Атаксия-телеангиэктазия. Синдром Вискотта—Олдрича.
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Краткое описание:
Иммуноглобулины класса Е (реагины) участвуют в развитии атопических аллергических реакций (бронхиальной астмы, ринита, крапивницы, атопического дерматита и др.). Они способны быстро присоединяться к поверхности тучных клеток и базофилов кожи и слизистых оболочек. Поэтому повторный контакт реагиновых IgE с антигеном (аллергеном) происходит на поверхности этих клеток, что приводит к высвобождению из них вазоактивных веществ (гистамина, серотонина, гепарина и др.) и развитию клинических проявлений реакции гиперчувствительности 1-го типа. Кроме того, IgE играют существенную роль в формировании антипаразитарного иммунитета к аскаридам, токсоплазмам, нематодам, эхинококкам, трихинеллам и другим паразитам, что обусловлено способностью IgE взаимодействовать с антигенами гельминтов. Поэтому повышение содержания в плазме крови общего IgE может свидетельствовать о наличии паразитарной инвазии.
Синонимы (rus): Иммуноглобулины класса Е.
Синонимы (eng): Immunoglobulin E, IgE, total (serum).
Метод исследования: Твердофазный хемилюминесцентный иммуноферментный анализ («сэндвич»-метод).
Подготовка к исследованию:
• Не принимать пищу в течение 2-3 часов перед сдачей анализа, можно пить чистую негазированную воду.
• Исключить физическое и эмоциональное перенапряжение в течение 30 минут до исследования.
• Не курить в течение 3 часов до исследования.
Единицы измерения: МЕ/мл (международная единица на миллилитр).
Тип биоматериала: Венозная кровь.
Тип пробирки: вакутейнер с активатором свертывания крови (цвет крышки: красный с желтым кольцом)
Цена услуги: 189 руб.
Срок выполнения: 3 рабочих дня, понедельник, четверг
Возраст Референсные значения
Новорожденные 0 - 1,5 МЕ/мл
Меньше 1 года 0 - 15 МЕ/мл
1-6 лет 0 - 60 МЕ/мл
6-10 лет 0 - 90 МЕ/мл
10-16 лет 0 - 200 МЕ/мл
Больше 16 лет 0 - 100 МЕ/мл
Причины повышения уровня суммарных IgE
• Паразитарные инвазии (аскаридоз, нематодоз кишечника, эхинококкоз, анкилостоматоз, амебиаз, синдром миграции личинок гельминтов).
• Аллергический бронхолегочный аспергиллез.
• Аллергические заболевания, обусловленные IgE-антителами: атопические (атопическая бронхиальная астма, аллергический ринит и синусит, атопический дерматит, лекарственная и пищевая аллергия) и анафилактические (анафилаксия, крапивница, ангионевротический отек).
• Иммунопатологические заболевания (IgE-миелома, узелковый периартериит, синдром гиперэозинофилии, дисплазия и аплазия тимуса (синдром Ди Джорджи), синдром Вискотта-Олдрича, гипер-IgE-синдром и возвратная пиодермия (синдром Джоба – Бакли), пузырчатка (синдром Неймана), реакция «трансплантат против хозяина».
Причины снижения уровня суммарных IgE
• Наследственная, сцепленная с полом или приобретенная гипогамма-глобулинемия.
• Атаксия-телеангиэктазия.
• Первичные или вторичные иммунодефициты
Атаксия-телеангиэктазия. Синдром Вискотта—Олдрича.
Атаксия-телеангиэктазия. Синдром Вискотта—Олдрича.
Внешние канцерогенные (ионизирующее излучение, некоторые химические вещества) и генетические факторы, предрасполагающие к повышению частоты хромосомных мутаций, одновременно предрасполагают и к возникновению новообразований. Это соответствует общепризнанной концепции клональной природы злокачественных опухолей и подтверждается высокой частотой онкологической патологии у больных с генетическими синдромами, в том числе кожными. Хотя каждый из приведенных здесь генодерматозов носит врожденный характер или проявляется в раннем детстве, многие из них не сопровождаются развитием злокачественных новообразований до наступления взрослого возраста.
Атаксия-телеангиэктазия (син.: синдром Луи-Бар) — редкое системное заболевание, характеризующееся сочетанием хромосомной нестабильности, мозжечковой атаксии, кожно-глазных телеангиэктазий, комбинированным иммунодефицитом и повышенным риском развития опухолей. Тип наследования аутосомно-рецессивный. Мугантный ген локализуется в 11-й хромосоме на участке q22-q23. У гетерозиготных носителей иногда отмечаются дефицит IgE и телеангиэктазий на коже и слизистых оболочках.
Атаксия-телеангиэктазия начинается в раннем детстве и проявляется мозжечковой атаксией (в 100% случаев), дрожанием головы и туловища, нарушением походки, интенционным тремором и хореоатетозом (в 90-100% случаев). Характерны нарушения движения глазных яблок (у 80-90% больных), нистагм (у 90-100%) и косоглазие, а также гипотония мышц, гипорефлексия, дизартрия. Дети отстают в умственном развитии.
В возрасте от 2 до 6 лет появляются телеангиэктазии на конъюнктиве, сгибательных поверхностях конечностей, слизистых оболочках мягкого и твердого нёба, склеродермия, прогерические изменения кожи и волос. Гистологические изменения в коже неспецифичны, отмечаются повышенное содержание пигмента в эпидермисе, расширение капилляров с периваскулярной инфильтрацией из нейтрофилов и лимфоцитов. Также описаны гирсутизм и acantosis nigricans.
Атаксия-телеангиэктазия сопровождается нарушением Т-и В-систем иммунитета, отсутствием сывороточных IgA, а иногда также IgG и IgE. Этим обусловлены хронические респираторные инфекции — синуситы и пневмонии (в 60-80% случаев).
Повышенная частота лейкозов и лимфом связана с хромосомной нестабильностью и другими нарушениями, например, повышенной чувствительностью к ионизирующей радиации. Недавние наблюдения указывают на 3-5-кратное повышение риска развития рака для гетерозиготных носителей гена атаксии-телеангиэктазии и на особенный риск развития рака молочной железы у женщин-носителей.
Диагноз атаксии-телеангиэктазии устанавливается на основании трех основных признаков: мозжечковой атаксии, телеангиэктазий и комбинированного иммунодефицита.
Дифференциальный диагноз атаксии-телеангиэктазии проводится с пигментной ксеродермой. пойкилодермией Ротмунда—Томсона, синдромом Блума, синдромом Коккейна, врожденным дискератозом.
Течение атаксии-телеангиэктазии прогрессирующее. Прогноз неблагоприятный вследствие тяжело протекающих легочных инфекций и повышенного риска злокачественных опухолей.
Лечение атаксии-телеангиэктазии в применении неспецифической иммунотерапии и общеукрепляющих средств, при необходимости — антибиотикотерапии. Рентгенотерапия новообразований противопоказана.
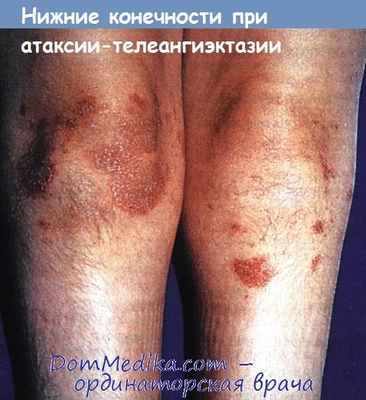
Гирсутизм нижних конечностей и экхимозы, развившиеся вторично вследствие атаксии и падений
Синдром Вискотта—Олдрича
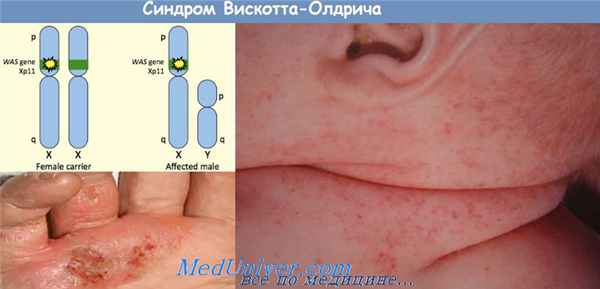
Синдром Вискотта—Олдрича — наследственный симптомокомплекс, характеризующийся тромбоцитопенической пурпурой, экземой и рецидивирующими инфекциями. Поражает мальчиков. Тип наследования рецессивный, связанный с Х-хромосомой. Установлено, что лимфоциты, нейтрофилы и тромбоциты больного изменяются под влиянием сильно гликозированной поверхности протеина сиалоформина (CD43), который не откладывается на других типах клеток. По-видимому, патологическое влияние измененного CD43 на клетки крови обусловлено повышенной чувствительностью этих клеток к протеолитическому расщеплению. Тромбоциты больных способны к нормальной агрегации, но их продукция и время существования сокращены, в результате чего развивается тромбоцитопения. В крови повышен уровень IgE и понижен уровень IgM. Содержание IgA и IgG в пределах нормы или повышено. Лимфоциты также аномальны, их чувствительность к антигенам in vitro снижена при сохранении чувствительности к митогенам. Предполагали, что первичным дефектом синдрома является дефицит синтеза CD43, однако выяснилось, что ген, контролирующий синтез этого протеина, находится в хромосоме 16, а не в Х-хромосоме, и, следовательно, его синтез не нарушен.
Патологический клинический комплекс синдрома вискотта - олдрича проявляется на первом году жизни классической триадой — иммунодефицитом, тромбоцитопенией и экземой.
Экзема при синдроме вискотта - олдрича развивается уже на 1 -й неделе жизни, локализуется на волосистой части головы, лице, в аногенитальной области, на сгибательных поверхностях конечностей и практически не отличается от атопической экземы, за исключением сочетания с пурпурой и возникновением интенсивных кровотечений из экскориаций. Кровотечения наиболее выражены на 6-м месяце жизни, возможны кишечные кровотечения, внутричерепные кровоизлияния. Дети склонны к бактериальным инфекциям (с развитием отита, пиодермии, менингита, септицемии), вирусным заболеваниям. Высока склонность к аутоиммунным процессам с развитием васкулитов, поражений почек (хронический гломерулонефрит), гемолитической анемии, артритов. Харакерны энтеропатии с большой потерей белка, бронхиальная астма, контактные уртикарные сыпи.
Большинство больных синдромом вискотта - олдрича умирают от инфекций или кровотечений в детстве, но у 5-12% в будущем развиваются злокачественные лимфопролиферативные заболевания, наиболее часты болезнь Ходжкина или острый лейкоз.
Лечение синдрома вискотта - олдрича: остановка кровотечений, переливание тромбоцитной массы, спленэктомия, антибактериальная терапия (у -глобулин, антибиотики и др.), трансплантация костного мозга.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
9. Атаксия-телеангиэктазия.
Атаксия-телеангиэктазия (Луи-Бар)— наследственное заболевание, при котором, как правило, тяжёлого иммунодефицита не возникает, поэтому больные в среднем живут до 30—40 лет. Наиболее постоянный признак — низкий уровень или отсутствие IgA — встречается примерно у 70% больных.
Причинойатаксии-телеангиэктазии является мутация гена, расположенного в 11-й хромосоме на участке q22-q23. Этот ген кодирует белок, участвующий в регуляции клеточного цикла. Однако патогенез заболевания до конца не изучен.
Помимо иммунодефицита развиваются следующие синдромы:
Недоразвитие ткани мозжечка проявляется нарушением координации движений (атаксией); нарушение походки развивается, как правило, с 4-летнего возраста и постепенно прогрессирует.
Телеангиэктазии — множественные очаги расширенных мелких сосудов конъюнктивы и кожи (обнаруживаются к концу первого года жизни).
Недоразвитие половых органов вследствие гипогонадизма (дефицита половых гормонов).
Раннее поседение волос.
Лечение.Этиотропного лечения не существует. Больным показано симптоматическое лечение: антибиотики, иммуноглобулины, общеукрепляющие средства.
10. Синдром Вискотта-Олдрича.
Наследственное заболевание, которое характеризуется сочетанием трех симптомов: кожной экземы, тромбоцитопении и иммунодефицита. Так как СВО характеризуется Х-сцепленным наследованием, болеют почти исключительно мальчики.
Признаки и симптомы:Тромбоцитопения проявляется возникновением синяков, подкожных кровоизлияний (петехий) и кровотечений – например, нередки кровавый понос, кровотечения из носа и десен. В большинстве случаев одним из симптомов является зудящая кожная сыпь, хотя у некоторых больных она может быть слабо выраженной или даже отсутствовать. Иммунодефицит связан как с нарушением функции В-лимфоцитов, так и с дефицитом Т-лимфоцитов, то есть речь идет о комбинированном иммунодефиците.
Диагностика:на основе клинических симптомов, результатов клинического анализа крови, микроскопического исследования мазка крови, а также измерения уровней различных иммуноглобулинов крови, которые изменены по сравнению с нормальными. Точное подтверждение диагноза может быть произведено на основе либо обнаружения мутации в соответствующем гене на Х-хромосоме больного.
Лечение:Для облегчения состояния больного могут применяться переливания компонентов крови. Так как тромбоциты разрушаются в селезенке, то иногда удаление селезенки может привести к улучшению состояния больного. Однако дети с удаленной селезенкой еще сильнее подвержены инфекциям. Кожная сыпь требует местного лечения и тщательного ухода за кожей.
11. Синдром Ди-Джорджи
Это иммунодефицитное заболевание, вызванное дефектом эмбриогенеза и состоящее в нарушении развития органов, дифференцирующихся из III, IV жаберных карманов.
Характеристика:отсутствие тимуса или резкое уменьшение его массы; отсутствием паращитовидных желез с явлениями гипокальциемии; пороками развития сердца и крупных сосудов; атрезией пищевода; дефектами развития лица. Генетические нарушения при синдроме Ди-Джорджи не выявлены. Все эти патологии могут наблюдаться в различных комбинациях и проявляются такими симптомами как деформация глаз (измененный разрез, большое расстояние между глазами из-за широкой переносицы), носа, рта («рыбий» рот), ушей. Отсутствие или недоразвитие тимуса – вилочковой железы – является причиной иммунодефицита с преимущественным поражением его клеточного звена (Т-лимфоциты) и сохранением функций гуморального иммунитета. Врожденные пороки сердца и аномалии дуги аорты являются причиной проявления таких признаков как цианоз – синюшность кожи и слизистых оболочек, слабость и головокружение по причине гипоксии тканей.
Диагностика.ОАК, ОАМ, б/х анализ крови с определением уровня кальция в крови для выявления гипокальциемии, указывающей на гипоплазию или аплазию паращитовидных желез. К необходимым инструментальным исследованиям относятся рентгенография, ЭКГ и УЗИ. На рентгеновских снимках грудной клетки определяются признаки аплазии или гипоплазии тимуса, также могут быть отмечены характерные для пороков сердца и дуги аорты признаки.
Лечение. При врожденных пороках сердца, если возможно, проводится их хирургическая коррекция, обычно на первом году жизни. В случае диагностирования гипотиреоза по причине гипоплазии или аплазии паращитовидных желез назначается лечение для устранения гипокальциемии и терапия, направленная на долговременную нормализацию в крови уровня кальция. В случае аплазии тимуса и при отсутствии угрожающих жизни состояний иногда проводится трансплантация фетального тимуса, что позволяет ликвидировать иммунную недостаточность. Могут быть назначены иммуноглобулины, переливание компонентов крови и иммуномодулирующие препараты. Важным компонентом лечения является профилактика и лечение вторичных инфекций, возникающих на фоне иммунодефицитного состояния.
Атаксия-телеангиэктазия (синдром Луи-Барр)
1. Атаксия-телеангиэктазия(синдром Луи-Барр)
Федеральное государственное бюджетное образовательное
учреждение высшего образования «Тюменский Государственный
Медицинский Университет» Министерства здравоохранения
Российской Федерации
(ГБОУ ВПО ТюмГМУ Минздрава России)
Кафедра детских болезней лечебного факультета с
курсом иммунологии и аллергологии - клиническая
Атаксия-телеангиэктазия(синдром Луи-Барр)
Проверила:
К.м.н,доцент кафедры
Сагитова А.С
Выполнила:
студентка 3 курса
педиатрического факультета
340 группы
Ханмурзаева Залина Муратовна
2. Содержание
1.Понятие о синдроме;
2.Патогенез;
3.Клиническая картина;
4.Диагностика;
5.Лечение;
6.Список используемой литературы.
3. Синдром Луи-Бар(атаксия телеангиэктазия)
Это редкое иммунодефицитное
нейродегенеративное аутосомно-рецессивное
заболевание, которое проявляет себя в виде
мозжечковой атаксии, телеангиэктазии кожи и
конъюнктивы глаз, недостаточностью Тклеточного звена иммунитета,вызывает
тяжелые формы паралича.
По статистике распространённость
заболевания приходится на одного человека
из сорока тысяч.
4. Атаксия характеризуется нарушениями координации движений, а телеангиэктазия — расширением кровеносных сосудов.
5. Патогенез
Генетические нарушения,
приводящие к развитию врожденной
нейроэктодермальной дисплазии:дефект
локализован на хромосоме 11q22-23 –ATM.
Белковый продукт данного гена
вовлекается в контроль клеточного
роста, распознавание клеткой
поврежденной ДНК,ее репарацию или
блокирование клеточного цикла в
клетках.
Увеличено количество циркулирующих Тклеток с преобладанием незрелых Тлимфоцитов.
Феномен слияния теломер в Т-лимфоцитах
причина хромосомной нестабильности клеток
превращения их в опухолевые.
Нарушение репарации ДНК
снижение устойчивости клеток к радиации и
повышение риска малигнизации.
7. Клиническая картина
1.Мозжечковая атаксия(на первом году жизни):дети с не
могут стоять и нормально передвигаться.
В более благоприятных случаях наблюдается шаткость
походки и тремор конечностей.
Помимо этого, неврологическая симптоматика выражается
в слабости мышц, дизартрии различной степени (невнятная
речь) и косоглазии.
2.Нарушения психики и замедление или полная остановка
в развитии (после десяти лет);
3.Изменение цвета кожи под воздействием
ультрафиолетовых лучей;
4.Расширение кровеносных сосудов в области внутренней
стороны коленей и локтей, на лице, в белках глаз;
8. Телеангиоэктазии
9. Телеангиэктазии
Могут включать появление
веснушек и пятен цвета кофе с
молоком, участков обесцвеченной
кожи.
Наличие гипо- и
гиперпигментаций делает кожные
симптомы синдрома Луи-Бар
схожими с клиникой
пойкилодермии. У многих больных
отмечается сухость кожи и участки
гиперкератоза.
Может наблюдаться гипертрихоз,
кожные элементы, напоминающие
акне или проявления псориаза.
10. Клиническая картина
5.Ранняя седина;
6.Повышенная чувствительность к рентгеновским лучам;
7.Тяжелые инфекции дыхательных путей, ушей, склонные
к рецидиву (у 80% больных):хронические риниты,
фарингиты, бронхиты, пневмонии, отиты, синуситы. Их
особенностями являются: стертость границ между
периодом обострения и ремиссии, скудность физикальных
данных, плохая чувствительность к антибактериальной
терапии и длительное течение.
8.Отсутствие рефлексов в мышцах глаз;
9.Аномальное развитие вилочковой железы, а в некоторых
случаях и ее полное отсутствие;
10.Лимфоцитопения (примерно 1/3 всех случаев);
11.Задержка полового развития или неполное развитие и
ранняя менопауза.
12.У 10-15%-лейкозы и лимфомы.
11. Почти у 50% детей отклонения в ЦНС:
Заторможенность, нарушения когнитивных
функций, особенно внимания и памяти,
умственное отставание.
Речевые расстройства: речь невнятная,
замедленная, с элементами скандирования.
Поражение подкорковых ядер проявляется
в виде гиперкинезов, навязчивого
гримасничанья.
При внешнем осмотре следует обратить
внимание на позу больного ребенка: голова
и плечи опущены, руки немного согнуты в
локтевых суставах и ротированы наружу.
Дети малоподвижны, с бедной мимикой,
вплоть до амимии.
Развиваются нарушения функций черепных
нервов, а именно: глазодвигательного,
отводящего, зрительного, тройничного,
лицевого, языкоглоточного.
12. Иммунные проявления:
Т-лимфопения сопровождается нарушением
функциональных свойств клеток в реакциях
бласттрансформации на антигены и митогены.
Гипоплазия тимуса, лимфатических узлов, селезенки.
Выработка специфических антител дефектна.
Более чем у половины детей имеются умеренные
изменения в иммунной системе или вовсе отсутствует
иммунодефицит.
У детей постепенно развиваются изменения в
иммунной системе, характерные для старческого
возраста.
13. Иммунные проявления:
Уникальная особенность иммунной системы больных
атаксией- телеангиэктазией состоит в том, что в
сыворотке у них выявляют а-фетопротеин,
характерный для эмбрионального периода развития
человека и для больных первичным раком печени.
14. Диагностика
При общем осмотре необходимо акцентировать внимание
на:
задержке полового развития;
пигментации кожных покровов;
нарушении или отсутствии сухожильных рефлексов;
нарушении роста;
уменьшенных размерах миндалин, лимфатических
узлов.
15. Диагностика
Определение уровня белка αфетопротеина (уровень
повышен).
ОАК(снижение уровня
лейкоцитов).
Определение концентрации
антител в крови (количество
антител уменьшается).
Иммунограмма (нижение
иммуноглобулина А и Е).
Выявление генетических
мутаций(11 хромосомы).
Тест на переносимость
глюкозы.
16. Диагностика
МРТ головного мозга и мозговых
структур (увеличение четвертого
желудочка и патологические
изменения в мозжечке —
дегенерация мозжечковых
клеток).
Рентген грудной клетки для
исключения пневмонии,
выявления изменения размеров
бронхов.
Анализ пигментных пятен
(наличие гиперкератоза,
отложение меланина в
эпидермисе, воспалительная
реакция в дерме).
17. Прогноз
Прогноз для жизни относительно неблагоприятный.
В среднем пациенты доживают до 14-20 лет.
Основные причины смерти:
опухоли, в основном лимфоидного происхождения:
-лимфома;
-лимфогранулематоз;
-лейкоз;
-ретикулосаркома;
тяжелые инфекционные заболевания с полиорганной
недостаточностью;
неврологические нарушения.
18. Лечение
Симптоматическая терапия:
гамма-глобулин и препарат «Т-активин»;
Витаминотерапия;
Антибиотикотерапия для борьбы с вторичной инфекцией
бактериального
характера(противовирусные,противогрибковые);
Физиотерапевтические мероприятия.
19. Список используемой литературы
1)Клиническая иммунология и аллергология,Г.Н.Дранник
2)Клиническая иммунология и аллергология,под ред.
Л.АГорячкиной,К.П.Кашкина
3)Феденко Е.С,Ильина Н.И. Аллергические заболевания
кожи в клинической практике.-Российский
аллергологический журнал
4) Шанин В. Ю.. Клиническая патофизиология. Учебник
для медицинских вузов.— СПб: «Специальная
Литература»,1998
Иммунодефициты: насколько это серьезно?
Иммунодефицит – недостаточность иммуной системы. Пациентов, у которых наблюдается такое состояние, также называют иммунокомпрометированными. У таких детей повышены риски развития и тяжелого течения инфекционных болезней.
Иммунодефициты делятся на:
Первичные иммунодефициты (ПИД) – это всегда врожденные болезни. Они обусловлены генетически: или наследуются от родителей, или возникают из-за мутаций. Каждый первичный иммунодефицит – это очень редкое заболевание. Но их, таких заболеваний, множество – в 2014 г. было известно более 200, в 2016 – уже более 300 разных первичных иммунодефицитов. Исследователи продолжают открывать новые. Даже в странах с высоким уровнем медицины не все дети с ПИД попадают к нужному специалисту, а в среднеразвитых, низкоразвитых странах выявляемость ПИД в десятки или сотни раз ниже. То есть такие дети умирают от разных инфекций, а настоящая причина, которую можно было бы лечить, так и остается вне поля зрения.
Вторичные иммунодефициты обусловлены неким другим состоянием. Есть какая-то первичная причина, которая уже вторично угнетает иммунную систему. Это не врожденное, а приобретенное состояние. Из инфекционных заболеваний вторичный иммунодефицит вызывается только ВИЧ / СПИД. Вирус иммунодефицита человека вызывает синдром приобретенного иммунодефицита.
Основные типы иммунокомпрометированных состояний
Первичные иммунодефициты
- B-клеточные дефекты
- X-связанная агаммаглобулинемия
- Дефицит субклассов IgG
- Общий вариабельный иммунодефицит
- Селективный дефицит IgA
- Гипер-IgM синдром
- Синдром Ди-Джорджи
- CD8 лимфоцитопения
- Дефицит цитокинов
- Дефективные T-клеточные рецепторы
- Комбинированные B- и T-клеточные дефекты
- Тяжелый комбинированный иммунодефицит
- Синдром Вискотта-Олдрича
- Атаксия-телеангиэктазия (синдром Луи-Барр)
- Гипер-IgE синдром
- Синдром Оменна
- Нарушение адгезии лейкоцитов
- Синдром Чедиака-Хигаси
- Дефицит миелопероксидазы
- Хроническая гранулематозная болезнь
- Лейкопения
- Синдром Костманна
- Синдром Швахмана-Даймонда
- Дефициты дефективной опсонизации системы комплемента
На сегодня известно более 300 первичных иммунодефицитов
Вторичные иммунодефициты
Вызываются такими факторами:
- Вирус иммунодефицита человека (ВИЧ)
- Онкопатология
- Трансплантация стволовых клеток костного мозга или целых органов
- Химиотерапия опухолей
- Ожоги
- Серповидно-клеточная анемия
- Кистозный фиброз
- Иммуносупрессиваные препараты
- Аспления
- Голодание
- Имплантация инородного тела
Некоторые педиатры ошибочно считают, что частые респираторные инфекции и вызываемые ими состояния (насморк, бронхиты) приводят к иммунодефициту. Об этом уже писалось в статье 10 вопросов педиатрам про иммунную систему.
Пониженный иммунитет из-за нехватки витаминов, неправильного питания, состояния окружающей среды – это ошибочные суждения, которые по разным причинам укоренились в сознании народных масс. Кроме того, миф о якобы слабом иммунитете порождает другие медицинские искажения – узнайте о них: Мифы о детских болезных.
Нужны достаточно веские причины для выведения иммунной системы из строя с возникновением недостаточности. Других иммунодефицитов, кроме первичных и вторичных, не бывает. Иммуная система или работает, или не работает.
Читайте также: