Врожденная колобома: причины, диагностика, лечение
Добавил пользователь Евгений Кузнецов Обновлено: 22.01.2026
Офтальмологическое заболевание, при котором возникает дефект тканей оболочек глаза в виде расщелины, называется колобомой. Название происходит от древнегреческого слова κολόβωμα - в переводе - «увечье», «отнятая часть».
Существует несколько разновидностей заболевания. Одни из них можно определить на вид. Так, дефект при врожденной колобоме радужки напоминает щель, грушевидное или в виде отверстия для ключа пятно черного цвета, которое как бы стекает вниз от зрачка. Направление отверстия идет обычно в сторону аномалии. Такая щель образовывается из-за отсутствия тканей. Дефекты же при более глубоких поражениях - например, при колобоме зрительного нерва, - увидит только специалист.
В данной статье рассмотрим виды, причины появления и возможные способы лечения колобомы.
Причины колобомы
Причины заболевания различаются в зависимости от того, к какому типу оно относится. Колобома может быть врожденной: тогда ее появление обусловлено:
- хромосомными аномалиями (в основном - синдромами Патау, Эдвардса и Дауна);
- наследственностью;
- инфекционными заболеваниями беременной (в частности, к патологии может привести инфицирование матери цитомегаловирусом, особенно произошедшее в первые три месяца гестации);
- злоупотребление беременной алкоголем;
- гормональные сбои.
Врожденная колобома встречается в 0,5-0,7 случаях на 10 000 младенцев и часто сопровождается такими дефектами как заячья губа и волчья пасть (расщелина верхнего неба).
При врожденной колобоме зрачок может сокращаться и расширяться под воздействием света, однако нужного размера он не достигает.
Приобретенная колобома встречается гораздо реже врожденной. Она может появиться вследствие:
- механических травм;
- осложнений офтальмологических операций;
- онкологии органов зрения;
- некрозов тканей глазного яблока различной этиологии;
- появления рубцов.
Сокращения зрачка пациента с приобретенной колобомой нарушены. Это происходит при поражении сфинктера, регулирующего ширину отверстия, из-за шрама, опухоли, травмы. Следовательно, нарушена и регуляция количества проникающих в глаз лучей. При таком заболевании есть риск получить серьезные зрительные нарушенияослепнуть, если освещение будет слишком ярким.
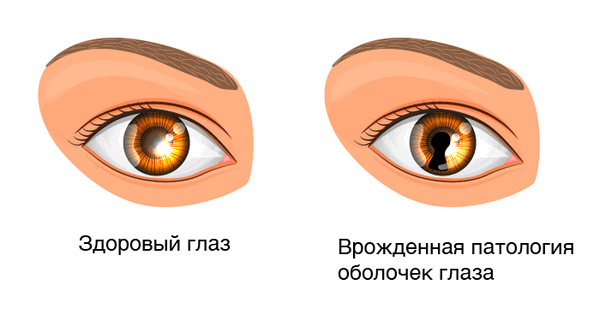
Симптомы колобомы радужки
При любом из видов колобомы можно наблюдать следующую симптоматику:
- повышенная утомляемость;
- фоточувствительность: пациент остро реагирует даже на незначительные изменения, неяркий свет;
- боль, неприятные ощущения в глазах;
- сокращения, подергивания зрачка, которые нельзя контролировать;
- размытость: воспринимаемая картинка теряет четкость;
- диплопия.
Виды колобомы
| Вид | Описание | Локализация | Симптомы |
|---|---|---|---|
| Колобома радужной оболочки глаза | Самый распространенный вид заболевания. Выглядит как грушевидная щель черного цвета. | Область зрачка одного или обоих глаз. | - небольшое снижение зрения; - появление в поле зрения скотом «слепых пятен»; - повышенная светочувствительность; - при двойной симметричной колобоме имеют место высокочастотные непроизвольные подергивания глаз - нистагм. |
| Колобома хориоидеи (сосудистой оболочки глаза) | Обнаруживается при исследовании глазного дна. Выглядит как окрашенный в белый цвет участок с неровными краями. | Нижний отдел глазного дна, иногда до зрительного нерва. | - значительное ухудшение зрения. |
| Ресничная колобома (колобома цилиарного тела) | У пациентов отсутствует часть сосудистой оболочки, к которой крепится хрусталик глаза | Ресничное тело. | - сложности с фокусировкой на расположенных близко предметах. Причина - плохая работа аккомодационного аппарата. |
| Колобома хрусталика | Отсутствие части хрусталика (к примеру, выемка на нижнем или нижне-внутреннем крае) или его деформации. Встречается очень редко.Иногда сопровождается гетерохромией. | Хрусталик | - симптомы, схожие с астигматизмом. Возникают из-за нарушения светопреломляющей функции хрусталика. |
| Колобома зрительного нерва | Особенность этого вида - изменение формы соска нерва, его деформация. На патологическом участке можно наблюдать выемки. Встречается очень редко. Может быть одно- и двусторонним. | Зрительный нерв | - двоение изображения; - головокружения; - появление косоглазия; - схожие с астигматическими нарушения, развивающиеся по миопическому типу. |
| Колобома века | Дефект нижнего или верхнего века - заметный косметический дефект. | Чаще всего - верхнее веко одного глаза. | - вторичное воспаление конъюнктивы; - потертости, эрозии - из-за контакта оболочек и ресниц; - неполное смыкание век. |
В зависимости от того, какой отдел глаза поражен, специалисты выделяют несколько видов заболевания.
Помимо локализации при разделении на виды учитываются:
- Количество пораженных слоев.
В этом случае колобома бывает полной и частичной. - Распространенность.
В соответствии с этим признаком болезнь делится на одностороннюю (когда поражен только один глаз) и двустороннюю. - Расположение в структурах глаза.
Выделяются типичная и атипическая формы. - Степень поражения.
Изолированная колобома (поврежден участок одной оболочки) и комбинированная (болезнь затрагивает несколько слоев, например, симметричная двухсторонняя колобома сетчатки глаза и радужной оболочки).
Диагностика и лечение колобомы
Подтвердить диагноз «колобома» в некоторых случаях не составляет труда. Так, при колобоме зрачка или века специалисту достаточно визуального осмотра. Врач расспросит о том, выявлялось ли заболевание у родственников пациента, как давно появились первые симптомы, в чем они заключаются.
Для дальнейшего исследования могут потребоваться такие методы как ультразвук, офтальмо- и биомикроскопия, периметрия, оптическая томография, КТ или МРТ, анализ на гистологию.
Консервативные методы лечения применяются ограниченно. Они могут дать эффект только при использовании у новорожденных: витаминизация, кератопротекторы и антисептические средства могут улучшить ситуацию, но косметический эффект останется.
В большинстве случаев для лечения колобомы показано хирургическое вмешательство. Врачи могут «сшить» края щели, вживить коллагеновую вставку, которая будет служить каркасом, провести коагуляцию некоторых участков. Эти методики применяются при ухудшениях состояния пациента, угрозах потерять зрение. Если же речь идет только о косметическом дефекте зрачка, хирургические операции не показаны.
Расщелины лица
Расщелины лица формируются в раннем внутриутробном периоде вследствие спонтанных мутаций, провоцируемых биологическими, химическими или физическими тератогенными факторами. Помимо эстетического дефекта внешности, у ребенка возникают множественные расстройства питания, речи, дыхания. Диагностика выполняется антенатально на УЗИ, постнатально с помощью КТ лицевой части черепа, орофарингоскопии, для уточнения причины патологии назначаются кордоцентез, амниоцентез, генетическое тестирование. Лечение расщелины лица предполагает разные типы пластических операций, помощь ортодонтов, логопедов, реабилитологов.
МКБ-10

Общие сведения
Частота встречаемости аномалий формирования лица — 0,6-1,6 случаев на 1000 новорожденных. Патология составляет 13% среди всех врожденных пороков, занимает среди них 2-3 место по распространенности. За последние 40 лет число больных возросло в среднем в 1,5-2 раза, что вызвано нарастанием числа тератогенных влияний, улучшением качества медицинской помощи, в результате чего такие дети имеют значительно большую ожидаемую продолжительность жизни. Расщелины лица — не только медицинская, но и социальная проблема, которая требует комплексных лечебных мероприятий.
Причины
Согласно схеме, предложенной в 1991 году белорусским генетиком Г.И. Лазюком с соавторами, в развитии врожденных аномалий строения лица участвуют экзогенные тератогенные влияния, эндогенные воздействия, которые включают мутации генетического материала и различные заболевания родителей. При комбинации этих факторов нарушается процесс эмбриогенеза, у плода деформируется лицевой скелет.
Точечные изменения генетического кода считаются самым частым этиологическим фактором, встречаются у 92-93% детей, имеющих расщелины. Оставшиеся 7-8% составляют грубые хромосомные аберрации, в том числе при синдромах Патау, Эдвардса, синдроме кошачьего крика. К возможным причинам повреждений генетического материала, сопровождающихся формированием расщелин лица, относят следующие:
- Повреждение половых клеток. Биологическая неполноценность родительского материала при зачатии связана с неправильным образом жизни (курение, алкоголь, наркотические вещества), воздействием профессиональных вредностей, факторов экологии. Чем старше родители, тем выше риск формирования расщелины у ребенка.
- Особенности течения беременности. Значимыми предпосылками порока развития являются: неправильное положение плода, опухоли матки, многоплодная беременность. Эти факторы мешают нормальному формированию скелета ребенка. Травмы живота в первые месяцы гестации также служат фактором риска.
- Экстрагенитальные болезни матери. Риск развития врожденных пороков повышается, если у беременной есть эндокринные нарушения: сахарный диабет, гипотиреоз, гиперкортицизм. Костные аномалии могут провоцироваться анемией у материи и сопутствующей ей хронической внутриутробной гипоксией на ранних сроках гестации.
- Химические факторы. Сильное тератогенное влияние оказывают яды (бензин, формальдегид, соли тяжелых металлов), лекарственные средства - цитостатики, кортикостероидные гормоны, избыточное потребление витамина А. Вероятность образования расщелины лица увеличивается, если беременная женщина страдает хроническим алкоголизмом, токсикоманией, наркоманией.
- Биологические факторы. В группе риска находятся дети, матери которых во время беременности переболели краснухой, корью, ветряной оспой либо являются носителями цитомегаловируса, вируса простого герпеса. Изредка нарушения эмбриогенеза провоцируются инфицированием бактериями, простейшими.
Патогенез
Комбинированные поражения лица, неба и верхней губы возникают у зародыша со 2 по 7 неделю внутриутробного развития. В это время закладываются зачатки будущего лицевого черепа, лобные, верхнечелюстные, нижнечелюстные бугры. Если тератогенный фактор влияет на раннем сроке беременности, возможны массивные расщелины костей, мягких тканей, вплоть до несовместимых с жизнью состояний.
После 7 недели основы челюстно-лицевой зоны сформированы, поэтому влияние экзогенных факторов провоцирует не такие тяжелые поражения. На этом этапе у эмбриона появляются изолированные расщелины неба. Опасный срок продолжается до 10-11 недели внутриутробного развития — к этому времени лицевой череп ребенка уже сформирован, поэтому тератогенные факторы, влияющие в более поздние стадии гестации, не вызывают костных аномалий.
Классификация
С учётом этиологического фактора выделяют наследственные, экзогенные, мультифакториальные аномалии, однако в 25% случаев точные причины установить не удается. По степени тяжести бывают грубые летальные и нелетальные пороки. В практической педиатрии, как правило, используется классификация по анатомической локализации дефектов строения лицевого черепа, согласно которой выделяют:
- Собственно расщелины лица: срединная, косая (колобома), поперечная (макростома).
- Расщелины верхней губы: врожденная скрытая, врожденная неполная (с/без деформации носа), врожденная полная.
- Расщелины неба: скрытые, полные, неполные.
Симптомы
Признаки расщелины лица выявляются сразу после рождения ребенка. Анатомические нарушения строения лицевого скелета различны и зависят от вида патологии. Наиболее типичные проявления аномалий развития лица у новорожденных: явное или скрытое расщепление с укорочением верхней губы, деформация кожно-хрящевого отдела носа, расщепление и укорочение неба.
Изменения лицевого скелета сопровождаются множественными функциональными расстройствами у новорожденного. Повреждения носа обуславливают нарушения дыхания, из-за деформации губ и отсутствия герметичности полости рта ребенок не может сосать материнскую грудь. Дефекты неба способствуют забрасыванию молока в верхние, а затем и нижние дыхательные пути.
Осложнения
При наличии расщелины лица непрогретый воздух вследствие ротового дыхания попадает в дыхательные пути, провоцируя назофарингиты, ларингиты, бронхиты. Воспаление носоглотки в 75% случаев осложняется евстахиитом и отитом, что приводит к стойкому снижению слуха у младенца. Аспирация пищи при сосании вызывает тяжелые пневмонии. Вследствие несостоятельности артикуляционного аппарата у ребенка отмечается слабый тихий крик, замедляется речевое развитие, развивается открытая ринолалия.
Большую опасность представляют нарушения потребления молока/смесей, из-за чего младенец недополучает нутриенты, отстает в физическом развитии, страдает от гиповитаминозов. По мере взросления беспокоят проблемы с прорезыванием зубов, аномалии прикуса, нарушения жевания. Вторичные функциональные осложнения появляются в раннем детстве и у дошкольников. Они включают снижение иммунитета, психоневротические проявления.
Диагностика
Врожденные аномалии лица в основном определяются еще в антенатальном периоде при плановом УЗ-скрининге беременной. При этом врач оценивает размеры и глубину поражения, чтобы заранее спланировать ход реконструктивной операции, скорректировать протокол ведения родов. При подозрении на сочетанные нарушения проводится амниоцентез, кордоцентез с последующей генетической диагностикой. В постнатальном периоде назначается полный комплекс обследований:
- КТ лицевого черепа. Наиболее информативный метод для выявления деформаций костей и мягких тканей, определения степени их выраженности и глубины дефекта. Томография показывает сопутствующие патологии ЛОР-органов, которые зачастую сопровождают расщелины лица.
- Риноскопия. Детальный осмотр носовой полости проводится для диагностики незаращения неба, искривления носовой перегородки, деформации хрящевой части наружного носа. При исследовании врач исключает атрезию хоан, которая может возникать при комбинированном варианте порока.
- Орофарингоскопия. Обследование начинается с тщательного осмотра ротовой полости, в ходе которого педиатр диагностирует расщелины мягкого и твердого неба, деформацию альвеолярных отростков верхней челюсти. Затем врач изучает состояние глотки и миндалин.
- Телерентгенография. Специальный метод диагностики рекомендован перед ортодонтической коррекцией. Он дает стоматологу детальную информацию про аномалии развития зубочелюстного ряда, показывает состояние прикуса, выявляет воспаления пародонта и другие осложнения. В комплексном обследовании метод дополняется ортопантомографией.
Лечение расщелин лица
Коррекция аномалий лицевого скелета представляет сложною задачу, требует участия неонатолога, ортодонта, челюстно-лицевого хирурга. При выявлении показаний к лечению новорожденного привлекают нейрохирурга, отоларинголога, генетика. На первом этапе (0-1 мес. жизни) обеспечивается тщательная диагностика порока и планирование операции, назначается поддерживающая терапия, подбирается оптимальный метод вскармливания. Комплексное лечение включает следующие этапы:
- В грудном возрасте (1-12 месяцев) выполняют начальную ортодонтическую коррекцию съемными или несъемными начелюстными аппаратами. Хирургическая помощь включает первичную хейлопластику, хейлоринопластику, первый этап двухэтапной уранопластики. В этом периоде хирурги также проводят первичную коррекцию колобомы, макростомии.
- В период раннего детства (1-3 года) продолжается ортодонтическое лечение, начинается логопедическое обучение ребенка для правильного формирования речи. Производится второй этап уранопластики, краниопластика, восстановление назоорбитальной области. При необходимости осуществляются костно-пластические реконструкции, дистракционный остеосинтез.
- В дошкольном возрасте (3-7 лет) обеспечиваются повторные реконструктивно-восстановительные вмешательства, чтобы окончательно устранить видимый дефект, достичь максимально возможного эстетического результата. Проводятся речеулучшающие и слухоулучшающие операции. Для исправления прикуса используется различная ортодонтическая аппаратура.
- В школьном возрасте выполняется остеопластика альвеолярного отростка для формирования правильного постоянного прикуса, зубы выравниваются брекет-системой. Повторную открытую ринопластику делают в подростковом периоде для улучшения симметричности и эстетики лица. По показаниям пациенту рекомендуют другие эстетические пластические операции.
Основной массив реабилитационных мероприятий планируется на ранний детский и дошкольный возраст, чтобы к моменту поступления в школу ребенок был максимально адаптирован, не имел грубых деформаций внешности. Родителям необходимо настроиться на продолжительное лечение, поскольку среднее число действий при расщелинах составляет около 4-7 операций, до 65 приемов у ортодонта, до 60 посещений детского психолога.
Прогноз и профилактика
Результаты лечения определяются локализацией расщелины, степенью повреждения артикуляционного аппарата, своевременностью проведения операции. В дальнейшем ребенку могут потребоваться услуги логопеда, психолога, реабилитолога. Профилактика заболевания включает медико-генетическое консультирование, исключение влияния тератогенных факторов при беременности, соблюдение родителями здорового образа жизни при планировании зачатия.
1. К вопросу детализации классификаций врожденной расщелины верхней губы и неба. Ю.С. Рогожина, С.И. Блохина// Проблемы стоматологии. — 2019.
2. Врожденные пороки развития лица, врожденные расщелины верхней губы и неба у детей (методические рекомендации)/ О.Ю. Ершова, Е.В. Меньшикова. — 2016.
3. Ортодонтия: учебное пособие для студентов стоматологического факультета, врачей-ортодонтов, врачей-интернов/ В.И. Куцевляк, А.В. Самсонов. - 2016.
4. Врожденные пороки развития челюстно-лицевой области у детей/ А.К. Корсак, Т.Н. Терехова, А.Н. Кушнер. — 2005.
Колобома
Колобома - это полиэтиологическое заболевание, для которого характерно изолированное или комбинированное расщепление радужки, сетчатки, сосудистой оболочки, зрительного нерва или века. Общими клиническими проявлениями всех форм колобомы являются снижение остроты зрения, чувство боли в глазах, астенопические жалобы. Специфическая диагностика зависит от типа поражения и может включать в себя наружный осмотр, визометрию, офтальмоскопию, биомикроскопию, УЗИ в В-режиме, КТ, МРТ. Консервативных методов лечения колобомы не разработано. Хирургическая тактика зависит от формы заболевания и может включать в себя перитомию, коллагенопластику, лазерокоагуляцию, витрэктомию.
Колобома - это приобретенная или врожденная патология органа зрения, которая проявляется полным или частичным расщеплением его структур. Термин колобома был введен Вальтером в 1821 году, в переводе с греческого значит «отсутствующая часть». Частота колобомы в популяции составляет около 0,5-0,7 на 10000 новорожденных. Согласно статистическим данным, среди всех форм больше всего распространена колобома радужки (1:6000). Патология с одинаковой частотой встречается среди лиц мужского и женского пола. Врожденное расщепление сосудистой оболочки является одной из причин слепоты и нарушения зрения в детском возрасте. Заболевание наиболее часто встречается в Китае (7,5:10000), США (2,6:10000) и Франции (1,4:10000).
Врожденная колобома является генетически детерминированным заболеванием с преимущественно аутосомно-доминантным типом наследования. Вариабельные мутации гена РАХ6 ассоциированы с большим количеством пороков развития глаз, в том числе и колобомой. В тоже время доказано, что аномалии метилирования ДНК отдельных гистонов под действием факторов внешней среды приводят к эпигенетическим изменениям, которые также могут быть триггером данной патологии. Расщепление структур глазного яблока является следствием нарушения закрытия зародышевой щели на 4-5 неделе эмбрионального развития. Причиной врожденного поражения может быть инфицирование матери цитомегаловирусом на ранних сроках гестации.
В отличие от других форм, колобома радужной оболочки может наследоваться как по аутосомно-доминантному, так и по аутосомно-рецессивному типу. Аутосомно-доминантный тип ассоциирован с поражением гена РАХ6 с локализацией в коротком плече 11 хромосомы. Тип мутированного гена при аутосомно-рецессивном пути передачи не установлен. При возникновении делеции 24-DEL, NT1353 расщепление радужки, как правило, сочетается с микрофтальмией. На развитие приобретенной формы влияют такие факторы, как злоупотребление спиртными напитками и гормональный дисбаланс. Иридоретинальная колобома формируется при мутации гена SHH длинного плеча 7 хромосомы, хориоретинальная - при поражении гена GDF6 8q22 или РАХ6 11р13. Причиной комбинированного расщепления диска зрительного нерва, сетчатки и хориоидеи является мутация гена SHH с локализацией на хромосоме 7q36.
Колобома век приобретенного генеза зачастую возникает вследствие некроза тканей или образования рубцов при травматических повреждениях. Провоцировать данную патологию также могут послеоперационные дефекты на фоне иридэктомии при патологических новообразованиях радужки.
Симптомы колобомы
С клинической точки зрения в офтальмологии выделяют колобому радужки, сетчатки, сосудистой оболочки, диска зрительного нерва, хрусталика и век. При вовлечении в патологический процесс всех вышеуказанных структур развивается полная колобома, при менее обширном поражении - частичная. Расщепление может быть односторонним или поражать оба глаза. При типичной форме дефект локализируется в нижненазальном квадранте, что обусловлено топографией щели глазного канала. Атипической называют колобому с локализацией в других отделах органа зрения. Клинические проявления зависят от формы заболевания.
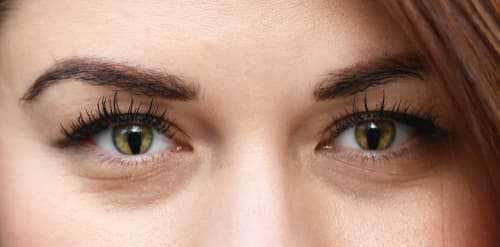
При колобоме радужки образуется выраженный косметический дефект, напоминающий грушу или замочную скважину. На фоне нормального или незначительного снижения остроты зрения пациенты неспособны регулировать объем поступления лучей света к сетчатке. Результатом является патология световосприятия. При поражении всей толщи сосудистой оболочки присоединяется симптоматика скотомы (появление участков затемнения перед глазами). Двухсторонняя колобома приводит к развитию нистагма. Заболевание может протекать изолированно или быть одним из проявлений Чардж или Экарди синдромов.
Степень снижения остроты зрения при колобоме зрительного нерва зависит от объема поражения. При изолированном дефекте зрение может соответствовать норме, в случае сочетания с микрофтальмом или расщеплением других структур глазного яблока возможна полная слепота. Большинство пациентов отмечают у себя двоение перед глазами, головокружение, нарушение бинокулярного зрения в виде страбизма. Заболевание часто сопровождается развитием клинической картины астигматизма по миопическому типу (снижение остроты зрения, искажение изображения перед глазами). Врожденная форма патологии часто сочетается с синдромами Гольденхара, Дауна и Эдвардса, а также эпидермальным невусом.
Для изолированной колобомы сетчатки характерно латентное течение. Пациенты предъявляют жалобы только при развитии вторичных осложнений в виде разрывов, сопровождающихся отслоением сетчатки. Длительное существование патологии приводит к формированию скотом. Аномалия клинически проявляется участками затемнения перед глазами. При расщеплении хрусталика нарушается его преломляющая функция, что становится причиной формирования зон с разным светопреломлением. Клинически заболевание проявляется симптоматикой астигматизма. Колобома век, как правило, сопровождается неполным смыканием, травмированием конъюнктивы ресницами, развитием эрозивных дефектов. Данная патология зачастую осложняется вторичным конъюнктивитом.
Диагностика колобомы
Методы диагностики колобомы зависят от формы заболевания. Колобома радужной оболочки характеризуется появлением дефекта в форме груши или замочной скважины, которые визуализируются при наружном осмотре. При проведении ультразвуковой биомикроскопии врожденная форма в большинстве случаев сопровождается гипоплазией цилиарного тела. Цилиарные отростки имеют меньшую длину и большую ширину по сравнению с нормой. Хаотичность волокон и нечеткость структуры цинновой связки указывает на ее недоразвитие. Степень снижения остроты зрения определяется методом визиометрии.
При колобоме зрительного нерва методом офтальмоскопии определяется незначительное увеличение его диаметра. Визуализируются округлые светлые углубления с четко ограниченными краями. Метод ультразвукового сканирования в В-режиме или компьютерная томография (КТ) позволяют обнаружить глубокие дефекты на заднем полюсе глазного яблока. В ряде случаев при магнитно-резонансной томографии (МРТ) определяется гипоплазия внутричерепного отдела зрительного нерва. В возрасте после 20 лет часто образуется регматогенная отслойка сетчатки. При прогрессировании у пациентов с патологическими углублениями в диске зрительного нерва определяются МРТ признаки макулярного отека, что зачастую приводит к разрыву и отслойке сетчатки. Гистологическое исследование позволяет выявить концентрически ориентированные волокна гладкой мускулатуры.
Колобома сосудистой оболочки при офтальмоскопии представляет собой образование белого цвета с фестончатыми краями. Как правило, дефект локализируется в нижних отделах глазного дна. При визометрии определяется миопия, степень которой зависит от объема поражения. При биомикроскопии колобома хрусталика имеет вид расщепления, которое расположено в нижневнутреннем квадранте. Прогрессирование патологии приводит к выраженной деформации экватора хрусталика.
Лечение колобомы
Тактика лечения колобомы зависит от формы и обширности поражения. При незначительном расщеплении радужки и отсутствии нарушений функции органа зрения оперативное вмешательство не показано. При снижении остроты зрения необходимо выполнить перитомию с последующим сшиванием краев радужной оболочки. С целью предупреждения дальнейшего прогрессирования колобомы рекомендовано поведение коллагенопластики. Целью оперативного вмешательства является формирование коллагенового каркаса, который препятствует прогрессированию колобомы.
При поражении зрительного нерва лазерокоагуляция показана только пациентам с формированием субретинальной неоваскулярной мембраны. Витрэктомия с последующей лазерной коагуляцией сетчатки рекомендована при снижении остроты зрения до 0,3 диоптрий с сопутствующей макулярной отслойкой сетчатки. Помимо этого, методом лечения хориоретинальной колобомы является эндодренирование сквозь промежуточную мембрану с дальнейшей лазерной фотокоагуляцией сетчатой оболочки вокруг краевой зоны. При выраженной колобоме хрусталика необходимо его удаление с последующей имплантацией интраокулярной линзы. В свою очередь, дефект в виде расщепления века устраняется при помощи блефаропластики.
Прогноз и профилактика
Специфических превентивных мер для предупреждения развития колобомы не разработано. Для профилактики нарушений световосприятия при колобоме радужки рекомендовано использование сетчатых очков или затемненных контактных линз с прозрачным центром. Пациентам с первичными проявлениями данной патологии необходимо два раза в год проходить осмотр у офтальмолога с обязательным проведением визометрии, биомикроскопии и офтальмоскопии глазного дна. При незначительном расщеплении структур глазного яблока прогноз для жизни и трудоспособности благоприятный. Обширное поражение может осложняться тотальным снижением остроты зрения вплоть до полной слепоты, что приводит к инвалидизации пациента.
Синдром Тричера-Коллинза ( Мандибулофациальный дизостоз , Синдром Тричера Коллинза-Франческетти , Челюстно-лицевой дизостоз )
Синдром Тричера Коллинза - это генетическая (иногда наследственная) болезнь, сопровождающаяся деформациями костей и мягких тканей лица. К симптомам относятся грубые дефекты строения лица: антимонголоидный разрез глаз, вырезки ткани век (колобомы), уменьшенные размеры челюсти и скул, гипоплазия и аномалии структур уха, расщелина или арковидная форма неба, увеличенные размеры ротовой щели и языка, слаборазвитые кости лица. Диагноз устанавливается по данным клинического осмотра, биогенетического теста и семейного анамнеза. Лечение симптоматическое, направлено на улучшение слуха, устранение жизнеугрожающих деформаций и косметических дефектов хирургическим способом.
У синдрома Тричера Коллинза есть несколько синонимов: челюстно-лицевой дизостоз, синдром Тричера Коллинза-Франческетти, мандибулофациальный дизостоз. Впервые патологию описал офтальмолог из Великобритании Эдвард Тричер Коллинз в 1900 году, поэтому наиболее распространено название, соответствующее его имени. Обширный обзор заболевания был сделан в 1949 году европейскими исследователями Э. Франческетти и Д. Клейном. В настоящее время понятие «синдром Тричера Коллинза» более распространено в Великобритании и США, а термин «синдром Франческетти-Клейна» чаще используется в странах Европы. Эпидемиология болезни составляет 1:50 000. Среди мальчиков и девочек заболеваемость одинакова.
При мутациях в гене TCOF1 тип наследования синдрома аутосомно-доминантный с показателем пенетрантности 90%. Это означает, что при мутации в одной хромосоме из пары вероятность проявления болезни очень высока. У больного родителя риск рождения ребенка с синдромом Тричера Коллинза составляет 50%. Возможна наследственная передача дефекта и спорадические генетические изменения (новые мутации). Экспрессивность мутации переменная - в пределах одной семьи вероятно как ослабление, так и усиление симптомов заболевания у последующих поколений. При дефектах генов POLR1C и POLR1D наследование происходит по аутосомно-рецессивному типу. В парах, где родитель имеет синдром, вероятность рождения больного малыша составляет 25%.
Пятая хромосома ответственна за правильное формирование скелета в период внутриутробного развития. Локализованный в ней ген TCOF1 кодирует структуру и синтез ядерного транспортного белка Treacle. Данный протеин экспрессируется в большинстве тканей организма в эмбриональном и постэмбриональном периоде, участвует в переносе генетической информации с ДНК на РНК.
В основе синдрома чаще всего лежит нонсенс-мутация, приводящая к образованию преждевременного кодона терминации и развитию гаплонедостаточности - дефицита белка, необходимого для нормального формирования лицевой части черепа. Здоровый ген обеспечивает организм белком Treacle наполовину, но такого количества недостаточно для правильного развития лицевых структур. При изменениях в генах POLR1D и POLR1C процесс транскрипции ДНК нарушается из-за недостаточности фермента-катализатора ДНК-зависимой РНК-полимеразы. Клинические проявления синдрома такие же, как и при первичной недостаточности Treacle-протеина.
У больных наблюдаются аномалии в строении лица. Распространенным признаком, встречающимся в 80% случаев, является двусторонняя симметричная гипоплазия скуловых костей, инфраорбитального края и нижней челюсти. Внешне это проявляется своеобразным уплощенным бесформенным лицом, на котором выделяется нос, а остальные части «утоплены» в мягких тканях. Деформация челюсти обуславливает нарушение прикуса, формирование ортогнатии (постоянно приоткрытого рта). 89% больных имеют ограниченную возможность открывания рта и антимонголоидный тип разреза глаз с заметным опущением внешнего уголка. Данные особенности частично обусловлены патологическим строением височно-нижнечелюстного сустава.
У 69% пациентов определяется колобома радужки и нижних век в промежутке между средней и внешней третью, чаще она имеет треугольную форму. Ресницы на внешнем крае нижнего века отсутствуют. Небо арковидной формы, иногда сформирована расщелина (у 28% больных). Аномалии наружного уха представлены недоразвитием или полным отсутствием ушной раковины (микротией, анотией), атрезией наружного слухового прохода и деформацией слуховых косточек. Зачастую пациенты имеют кондуктивную тугоухость. В редких случаях диагностируется энхондрома, предкозелковые фистулы, аномальное строение сердца и позвоночника.
Микрогнатия и стеноз верхних дыхательных путей уже в первые годы жизни могут спровоцировать проблемы при приеме пищи и трудности дыхания вплоть до удушья. Своевременная диагностика заболевания позволяет спрогнозировать эти осложнения и предпринять меры по их предупреждению. Как правило, пациенты не имеют врожденных интеллектуальных расстройств, но при отсутствии коррекции нарушений слуха становится невозможным правильное формирование речи и обучение в обычных условиях. Дети начинают отставать в умственном развитии от сверстников, имеют задержку психического развития различной степени выраженности. В связи с наличием дефектов внешности и негативным отношением окружающих больные всех возрастов относятся к группе риска по возникновению депрессии, ипохондрии, тревожности и иных невротических расстройств.
Диагноз может быть установлен во время беременности или сразу после рождения. Обследование показано женщинам из группы риска и детям с врожденными лицевыми деформациями. В процессе диагностики принимают участие врачи-генетики и педиатры. Синдром Тричера-Коллинза необходимо дифференцировать с другими генетическими заболеваниями, при которых существует деформация лицевой части черепа, например, с синдромом Нагера и синдромом Гольденхара. Используются следующие методы:
Дополнительно назначаются обследования, позволяющие своевременно обнаружить жизнеугрожающие состояния, оценить степень деформации костей черепа. Определяется эффективность кормления ребенка, уровень насыщения гемоглобина кислородом, ритмичность и глубина дыхания. Для диагностики сохранности слуха на 5-6 день жизни проводится аудиологическое тестирование. Инструментальная диагностика включает рентгенографию черепа, КТ и МРТ головного мозга.
Лечение синдрома
Специфической терапии не существует. Лечение нацелено на устранение симптомов и последствий заболевания, предполагает проведение хирургических операций и реабилитационных мероприятий. Объем процедур и сроки их выполнения устанавливаются индивидуально с учетом наличия угрозы для жизни больного, противопоказаний и рисков, связанных с оперативным вмешательством. Общая схема лечения включает:
- Восстановление глотания и дыхания. При развитии респираторного дистресс-синдрома осуществляется трахеостомия, дистракция подвижной челюсти, неинвазивная вентиляция легких. При невозможности потребления пищи устанавливается гастростома.
- Восстановление слуха. Деформация наружного и среднего уха устраняется хирургическим путем, но потеря слуха чаще обусловлена повреждением слуховых мелких косточек, поэтому оперативные вмешательства с целью устранения тугоухости неэффективны. Предпочтительна реабилитация слуховыми аппаратами.
- Устранение внешних дефектов. Деформации корректируются методами пластической и нижнечелюстно-лицевой хирургии. Применяется липоскульптурирование, хирургическая дистракция костей, установка трансплантатов и хирургическое восстановление неба.
Комплексное лечение и реабилитация значительно улучшают качество жизни больных. При легкой и умеренной выраженности синдрома прогноз благоприятный. Профилактика затруднена, поскольку заболевание является генетическим, а мутации способны возникать спонтанно. Супружеским парам, в которых один родитель болен, необходимо медико-генетическое консультирование и перинатальная диагностика синдрома на ранних сроках беременности. Для снижения риска вынашивания больного ребенка рекомендуется процедура экстракорпорального оплодотворения с предварительным отбором генетически здоровых эмбрионов.
1. Медицинская и клиническая генетика для стоматологов: учебник для вузов. Под ред. О. О. Янушевича - 2009.
2. Особенности стоматологической патологии при некоторых наследственных заболеваниях / Шишкова О.В., Максимова Ю.В. // Медицина и образование в Сибири - 2007 - №3.
Что такое колобома: диагностика и лечение
Колобома - это врожденная (присутствующая при рождении) аномалия глаза. Колобомы - это недостающие кусочки ткани, которые могут выглядеть как зазоры или вырезы. Когда колобома поражает радужную оболочку глаза, она проявляется в виде замочной скважины или кошачьего глаза в зрачке. По оценкам, от колобомы страдает каждый десятитысячный человек. Это заболевание не всегда меняет внешний вид глаза и не во всех случаях влияет на зрение человека. Поэтому считается, что у некоторых людей оно, скорее всего, не диагностируется.

Колобомы могут включать в себя одну или несколько структур глаза, в том числе:
Симптомы колобомы могут включать в себя:
- отсутствие кусочков ткани в одной или нескольких структурах, образующих глаз (радужка, сосудистая оболочка глаза, зрительный нерв или сетчатка глаза);
- замочная скважина или появление зрачка с кошачьим глазом (когда колобома поражает радужную оболочку глаза);
- пропавшие кусочки ткани на одном или обоих глазах;
- чувствительность к свету;
- Заметный дефект или выемка в веке(в результате колобомы века).
Важно различать колобомы глазного яблока и промежутки, которые образуются в веках (которые также называются колобомами). Колобомы век возникают из-за отклонений во время развития плода. - нарушения зрения(в зависимости от размера и расположения колобомы);
- дефект поля (потеря зрения в определенной части поля зрения - например, в верхней части поля зрения). Обычно это результат колобом, которые поражают часть сетчатки;
- снижение зрения, связанное с невозможностью коррекции зрения с помощью корректирующих линз; обычно это результат воздействия колобом на зрительный нерв.
Колобомы, как правило, не ухудшают зрения, если только они не поражают сетчатку или зрительный нерв.
Колобомы часто сопутствуют другим заболеваниям глаз, в том числе таким заболеваниям, как:
- катаракта (помутнение хрусталика),
- близорукость,
- нистагм (непроизвольные движения глаз),
- глаукома (увеличенное давление внутри глаза, которое может повредить зрительный нерв),
- микрофтальмия (одно или оба глазных яблока аномально малы), .
Колобомы могут возникать сами по себе (так называемые несиндромные или изолированные колобомы) или они могут быть частью синдрома, поражающего другие органы и ткани (так называемые синдромные колобомы).
Причиной колобомы является аномальное развитие глаза в матке (в утробе), в частности, во второй месяц развития плода. Дефект является результатом неправильного закрытия шва (называемого фиссурой зрительного нерва) во время развития плода. При развитии плода зрительная трещина образует нижнюю часть глазного яблока, поэтому в нижней части глаза проступают колобомы. Точное строение глаза, которое в конечном итоге подвергнется воздействию колобомы, зависит от того, какой участок фиссуры зрительного нерва не закрылся должным образом.
Генетическое воздействие
Изменения в генах, влияющие на раннее развитие глаза, могут быть связаны с колобомой. Фактически, по данным Дома генной инженерии, многие из этих генов были идентифицированы, но только у очень небольшого числа людей. Необходимы дополнительные исследования для окончательной связи специфических генетических дефектов с развитием колобомы.
Экологические факторы
Экологические факторы (такие как употребление алкоголя матерями во время беременности) могут повысить риск возникновения колобомы у плода
Диагноз
Офтальмолог будет использовать инструмент, называемый офтальмоскопом, для обследования глаз младенца при подозрении на колобому. По мере того, как ребенок становится старше, могут быть проведены другие тесты для измерения масштаба проблемы. Эти тесты могут включать тесты остроты зрения (для измерения наличия и тяжести потери зрения).
Лечение
Хотя в настоящее время нет лекарства от колобомы, существуют некоторые варианты лечения.
Для людей с нарушениями зрения лечение направлено на то, чтобы помочь ребенку адаптироваться. Некоторые из сопутствующих заболеваний колобомы (например,катаракта) могут быть вылечены. Лечение катаракты, глаукомы или других сопутствующих заболеваний глаз поможет улучшить состояние зрения, но не решит проблему полностью.
Проблемы со зрением
Лечение для тех, у кого проблемы со зрением из-за колобомы, может включать в себя:
- использование очков или линз во избежание ухудшения зрения и услуги по реабилитации зрения (услуги, которые помогают людям с плохим зрением жить как можно более независимо и поддерживать высокое качество жизни)
- лечение «ленивого глаза» у детей;
- лечение микрофтальмии у детей
- лечение сопутствующих глазных заболеваний, таких как катаракта, глаукома или отслоение сетчатки.
Профилактическое лечение
Для предотвращения «ленивого глаза» (для ребенка с колобомой только на одном глазу) лечение может включать в себя терапию незатронутого глаза, например:
- использование повязки,
- специальные глазные капли,
- очки.
По данным Американской академии офтальмологии, «иногда это лечение (глазная повязка, глазные капли или очки для незатронутого глаза) может улучшить зрение в глазах даже при сильных колобомах».
Если у Вас есть ребенок с диагнозом колобома, вы, вероятно, знаете,что у вашего ребенка может быть нормальное зрение или потеря зрения, но вы все равно можете беспокоиться о том, что ждет его в будущем. В этом могут помочь знания о колобоме, а также ознакомление с текущими медицинскими исследованиями, обращение за помощью к другим родителям и участие в системе поддержки (например, индивидуальная или групповая терапия). Одним из лучших способов помочь ребенку, страдающему колобомой, является регулярное посещение офтальмологической клиники и офтальмологическое обследование. Регулярное посещение офтальмологической клиники и офтальмологические обследования повысят шансы ребенка на раннее выявление новых проблем. Поиск любых проблем или обнаружение ухудшения зрения ребенка на ранней стадии приведет к наилучшему долгосрочному результату.
Читайте также: