех случаев МВПР встречаются хромосомные синдромы, достаточно широко изученные и хорошо диагностируемые. Для лабораторной диагностики этих повреждений разработаны разнообразные и хорошо известные методы: цитогенетический, молекулярно-цитогенетический и молекулярно-генетический. МВПР нехромосомного генеза встречаются в 40% случаев. Среди них выделяют синдромы, ассоциации и неклассифицированные комплексы. Применительно к нехромосомным синдромам цитогенетическое исследование позволяет лишь отвергнуть хромосомную природу данного комплекса пороков развития, но не установить диагноз.
Сегодня имеется определенное количество публикаций о выявлении некоторых синдромов и ассоциаций при пренатальной эхографии [6, 7]. Однако дородовая идентификация нозологических форм нехромосомных синдромов пока не вошла в рутинную практику врачей в области пренатальной диагностики и носит случайный характер. В литературе отсутствуют четкие рекомендации о системном подходе к диагностике наследственных синдромов различной этиологии, нет диагностических алгоритмов и определенных схем взаимодействия врачей ультразвуковой пренатальной диагностики, генетиков, акушеров-гинекологов.
Использование эхографии делает возможным пренатальное выявление не менее 80% врожденных дефектов во II, III триместрах беременности и свыше 50% значимых нарушений анатомии плода уже в I триместре начиная со сроков 11-12 нед. Весь комплекс диагностированных пороков развития и различных микроаномалий в совокупности составляет фенотипический спектр синдрома, который состоит из «ядерных» (главных, ключевых) признаков и дополнительных аномалий, наличие которых не является обязательным для диагностики клинической нозологии синдрома.
Революцией в пренатальной ультразвуковой диагностике явилось появление объемной эхографии, которая, обладая такими качествами, как неинвазивность, безопасность и возможность многократного применения у одной пациентки, имеет высокую информативность в исследовании анатомии плода и изучении его фенотипа. При применении различных режимов объемной эхографии абсолютно очевидно их преимущество по сравнению с обычным сканированием. Детально можно изучить лицо плода (рис. 1-4) в различные сроки беременности, начиная со сроков первого пренатального скрининга в 11-14 нед, конечности плода, причем не только их наличие и положение (рис. 5, 6), но и состояние и количество пальцев (рис. 7-9) как на руках, так и на ногах. Также можно изучить позвонки плода (рис. 10), состояние твердого нёба (рис. 11, 12), строение наружного уха (ушной раковины) (рис. 13), состояние основных швов черепа и родничков, исключая их преждевременное закрытие при кранисиностозах (рис. 14, 15).
Ультразвуковое исследование лопаток и ключиц плода
Научно-исследовательский институт акушерства и педиатрии, Ростов-на-Дону.
Институт повышения квалификации ФМБА, Москва.
УЗИ сканер RS80
Эталон новых стандартов! Беспрецедентная четкость, разрешение, сверхбыстрая обработка данных, а также исчерпывающий набор современных ультразвуковых технологий для решения самых сложных задач диагностики.
Введение
Врожденные пороки развития опорнодвигательной системы - это многочисленная группа пороков различных по этиологии, патогенезу и клиническим проявлениям. Пренатальная диагностика врожденных пороков развития опорнодвигательной системы имеет важное практическое значение, поскольку многие из этих пороков имеют неблагоприятный прогноз для жизни и здоровья. Около 25% плодов с врожденными пороками развития опорнодвигательной системы рождаются мертвыми, 30% детей с этой патологией умирают в неонатальном периоде [1].
Ультразвуковая оценка опорнодвигательной системы плода становится возможной с конца I триместра беременности. Отдельные элементы скелета начинают визуализироваться с самых ранних сроков беременности и к 12-14 нед все его основные структуры становятся доступными для оценки [2]. Основу пренатальной диагностики врожденных пороков развития опорнодвигательной системы составляет методически правильная фетометрия всех составных компонентов скелета плода. Отсутствие или гипоплазия некоторых из них свидетельствует о наличии у плода серьезного заболевания. Например, изменения лучевых костей требуют исключения у плода ряда синдромов (Холт - Орама, Баллера - Герольда, Леви - Холистера, VACTERAL, анемия Фанкони, синдром TAR и др.).
Помимо оценки размеров костей и их количества, врач ультразвуковой диагностики должен обращать внимание на форму костей, их эхогенность, оценивать структуру позвоночника, а также состояние мягких тканей конечностей и подвижность крупных суставов. Дополнительно необходимо изучать строение кистей и стоп, поскольку при врожденных пороках развития опорнодвигательной системы нередко регистрируются их аномалии.
Если изменение длинных трубчатых костей плода при основных врожденных пороках развития опорнодвигательной системы к настоящему времени уже достаточно подробно исследовано, то оценка ключиц и лопаток плода до сих пор остается мало изученной, а ведь их гипоплазия нередко регистрируется при различных скелетных дисплазиях и может использоваться в качестве одного из дифференциальнодиагностических критериев. Так, гипоплазия ключиц часто регистрируется при клейдокраниальной дисплазии, синдроме Эдвардса, синдроме Холт - Орама, церебро- косто-мандибулярном синдроме, синдроме Мардена - Уокера и других аномалиях опорнодвигательной системы [3].
Для правильной оценки размеров ключиц и лопаток плода необходимо разработать их нормативные показатели в зависимости от срока беременности на базе процентильных значений, так как размеры ключицы и лопатки постоянно увеличиваются на протяжении гестационного периода.
Ключица является парной костью плечевого пояса, которая имеет форму вытянутой буквы S (рис. 1). Наружным концом она сочленяется с отростком лопатки (акромиально-ключичное сочленение), а внутренним (грудинным) - с грудиной (грудино-ключичное сочленение). Оба сочленения укреплены мощными связками. Ключица является единственной костью, скрепляющей верхнюю конечность со скелетом туловища. Функциональное значение ее велико: она отставляет плечевой сустав на должное расстояние от грудной клетки, обусловливая большую свободу движений конечности.
Рис. 1. Ключица. Вид сверху.
Ключица получает первичную точку окостенения раньше всех других костей - на 6-й нед внутриутробного развития. Вторичная точка окостенения на грудинном конце ключицы появляется на 12-й нед беременности [4]. Поэтому идентификация ключиц при ультразвуковом исследовании плода удается к концу I триместра беременности.
Лопатка представляет плоскую треугольную кость, прилегающую к задней поверхности грудной клетки на пространстве от II до VII ребра (рис. 2). Сообразно форме кости в ней различают три края: медиальный, обращенный к позвоночнику; латеральный и верхний, на котором находится вырезка лопатки. Перечисленные края сходятся друг с другом под тремя углами, из которых один направлен книзу, а два других находятся по концам верхнего края лопатки. Первичная точка окостенения появляется в теле лопатки в конце I триместра беременности. Поэтому ее визуализация при ультразвуковом исследовании потенциально возможна с 12 нед беременности. Следует отметить, что к моменту рождения из костной ткани состоит только тело и ость лопатки.
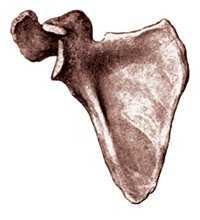
Рис. 2. Лопатка. Вид спереди.
Целью настоящего исследования явилась разработка нормативных процентильных показателей размеров ключиц и лопаток плода на основе данных пренатального ультразвукового обследования.
Материал и методы
Для разработки нормативов размеров ключицы и лопатки плода были отобраны результаты эхографических исследований при сквозном наблюдении за 446 пациентками в сроки от 14 до 34 нед беременности.
Критериями отбора пациенток в данную группу явились: 1) известная дата последней менструации при 26-30-дневном цикле; 2) неосложненное течение беременности; 3) наличие одноплодной беременности без признаков какой-либо патологии у плода; 4) отсутствие факта приема оральных контрацептивов в течение 3 мес до цикла зачатия; 5) срочные роды нормальным плодом с массой при рождении в пределах нормативных значений (более 10-го и меньше 90-го процентиля по массе и длине тела в зависимости от гестационного возраста).
Всего для решения поставленной цели было проведено 698 ультразвуковых обследований плодов в сроки от 14 до 34 нед беременности в двух отделениях пренатальной диагностики Москвы и Ростова- на-Дону по единому протоколу. Для каждого 7-дневного интервала использовались данные, полученные не менее чем у 31 женщины.
Каждое эхографическое исследование наряду с обязательной фетометрией дополнительно включало оценку длины ключицы и ширину лопатки плода. По результатам измерений, полученным при ультразвуковом исследовании, для каждого 7-дневного интервала по каждому из изучаемых фетометрических параметров были созданы базы данных. По этим данным рассчитывались МЕ (медиана, значение 50-го процентиля), а также численные значения 5-го и 95-го процентиля для каждого 7-дневного интервала.
Результаты и обсуждение
В ходе проведенных исследований нами было установлено, что визуализация ключиц и лопаток достигается достаточно легко и занимает в среднем не более одной минуты. По нашим данным, при использовании трансвагинальной эхографии визуализация ключиц и лопаток удается у большинства плодов с 12 нед беременности. Однако в нашем исследовании для разработки нормативных значений размеров ключиц и лопаток плода было использовано только трансабдоминальное сканирование, при котором наиболее отчетливое изображение с точным измерением этих костей было достигнуто с 14 нед беременности. В целом успешная визуализация ключиц и лопаток при трансабдоминальном сканировании в сроки от 14 до 34 нед беременности была достигнута более чем у 97% плодов.
Для обеспечения быстрой оценки ключиц и лопаток в ходе ультразвукового обследования плода преимущественно выбиралась его позиция, при которой поперечный срез позвоночника располагался в интервале от 10 до 2 часов. При этом передвигая датчик от головы к грудной клетке плода осуществлялась последовательная идентификация первоначально его ключиц, а затем лопаток.
При визуализации ключиц плода было установлено, что их типичная вытянутая S-образная форма идентифицируется уже с начала II триместра и практически не изменяется на протяжении второй половины беременности (рис. 3, 4). При невозможности одновременной визуализации обеих ключиц осуществляли их раздельную идентификацию (рис. 5).
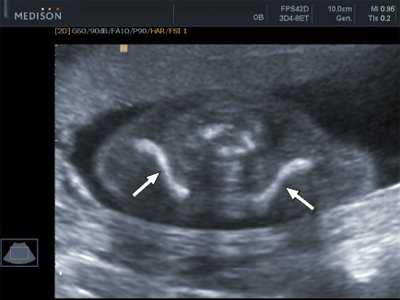
Рис. 3. Эхограмма ключиц (стрелки) плода в 16 недель беременности. Длина ключиц составляет 15 мм.
Читайте также: