Болезнь Розаи-Дорфмана
Добавил пользователь Morpheus Обновлено: 27.01.2026
Синдром Вискотта–Олдрича (СВО) – первичное иммунодефицитное состояние (ПИДС), характеризующееся типичными симптомами разной степени тяжести: тромбоцитопения, инфекция, иммунная дизрегуляция и склонность к онкологическим заболеваниям. В связи с этим при любом лимфопролиферативном состоянии при СВО необходима биопсия лимфоузлов. Однако трактовка гистологической картины требует знания особенностей патоморфологии лимфоузлов у пациентов с иммунодефицитом и нередко представляет сложности. В данной статье представлено клиническое наблюдение с описанием редкого сочетания поздно диагностированного СВО с лимфаденопатией, клинически и морфологически напоминающей болезнь Розаи–Дорфмана в контексте ПИДС. Родители дали согласие на использование информации, в том числе фото ребенка, в научных исследованиях и публикациях.
Ключевые слова
Об авторах
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
канд. мед. наук, врач – аллергологиммунолог,
117997, Москва, ГСП-7, ул. Саморы Машела, 1
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
Москва
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
Москва
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
Москва
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
Москва
ФГБУ «Национальный медицинский исследовательский центр детской гематологии, онкологии и иммунологии им. Дмитрия Рогачева» Минздрава России
Россия
Москва
Список литературы
1. Jin Y.Y., Wu J., Chen T.X., Chen J. When WAS Gene Diagnosis Is Needed: Seeking Clues Through Comparison Between Patients With Wiskott–Aldrich Syndrome and Idiopathic Thrombocytopenic Purpura. Front Immunol 2019; 10: 1549.
2. Derry J.M., Ochs H.D., Francke U. Isolation of a novel gene mutated in Wiskott– Aldrich syndrome. Cell 1994; 78: 635–44.
3. Snapper S.B., Rosen F.S., Mizoguchi E., Cohen P., Khan W., Liu C.H., et al. Wiskott–Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 1998; 9: 81–91.
4. Notarangelo L.D., Miao C.H., Ochs H.D. Wiskott–Аldrich syndrome. Curr Opini Hematol 2008; 15: 30–6.
5. Imai K., Nonoyama S., Ochs H.D. WASP (Wiskott–Aldrich syndrome protein) gene mutations and phenotype. Curr. Opin. Allergy Clin Immunol 2003; 3: 427–36.
6. Aldrich R.A., Steinberg A.G., Campbell D.C. Pedigree demon- strating a sex-linked recessive condition characterized by draining ears, eczematoid dermatitis, and bloody diarrhea. Pediatrics 1954; 13: 133–9.
7. Albert M.H., Bittner T.C., Nonoyama S., Notarangelo L.D., Burns S., Imai K., et al. X-linked thrombocytopenia (XLT) due to WAS mutations: clinical characteristics, long-term outcome, and treatment options. Blood 2010; 115: 3231–8.
8. Mahlaoui N., Pellier I., Mignot C., Jais J.P., Bilhou-Nabera C., Moshous D., et al. Characteristics and outcome of early-onset, severe forms of Wiskott– Aldrich syndrome. Blood 2013; 121: 1510–6.
9. Zhu Q., Zhang M., Blaese R.M., Derry J.M., Junker A., Francke U., et al. The Wiskott– Aldrich syndrome and X-linked congenital thrombocytopenia are caused by mutations of the same gene. Blood1995; 86: 3797–804.
10. Massaad M.J., Ramesh N., Geha R.S. Wiskott–Aldrich syndrome: a comprehensive review. Annals of the New York Academy of Sciences 2013; 1285 (1): 26–43.
11. Worth A.J., Thrasher A.J. Current and emerging treatment options for Wiskott– Aldrich syndrome. Expert Rev Clin Immunol 2015; 11 (9): 1015–32.
12. Candotti F. Clinical manifestations and pathophysiological mechanisms of the Wiskott–Aldrich syndrome. J Clin Immunol 2018; 38: 13–27.
13. Zhang X., Dai R., Li W., Zhao H., Zhang Y., Zhou L., et al. Abnormalities of follicular helper T-cell number and function in Wiskott–Aldrich syndrome. Blood 2016; 127 (25): 3180–91.
14. Perry G.S., Spector B.D., Schumann L.M., Mandel G.S., Anderson V.E., McHugh R.B., et al. The Wiskott–Aldrich syndrome in the United States and Canada (1892- 1979). J Pediatr 1980; 97: 72–7.
15. Cotelingam J.D., Witebsky F.G., Hsu S.M., Blaese R.M., Jaffe E.S. Malignant lymphoma in patients with the Wiskott– Aldrich syndrome. Cancer Invest 1985; 3: 515–22.
16. Meropol N.J., Hicks D., Brooks J.J., Siminovitch K.A., Fishman N.O., Kant J.A., et al. Coincident Kaposi sarcoma and T-cell lymphoma in a patient with the Wiskott–Aldrich syndrome. American Journal of Hematology 1992; 40 (2): 126–34.
17. Cooper M.D., Chase H.P., Lowman J.T., Krivit W., Good R.A. Wiskott–Aldrich syndrome: an immunologic deficiency disease involving the afferent limb of immunity. Am J Med 1968; 44 (4): 499–513.
18. Wolff J.A. Wiskott–Aldrich syndrome: clinical, immunologic and pathologic observations. J Pediatr 1967; 70 (2): 221–32.
19. Blaese R.M., Strober W., Waldmann T.A. lmmunodeficiency in the Wiskott–Aldrich syndrome. Birth Defects 1975; 11 (1): 250–4.
20. Berglund G., Finnstrom O., Johansson S.G., Moiler K.L. Wiskott–Aldrich syndrome: a study of 6 cases with determination of the immunoglobulins A, D, G, M and ND. Acta Paediatr Scand 1968; 57 (2): 89–97.
21. Blaese R.M., Strober W., Levy A.L., Wakhnann T.A. Hypercatabolism of IgG, IgA, IgM and albumin in the Wiskott– Aldrich syndrome: a unique disorder of serum protein metabolism. J Clin Invest 1971; 50 (11): 2331–8.
22. Snover D.C., Frizzera G., Spector B.D., Perry III G.S., Kersey J.H. Wiskott–Aldrich syndrome: histopathologic findings in the lymph nodes and spleens of 15 patients. Hum Pathol 1981; 12 (9): 821–31.
23. Hastrup J., GrahI-Madsen R. Wiskott– Aldrich syndrome: thrombocytopenia, eczema and recurrent infection. Dan Med Bull 1965; 12: 99–102.
24. Ballow M., Dupont B., Good R.A. Autoimmune hemolytic anemia in Wiskott– Aldrich syndrome during treatments with transfer factor. J Pediatr 1973; 83: 772–80.
25. Spitler L.E. Transfer factor therapy in the Wiskott–Aldrich syndrome: results of long-term follow-up in 32 patients. Am J Med 1979; 67 (1): 59–66.
26. Risdall R.J., McKenna R.W., Nesbit M.E., Krivit W., Balfour Jr.H.H., Simmons R.L., Brunning R.D. Virus-associated hemophagocytic syndrome: a benign histiocytic proliferation distinct from malignant histiocytosis. Cancer 1979; 44 (3): 993–1002.
27. Foucar E., Rosai J., Dorfman R.F., Eyman J.M. Immunologic abnormalities and their significance in sinus hisiocytosis with massive lymphadenopathy. Am J Pathol 1984; 82 (5): 515–25.
28. Goyal G., Ravindran A., Young J.R., Shah M.V., Bennani N.N., Patnaik M.M., et al. Mayo Clinic Histiocytosis Working Group. Clinicopathological features, treatment approaches, and outcomes in Rosai–Dorfman disease. Haematologica 2019; 219626. DOI: 10.3324/haematol.2019.219626
29. Rosai J., Dorfman R.F. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol 1969; 87 (1): 63–70.
30. Foucar E., Rosai J., Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): review of the entity. Semin Diagn Pathol 1990; 7 (1): 19–73.
31. Paulli M., Bergamaschi G., Tonon L., Viglio A., Rosso R., Faccetti F., et al. Evidence for a Polyclonal Nature of the Cell Infiltrate in Sinus Histiocytosis with Massive Lymphadenopathy (Rosai–Dorfman Disease). Br J Haematol 1995; 91 (2): 415–8.
32. Young J.R., Johnson G.B., Murphy R.C., Go R.S., Broski S.M. (18)F-FDG PET/CT in Erdheim-Chester Disease: Imaging Findings and Potential BRAF Mutation Biomarker. J Nucl Med 2018; 59 (5): 774–9.
33. Абрамов Д.С., Мякова Н.В., Абугова Ю.Г., Грачев Н.С, Дьяконова Ю.Ю., Калинина М.П. и др. Синусовый гистиоцитоз с массивной лимфаденопатией (болезнь Розаи–Дорфмана). Вопросы гематологии/онкологии и иммунопатологии в педиатрии 2014; 4 (13): 63–8.
34. Fu W.J., Du J., Lu J., Wang L.Z., Yang J.M., He M.X., et al. Rosai–Dorfman disease: a clinicopathologic analysis and whole exome sequencing in 23 cases. Zhonghua Xue Ye Xue Za Zhi 2019; 40 (8): 656–61.
35. Maric I., Pittaluga S., Dale J.K., Niemela J.E., Delsol G., Diment J., et al. Histologic features of sinus histiocytosis with massive lymphadenopathy in patients with autoimmune lymphoproliferative syndrome. Am J Surg Pathol 2005; 29 (7): 903–11.
36. Gualco G., van den Berg A., Koopmans S., Bacchi L.M., Carneiro S.S., Ruiz E.Jr., et al. Autoimmune lymphoproliferative syndrome in a patient with a new minimal deletion in the death domain of the FAS gene. Hum Pathol 2008; 39 (1): 137–41.
37. Xie Y., Pittaluga S., Price S., Raffeld M., Hahn J., Jaffe E.S., et al. Bone marrow findings in autoimmune lymphoproliferative syndrome with germline FAS mutation. Haematologica 2017; 102 (2): 364–72.
38. Venkataraman G., McClain K.L., Pittaluga S., Rao V.K., Jaffe E.S. Development of disseminated histiocytic sarcoma in a patient with autoimmune lymphoproliferative syndrome and associated Rosai– Dorfman disease. Am J Surg Pathol 2010; 34 (4): 589–94.
39. Gupta N., Verma R., Belho E.S., Manocha A. Isolated Extranodal Rosai–Dorfman Disease on 18F-FDG PET-CT Scan. Indian J Nucl Med 2019; 34 (4): 319–20.
40. Jin Y., Mazza C., Christie J.R., Giliani S., Fiorini M., Mella P., et al. Mutations of the Wiskott–Aldrich Syndrome Protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood 2004; 104 (13): 4010–9.
41. Нестеренко З.А., Кузьменко Н.Б., Бурлаков В.И., Викторова Е.А., Ведмедская В.А., Першин Д.Е. и др. Феномен реверсной мутации у пациента с синдромом Вискотта–Олдрича. Вопросы гематологии/онкологии и иммунопатологии в педиатрии 2019; 18 (3): 104–11.
42. Мухина А.А., Кузьменко Н.Б., Родина Ю.А., Кондратенко И.В., Бологов А.А., Латышева Т.В. и др. Характеристика пациентов с первичными иммунодефицитными состояниями в Российской Федерации: от рождения до старости. Педиатрия 2019; 98 (3): 24–31.
43. Балашов Д.Н., Козловская С.Н., Радыгина С.А., Лаберко А.Л., Султанова Э.Р., Шелихова Л.Н. и др. Успехи проведения трансплантации гемопоэтических стволовых клеток при синдроме Вискотта– Олдрича. Педиатрия 2019; 98 (3): 54–9.
44. Menotti M., Ambrogio C., Cheong T.C., Pighi C., Mota I., Cassel S.H., et al. Wiskott–Aldrich syndrome protein (WASP) is a tumor suppressor in T-cell lymphoma. Nat Med 2019; 25 (1): 130–40.
45. Facchetti F., Blanzuoli L., Ungari M., Alebardi O., Vermi W. Lymph node pathology in primary combined immunodeficiency diseases Springer Semin Immunopathol 1998; 19: 459–78.
Болезнь Розаи-Дорфмана
Морозовская детская городская клиническая больница Департамента здравоохранения Москвы, Москва, Россия, 119049
Научная группа при кафедре болезней уха, горла и носа Первого МГМУ им. И.М. Сеченова Минздравсоцразвития России
Морозовская детская городская клиническая больница Департамента здравоохранения Москвы, Москва, Россия, 119049
Кафедра оториноларингологии педиатрического факультета Российского национального исследовательского медицинского университета им. Н.И. Пирогова, Москва, Россия, 117997
Морозовская ДГКБ Департамента здравоохранения Москвы, Москва, Россия, 119049
Болезнь Розаи—Дорфмана: обзор литературы и клиническое наблюдение экстранодальной формы заболевания с поражением слизистой оболочки носа и околоносовых пазух
Журнал: Вестник оториноларингологии. 2018;83(6): 72‑75
Морозовская детская городская клиническая больница Департамента здравоохранения Москвы, Москва, Россия, 119049
Синусный гистиоцитоз с массивной лимфаденопатией (болезнь Розаи—Дорфмана) представляет собой редкое неопухолевое заболевание неизвестной этиологии с доброкачественным течением. В статье представлены обобщенные данные литературы и клиническое наблюдение экстранодальной формы заболевания с поражением слизистой оболочки носа и околоносовых пазух.
Морозовская детская городская клиническая больница Департамента здравоохранения Москвы, Москва, Россия, 119049
Научная группа при кафедре болезней уха, горла и носа Первого МГМУ им. И.М. Сеченова Минздравсоцразвития России
Морозовская детская городская клиническая больница Департамента здравоохранения Москвы, Москва, Россия, 119049
Кафедра оториноларингологии педиатрического факультета Российского национального исследовательского медицинского университета им. Н.И. Пирогова, Москва, Россия, 117997
Морозовская ДГКБ Департамента здравоохранения Москвы, Москва, Россия, 119049
Описанный впервые в 1969 г. J. Rosai и R. Dorfman [1] синусный гистиоцитоз с массивной лимфаденопатией (СГМЛ) — болезнь Розаи—Дорфмана представляет собой редкое неизвестной этиологии доброкачественное неопухолевое заболевание, характеризующееся накоплением пролиферирующих гистиоцитов в синусах лимфатических узлов, в результате чего происходит их массивное увеличение. СГМЛ относится к группе так называемых заболеваний с атипичными клеточными нарушениями лимфатических узлов. Атипия в данном случае проявляется тем, что ненормальное увеличение количества гистиоцитов происходит в результате выраженного фагоцитоза ими клеток лимфоидного ряда. Редкость заболевания, противоречивость различных описаний, неопределенность специфических гистиоцитарных маркеров и четких методов определения клональности, а также существование ряда близких клинических и гистологических состояний реактивного инфекционного и опухолевого происхождения делают это заболевание сложным для диагностики. По данным Гематологического научного центра РАМН, более чем у 50% больных с синусным гистиоцитозом в дальнейшем выявляются онкогематологические заболевания [2].
Этиология СГМЛ остается неизвестной, и хотя его клинические проявления и гистологическая картина сходны с наблюдаемыми при инфекционных процессах, микроорганизмы при СГМЛ до настоящего времени не идентифицированы [3]. Предполагают, что в патогенезе СГМЛ имеют значение повсеместно распространенные вирус герпеса 6-го типа и вирус Эпштейна—Барр, которых обнаруживают более чем у 50% больных СГМЛ [3, 4]. Данные молекулярных исследований указывают на то, что СГМЛ — поликлональное заболевание [5].
После создания в 1990 г. регистра пациентов, страдающих СГМЛ, выяснилось, что клинические признаки достаточно разнообразны: если в первых описаниях упоминалось заболевание с выраженной двусторонней лимфаденопатией с поражением преимущественно шейных узлов у маленьких детей негроидной расы [1, 6], то анализ регистра показал, что возраст больных варьирует от новорожденности до 74 лет, мужчины заболевают в 1,5 раза чаще женщин, наряду с «излюбленной» локализацией в шейных лимфоузлах могут быть поражены и другие группы лимфатических узлов, а у 40% больных выявляют экстранодальные очаги поражения [3]. В большинстве случаев локализованная лимфаденопатия является первым и единственным проявлением заболевания, хотя описаны случаи СГМЛ в виде псевдоопухолей на коже, орбите, в среднем ухе, верхних дыхательных путях, желудочно-кишечном тракте, мозговых оболочках и др. [2, 7—14].
Чаще заболевание имеет доброкачественное течение, склонное к спонтанной регрессии, сопровождающееся частыми рецидивами, однако лимфаденит может персистировать годами. Клиническая картина СГМЛ характеризуется массивной лимфаденопатией с преимущественным поражением шейных лимфатических узлов, отсутствием симптомов интоксикации. Помимо увеличения лимфатических узлов и экстранодальных очагов, при обострении наблюдается лихорадка, лейкоцитоз, повышение СОЭ. У ряда больных отмечаются нарушения уровня сывороточных белков в виде умеренной поликлональной гипергаммаглобулинемии. У части пациентов встречаются те или иные признаки поражения иммунной системы: полиартралгия, бронхолегочные поражения или гемолитическая анемия, предшествующие развитию СГМЛ или развивающиеся в дебюте [2, 15].
Диагноз устанавливается исключительно на основании морфологического исследования биопсийного материала. Микроскопически структура лимфатического узла обычно нарушена за счет резко выраженного расширения синусов, стертости фолликулов и герминальных центров. Характерными признаками являются склерозирование капсулы и перикапсулярное разрастание соединительнотканных волокон и жировой ткани. Растянутые синусы и медуллярные тяжи содержат смешанную популяцию клеток, представленную полиморфно-клеточными лейкоцитами и лимфоцитами, однако преобладающей популяцией являются гистиоциты, характеризующиеся выраженным полиморфизмом. Это крупные клетки с большой светлой, иногда вакуолизированной цитоплазмой с одним или несколькими вогнутыми ядрами. Митозы наблюдаются редко. Внутрицитоплазменные вакуоли содержат фагоцитированные лимфоциты, эритроциты или нейтрофилы, они не подвергаются воздействию цитолитических ферментов. Некоторые клетки, особенно лимфоциты, способны жить в вакуолях (феномен эмпериополеза), другие постепенно деградируют, формируя ядерные фрагменты. Некоторые вакуоли содержат только остатки ядер деградировавших клеток или липиды, которые хорошо окрашиваются суданом. В мозговом слое отмечаются многочисленные плазматические клетки, часть из которых двуядерные. Эозинофилы встречаются редко. Некроза также не отмечается. Микроскопическая картина экстранодальных очагов СГМЛ сходна с наблюдаемой в лимфатических узлах, хотя экстранодальные очаги характеризуются более выраженным фиброзом, менее выраженным скоплением гистиоцитов и лимфоцитофагоцитозом [2].
Цитологические препараты лимфатического узла гиперклеточны, с обилием гистиоцитов, фагоцитированных лимфоцитов на фоне реактивации лимфоидной ткани. Гистиоциты обычно с рыхлым ядром и обильной бледной, часто вакуолизированной цитоплазмой и феноменом эмпериополеза. Атипичные гистиоциты содержат гиперхромные ядрышки, некоторые гистиоциты достигают гигантских размеров. На ранних стадиях СГМЛ в мазках можно увидеть множество лимфоцитов и иногда иммунобласты [16]. На более поздних стадиях доминируют многочисленные плазматические клетки и тельца Русселя [17]. В большинстве случаев имеется лизосомная активность гистиоцитов, хотя число лизосом может варьировать в широких пределах, вплоть до их отсутствия. При иммуногистохимическом исследовании установлена принадлежность клеток при СГМЛ к макрофагально-гистиоцитарной группе. Но неясно, к какому конкретно представителю этой группы они относятся, так как во всех случаях СГМЛ происходит экспрессия белка S100, который является специфическим маркером интердигитирующих клеток в лимфатических узлах и клеток Лангерганса в коже [18].
Дифференциальный диагноз необходимо проводить со следующими заболеваниями: синусный гистиоцитоз как неспецифическая реакция лимфатических узлов на инфекцию или опухоль, злокачественный гистиоцитоз, гранулематозное поражение; эозинофильная гранулема; болезнь Хенда—Шюллера—Крисчена (для нее характерно поражение скелета), болезнь Леттерера—Сиве (встречается у младенцев, характерны поражения кожи), болезнь Ходжкина [2, 3, 14].
В настоящее время методы лечения СГМЛ не разработаны [14, 19]. В большинстве случаев прогноз благоприятный [8].
В связи с редкостью болезни Розаи—Дорфмана считаем целесообразным привести казуистическое наблюдение экстранодальной ее формы с поражением слизистой оболочки носа и околоносовых пазух.
Родители мальчика К., 4 лет (2013 г. р.), впервые обратились в Морозовскую ДГКБ в сентябре 2016 г. с жалобами на наличие образования на конъюнктиве правого глаза. Офтальмологом обнаружен сосудистый невус конъюнктивы склеры правого глаза, рекомендовано местное лечение, динамическое наблюдение, при отсутствии положительной динамики — плановое хирургическое лечение. Через 3 мес после первичного обращения, в декабре 2016 г., родители обнаружили образование в области левого яичка. В связи с начавшимся через 4 мес после обнаружения ростом образования яичка (апрель 2017 г.) по месту жительства была выполнена МРТ: согласно заключению, картину необходимо дифференцировать между сосудистой опухолью и гамартомой левой половины мошонки. Лабораторные исследования крови на онкомаркеры: определение уровня АФП — 0,135 мМЕ/мл и β-ХГЧ — 0,92 МЕ/мл, убедительных данных за онкологический процесс не получено. Выполнено УЗИ мошонки (апрель 2017 г.): левое яичко в мошонке размером 1,0×0,7×0,7 см, выше него определяется образование узлового характера неоднородной структуры размером 0,8×0,6×1,5 см, рядом с ним расположены еще несколько узлов.
Ребенок консультирован онкологом Морозовской ДГКБ — рекомендовано удаление образования со срочным гистологическим исследованием и решением вопроса об объеме операции по результатам интраоперационного гистологического ответа. В июле 2017 г. выполнена поперечная скрототомия — при ревизии мошонки обнаружено объемное образование, плотное, желтоватого цвета, с неровными контурами, интимно прилежащее к нижнему полюсу левого яичка, размером 2×3 см. Дистальнее, по ходу элементов семенного канатика, обнаружено подобное образование размером 0,8×0,5 см. Удаленные объемные образования направлены на экспресс-биопсию. Интраоперационно получен ответ — рабдомиосаркома, в связи с чем онкологом было рекомендовано выполнение орхифуникулэктомии паховым доступом. Однако при дальнейшем гистологическом и иммуногистохимическом исследовании удаленного образования и регионарных лимфатических узлов у ребенка установлен диагноз синусного гистиоцитоза — болезни Розаи—Дорфмана. В дальнейшем рекомендованы консультация и наблюдение в центре детской гематологии и онкологии Морозовской ДГКБ.
В сентябре 2017 г. родители обратились к оториноларингологам Морозовской ДГКБ в связи жалобами на затруднение носового дыхания в течение нескольких месяцев. При осмотре — носовое дыхание через левую половину носа отсутствует, справа — свободное. При диагностической фиброэндоскопии полости носа и носоглотки обнаружено, что поверхность носовых раковин покрыта видоизмененной бугристой слизистой оболочкой бледно-розового, местами желтоватого цвета, при надавливании на которую появляется вязкое слизистое отделяемое. Через левую половину носа доступ к носоглотке невозможен в связи с наличием плотного, розово-серого цвета образования, исходящего из левого среднего носового хода, из левой верхнечелюстной пазухи и блокирующего левую половину носа в средних отделах; не кровоточащего при пальпации. Слизистая оболочка (СО) носовых раковин справа аналогично изменена, общий носовой ход достаточно широкий, хоана обычной формы, в куполе носоглотки лимфоидная ткань на уровне ½ сошника, трубные валики не увеличены.
Пациенту выполнена компьютерная томография полости носа и околоносовых пазух с контрастным усилением (ультравист) в условиях аппаратно-масочной анестезии (сентябрь 2017 г.) — отмечается тотальное заполнение патологическим содержимым левой верхнечелюстной пазухи, субтотальное заполнение клеток левого решетчатого лабиринта; медиальная стенка левой верхнечелюстной пазухи частично разрушена. Отмечается наличие периостальных наслоений по внутренней поверхности стенок пазухи. СО правой верхнечелюстной пазухи неравномерно утолщена, СО носовых раковин утолщена (рис. 1, 2). Рис. 2. Пациент К. Компьютерная томограмма полости носа и околоносовых пазух в мягкотканном режиме. а — аксиальная; б — коронарная проекции. Рис. 1. Пациент К. Компьютерная томограмма полости носа и околоносовых пазух в костном режиме. а — аксиальная; б — коронарная; в — сагиттальная проекции.
Ребенок консультирован онкологом (сентябрь 2017 г.), которым высказано предположение об экстранодальном проявлении болезни Розаи—Дорфмана. Рекомендовано удаление образования.
После предоперационной подготовки под интубационным наркозом выполнена левосторонняя эндоназальная эндоскопическая этмоидогайморотомия, удалены патологическое образование из верхнечелюстной пазухи, а также патологически измененная СО нижних носовых раковин и клеток решетчатого лабиринта с обеих сторон.
По данным патоморфологического исследования операционного материала (гистологическое и иммуногистохимическое исследование; окраска IHC: CD1a, CD3, CD38, CD68, CD79a, Macrophage, S100) определяются механически деформированные фрагменты СО околоносовых пазух с мелким участком костной ткани, частично покрытые многорядным призматическим реснитчатым эпителием, в собственной пластинке СО отек, очаговый склероз, диффузная инфильтрация большого количества крупных гистиоцитов с обильной вакуолизированной цитоплазмой, округлыми ядрами с крупным хроматином и различимым одним ядрышком и явлениями эмпериополеза (фагоцитированные лимфоциты, эритроциты и плазматические клетки). Тотальная экспрессия в воспалительном инфильтрате S100, CD68, Macrofage, CD79a, CD3, CD38. Реакция отрицательна с антителом к CD1a. Заключение: морфологическая картина соответствует болезни Розаи—Дорфмана, экстранодальный вариант.
Также проведено исследование на базе ФГБУ «Гематологический научный центр» Минздрава России.
Микроскопическое исследование: мелкие фрагменты СО, покрытой многоядерным мерцательным эпителием со слизистыми железами: фрагменты рыхлой соединительной ткани с пролифератом, представленным крупными скоплениями зрелых плазматических клеток, инфильтрацией из мелких лимфоидных клеток, макрофагов, наличием крупных гистиоидных клеток с пузырьковидным ядром, светлой/слабоэозинофильной широкой цитоплазмой, отдельные из них — с признаками эмпериополеза.
При иммуногистохимическом исследовании препаратов: крупные гистиоидные клетки с пузырьковидными ядрами, расположенные разрозненно и в виде рыхлых скоплений среди лимфоплазмоцитарного инфильтрата с примесью макрофагов, экспрессируют S-100, ядерно-цитоплазматическая реакция СD68, многочисленные зрелые плазматические клетки CD38 — позитивны. Реакция с антителом к CD7 позитивна в мелких В-клетках, в зрелых плазматических клетках многочисленная Т-клеточная популяция CD3+. Заключение: морфологическая картина и иммунофенотип характеризуют нелангергансоклеточный гистиоцитоз — экстранодальный субстрат болезни Розаи—Дорфмана (клональную гистиоцитарную пролиферацию).
В послеоперационном периоде производился туалет полости носа растворами антисептиков, в общих носовых ходах были установлены тампоны, которые были удалены на 5-е сутки, и на 7-е сутки ребенок выписан домой в удовлетворительном состоянии.
За истекший период — 6 мес рецидива данного процесса не отмечено.
Заключение
Болезнь Розаи—Дорфмана представляет собой одно из редких заболеваний неизвестной пока этиологии, характеризующееся выраженным синусным гистиоцитозом. Диагноз в данном случае был установлен на основании классических гистологических данных, в том числе явлений эмпериополеза, типичной иммуногистохимической характеристики гистиоцитов в синусах.
Особенностью данного случая является экстранодальное поражение СО носа и околоносовых пазух с частичным разрушением костных структур, сопровождавшееся яркими клиническими проявлениями. В связи с отсутствием разработанных методов химиотерапевтического воздействия особую важность в диагностике и терапии имеет правильное и эффективное хирургическое лечение.
Роль лучевой терапии в лечении болезни Розаи–Дорфмана: обзор литературы и клиническое наблюдение пациента с изолированным поражением кожи

Болезнь Розаи–Дорфмана (БРД) – редкий вариант нелангергансоклеточного гистиоцитоза. Широкий спектр проявлений, наличие системной и изолированной кожной форм, малое количество описаний обусловливают трудности диагностики.
Цель. Обзор литературы и описание пациента с кожной формой БРД.
Результаты. У мужчины 56 лет в левой височной области в октябре 2019 г. появилось опухолевидное образование. После радикального удаления в течение 2 нед отмечен повторный рост в зоне операции. По результатам гистологического и иммуногистохимического исследований с последующим стадированием верифицирована кожная форма БРД с изолированным поражением кожи лица и височной области. Проведено облучение в суммарной дозе 36 Гр. Получена положительная динамика в виде уменьшения образования. В течение 4 мес ответ сохраняется.
Заключение. Лучевая терапия у пациента с локализованной формой БРД привела к длительному противоопухолевому ответу.
Ключевые слова
Полный текст
Болезнь Розаи–Дорфмана–Дестомбса (БРД) – это редкий вариант нелангергансоклеточного гистиоцитоза, который впервые описан в 1960-х годах патологами Пьером Дестомбсом, Хуаном Розаи и Рональдом Дорфманом. В их работах представлены больные со значительным увеличением шейных лимфоузлов. При гистологическом анализе узла был выявлен выраженный гистиоцитоз синусов. Авторы назвали заболевание «синусовый гистиоцитоз с массивной лимфаденопатией» [1].
В соответствии с актуальной классификацией Международного гистиоцитарного общества БРД отнесена сразу к 2 группам: системные формы к R-группе, а вариант с изолированным кожным поражением – к С-группе (табл. 1) [2].
Таблица 1. Классификация гистицоитозов и новообразований макрофагально-дендритической линейности
Table 1. Classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages
L-группа:
• гистиоцитоз из клеток Лангерганса (ГКЛ)
• недетерминированный гистиоцитоз
• болезнь Эрдхейма–Честера (БЭЧ)
• смешанный ГКЛ/БЭЧ
С-группа:
• кожный нелангергансоклеточный гистиоцитоз:
– ксантогранулема (КГ): ювенильная КГ, КГ взрослых, солитарная ретикулогистиоцитома, доброкачественный цефалический гистиоцитоз, генерализованный эруптивный гистиоцитоз, прогрессирующий нодулярый гистиоцитоз
– нексантогранулемные: кожная форма БРД, некробиотическая КГ, другие кожные нелангергансоклеточные гистиоцитозы неуточненные
• кожный нелангергансоклеточный гистиоцитоз со значительным системным компонентом
R-группа:
• семейная БРД
• спорадическая БРД: классическая, экстранодальная, БРД с новообразованием или иммунным заболеванием, неклассифицируемая
М-группа:
• первичный злокачественный гистиоцитоз
• вторичный злокачественный гистиоцитоз (следующий за или ассоциированный с другим гемобластозом). Подтипы: гистиоцитарый, интердигитирующий, лангергансоклеточный, недетерминированный
H-группа:
• первичный гемофагоцитарный синдром: моногенно наследуемые состояния, ведущие к гемофагоцитарному лимфогистиоцитозу
• вторичный гемофагоцитарный лимфогистиоцитоз (наследуемый не по закону Менделя)
• гемофагоцитарный лимфогистиоцитоз неуточненного/неизвестного генеза
БРД встречается как самостоятельное заболевание, так и в ассоциации с аутоиммунными, наследственными и опухолевыми процессами [3, 4].
Заболевание считается незлокачественным, так как к настоящему времени клональности гистиоцитов при БРД не обнаружено [5, 6]. Прогноз БРД, как правило, благоприятный, особенно при локализованных формах [3]. Отмечены и спонтанные ремиссии с вероятностью до 50% у больных без выраженных симптомов [7–9].
Медиана возраста начала заболевания 56 (20–81) лет [10]. Обычно у больных БРД есть двустороннее массивное безболезненное увеличение шейных лимфоузлов. У 92% пациентов встречается экстранодальное поражение, в том числе поражение глаз, центральной нервной системы, органов грудной, брюшной полости, костей [3], иногда присутствуют также В-симптомы [11]. У 52% больных при системном процессе встречается поражение кожи [3, 12, 13].
Изолированное поражение кожи встречается редко и представляет собой отдельный вариант БРД, который принято называть кожной формой БРД [14]. Поражение кожи характеризуется как одиночными (40%), так и множественными (60%) высыпаниями и может быть представлено папулами, узлами, бляшками, редко – пигментированными пятнами. Заболевание обычно протекает бессимптомно, в редких случаях сопровождается зудом или изъязвлением высыпаний. Наиболее часто поражается кожа туловища и конечностей, реже – кожа лица, шеи, волосистой части головы и половых органов [15].
Гистологическая картина кожной формы БРД характеризуется дермальным инфильтратом из крупных гистиоцитов с обильной бледной цитоплазмой, крупными ядрами и типичным иммунофенотипом S100+CD68+CD1a-. Гистиоциты также могут экспрессировать другие макрофагальные маркеры – CD163 и CD14 [16]. Феномен эмпериополеза хоть и типичен для БРД, но встречается и при других гистиоцитозах [17]. Клиническая картина и патоморфологические проявления БРД варьируют от пациента к пациенту, поэтому при верификации диагноза необходимо учитывать весь спектр клинических и патоморфологических данных. Выраженный синусовый гистиоцитоз, в том числе с иммуногистохимическими (ИГХ) признаками БРД, может быть и реактивным процессом при злокачественных новообразованиях, после протезирования и по другим причинам [18–23].
Изменения крови, как правило, неспецифичны. Могут быть лейкоцитоз, поликлональная гипергаммаглобулинемия, повышение скорости оседания эритроцитов, а также гипо- или нормохромная анемия [24].
Терапия БРД в настоящее время не разработана. При запросе в поисковой системе Pubmed по ключевым словам Rosai–Dorfman disease индексируется 1927 статей, большинство из которых является описанием маленьких групп или отдельных пациентов. При локализованных формах оправдано хирургическое удаление или облучение. Описан положительный эффект при применении метотрексата, ритуксимаба, иматиниба, кладрибина, глюкокортикостероидов, леналидомида у больных с прогрессированием после местной терапии, а также при системных формах [8, 25, 26].
Лучевая терапия (ЛТ) используется обычно в качестве адъювантного лечения после оперативных вмешательств или для лечения местных рецидивов. При этом в согласительных рекомендациях Международного гистиоцитарного общества отмечается, что ЛТ имеет умеренную эффективность [8]. В табл. 2 обобщены случаи использования ЛТ в лечении БРД.
Таблица 2. Результаты ЛТ пациентов с локализованной формой БРД
Table 2. Results of the radiotherapy in patients with localized form of Rosai–Dorfman disease
Экстранодальная интраоссальная болезнь Розаи–Дорфмана в области нижней челюсти: описание клинического случая и обзор литературы
Болезнь Розаи–Дорфмана, или синусный гистиоцитоз, – редкое заболевание, которое поражает различные группы лимфатических узлов. Экстранодальная форма наблюдается в 45 % случаев, при этом кости лицевого черепа вовлекаются в процесс особенно редко. Мужчина, 62 лет, обратился с жалоб a ми на онемение подбородка и нижней губы. После клинического обследования и хирургического лечения в объеме расширенной биопсии была диагностирована экстранодальная интраоссальная форма болезни Розаи–Дорфмана в области нижней челюсти. Проведена отложенная реконструкция свободным васкуляризированным костным лоскутом малой берцовой кости. Спустя 2 года наблюдения признаков рецидива не отмечалось.
Цель публикации – описание редкого случая поражения нижней челюсти со значительной костной деструкцией. Обсуждаются клинические и микроскопические признаки данного патологического процесса.
Ключевые слова
Об авторах
Отделение онкологии полости рта и области головы и шеи, 9-й народный госпиталь, Медицинская школа Шанхайского университета Дзяо Тун; Шанхайская ключевая лаборатория стоматологии и Шанхайский исследовательский институт стоматологии; Национальный клинический исследовательский центр стоматологии.
Китай
200011, Шанхай, шоссе Джидзяодзю, 639
Отделение онкологии полости рта и области головы и шеи, 9-й народный госпиталь, Медицинская школа Шанхайского университета Дзяо Тун; Шанхайская ключевая лаборатория стоматологии и Шанхайский исследовательский институт стоматологии; Национальный клинический исследовательский центр стоматологии; ФГБУ «Центральный научно-исследовательский институт стоматологии и челюстно-лицевой хирургии» Минздрава России.
Россия
200011, Шанхай, шоссе Джидзяодзю, 639; 119991, Москва, ул. Тимура Фрунзе, 16
ФГБУ «Национальный медицинский исследовательский центр онкологии им Н. Н. Блохина» Минздрава России.
Россия
1154784, Москва, Каширское шоссе, 24
Отделение онкологии полости рта и области головы и шеи, 9-й народный госпиталь, Медицинская школа Шанхайского университета Дзяо Тун; Шанхайская ключевая лаборатория стоматологии и Шанхайский исследовательский институт стоматологии; Национальный клинический исследовательский центр стоматологии.
Китай
200011, Шанхай, шоссе Джидзяодзю, 639
Отделение онкологии полости рта и области головы и шеи, 9-й народный госпиталь, Медицинская школа Шанхайского университета Дзяо Тун; Шанхайская ключевая лаборатория стоматологии и Шанхайский исследовательский институт стоматологии; Национальный клинический исследовательский центр стоматологии.
Китай
200011, Шанхай, шоссе Джидзяодзю, 639
Отделение онкологии полости рта и области головы и шеи, 9-й народный госпиталь, Медицинская школа Шанхайского университета Дзяо Тун; Шанхайская ключевая лаборатория стоматологии и Шанхайский исследовательский институт стоматологии; Национальный клинический исследовательский центр стоматологии.
Китай
200011, Шанхай, шоссе Джидзяодзю, 639
Список литературы
1. Rosai J., Dorfman R. F. Sinus histiocytosis with massive lymphadenopathy: a newly recognized benign clinicopathologic entity. Arch Pathol 1969;87:63–70.
2. Rosai J., Dorfman R. F. Sinus histiocytosis with massive lymphadenopathy: a pseudolymphomatous benign disorder. Analysis of 34 cases. Cancer 1972;30(5):1174–88. PMID: 5083057.
3. Suster S., Cartagena N., Cabello-Inchausti B., Robinson M. J. Histiocytic lymphophagocytic panniculitis. An unusual extranodal presentation of sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease). Arch Dermatol 1988;124(8):1246–9. PMID: 3401030.
4. Wenig B. M., Abbondanzo S. L., Childers E. L. et al. Extranodal sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease) of the head and neck. Hum Pathol 1993;24(5):483–92. PMID: 8491488.
5. Aoyama K., Terashima K., Imai Y. et al. Sinus histiocytosis with massive lymphadenopathy. A histogenic analysis of histiocytes found in the fourth Japanese case. Acta Pathol Jpn 1983;34(2):375–88. PMID: 6741550.
7. Pendse A. A., Wobker S. E., Greene K. G. et al. Intraosseous Rosai–Dorfman disease diagnosed by touch imprint cytology evaluation: a case series. Diagn Cytopathol 2018;46(1):83–7. DOI: 10.1002/dc.23802. PMID: 28834636.
8. Naidu R. K., Urken M. L., Som P. M. et al. Extranodal head and neck sinus histiocytosis with massive lymphadenopathy. Otolaryngol Head Neck Surg 1990;102(6):764–7.
9. Foucar E., Rosai J., Dorfman R. F. Sinus histiocytosis with massive lymphadenopathy (Rosai–Dorfman disease): review of the entity. Semin Diagn Pathol 1990;7(1):19–73. PMID: 2180012.
11. Günhan O., Finci R., Günaydin Y., Somuncu I. Sinus histiocytosis with massive lymphadenopathy: a case with facial bones involvement. J Oral Maxillofac Surg 1991;49(2):205–9. PMID: 1990100.
12. Alawi F., Robinson B. T., Carrasco L. Rosai–Dorfman disease of the mandible. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2006;102(4):506–12. DOI: 10.1016/j.tripleo.2005.10.071. PMID: 16997119.
14. Xu Q., Fu L., Liu C. Multimodality imaging-based evaluation of Rosai–Dorfman disease in the head and neck: a retrospective observational study. Medicine (Baltimore) 2017;96(51):e9372. DOI: 10.1097/MD.0000000000009372. PMID: 29390533.
15. Humble J. G., Jayne W. H., Pulvertaft R. J. Biological interaction between lymphocytes and other cells. Br J Haematol 1956;2(3):283–94. PMID: 13342362.
17. Smith R. M., Hassan A., Robertson C. E. Numb chin syndrome. Curr Pain Headache Rep 2015;19(9):44. DOI: 10.1007/s11916-015-0515-y. PMID: 26210355.
18. Lai K. L., Abdullah V., Ng K. S. et al. Rosai–Dorfman disease: presentation, diagnosis, and treatment. Head Neck 2013;35(3):E85–8. DOI: 10.1002/hed.21930. PMID: 22083607.
19. Maklad A. M., Bayoumi Y., Tunio M. et al. Steroid-resistant extranodal Rosai–Dorfman disease of the cheek mass and ptosis treated with radiation therapy. Case Rep Hematol 2013;2013:428297. DOI: 10.1155/2013/428297. PMID: 23738161.
20. Shemen L., D’Anton M., Klijian A. et al. Rosai–Dorfman disease involving the premaxilla. Ann Otol Rhinol Laryngol 1991;100(10):845–51. DOI: 10.1177/000348949110001011. PMID: 1952653.
Журнал «Здоровье ребенка» 6 (57) 2014
Синусный гистиоцитоз с массивной лимфаденопатией встречается у детей редко. В статье приведено описание случая болезни Розаи — Дорфмана у мальчика 3 лет. Заподозрить болезнь Розаи — Дорфмана позволило возникновение лимфаденопатии, преимущественно цервикальной, с одновременным поражением экстранодальной зоны — селезенки. Для уточнения диагноза использовали УЗИ. Окончательный диагноз был установлен в результате хирургического удаления конгломерата лимфоузлов шеи, гистологического и гистохимического исследования. После проведенного оперативного вмешательства ребенок выписан для прохождения дальнейшего лечения в отделении онкогематологии.
Синусний гістіоцитоз із масивною лімфаденопатією зустрічається в дітей рідко. У статті наведено опис випадку хвороби Розаї — Дорфмана в хлопчика 3 років. Запідозрити хворобу Розаї — Дорфмана дозволило виникнення лімфаденопатії, переважно цервікальної, з одночасним ураженням екстранодальної зони — селезінки. Для уточнення діагнозу використовували УЗД. Остаточний діагноз був установлений у результаті хірургічного видалення конгломерату лімфовузлів шиї, гістологічного та гістохімічного дослідження. Після проведеного оперативного втручання дитина виписана для проходження подальшого лікування у відділенні онкогематології.
Sinus histiocytosis with massive lymphadenopathy is rare in children. The article describes a case of Rosai — Dorfman disease in 3-year-old boy. The emergence of lymphadenopathy, mainly cervical one, with synchronous lesion of extranodal area — spleen, enabled to suspect Rosai — Dorfman disease. To clarify the diagnosis, ultrasound has been used. The final diagnosis was established as a result of surgical removal of the lymph nodes of the neck, histological and histochemical study. Following the surgery, the child was discharged for further treatment at the department of oncohematology.
болезнь Розаи — Дорфмана, гистиоцитоз, дети.
хвороба Розаї — Дорфмана, гістіоцитоз, діти.
Rosai — Dorfman disease, histiocytosis, children.
Гистиоцитарные синдромы включают разнообразную группу заболеваний, которые характеризуются пролиферативными процессами в клетках системы фагоцитирующих мононуклеаров. В зависимости от степени зрелости и дифференцировки гистиоцитарных элементов существуют различные формы заболевания, имеющие особенности клинического течения, прогноза и лечения. В соответствии с данными Международного общества по изучению гистиоцитозов выделяют распространенную группу гистиоцитозов из клеток Лангерганса (I класс), существенно реже встречающиеся гистиоцитозы нелангергансового типа (II класс) и злокачественные гистиоцитарные заболевания (III класс) [1].
Синусный гистиоцитоз с массивной лимфаденопатией (СГМЛ) впервые описан в 1969 году Juan Rosai и Ronald Dorfman, относится к нелангергансоклеточным гистиоцитозам и представляет собой редкое заболевание, характеризующееся накоплением пролиферирующих гистиоцитов как в синусах лимфатических узлов с их массивным увеличением, так и в экстранодальных зонах [2]. До настоящего времени его этиология не установлена. Данное заболевание редко встречается в любом возрасте, но относительно часто выявляется у детей и подростков [5]. В процесс вовлекаются преимущественно шейные лимфатические узлы с их двусторонним поражением. Более редкой локализацией данного заболевания являются подмышечные, медиастинальные и паховые лимфоузлы [3, 4]. За последние 20 лет в нашей клинике лечился только один ребенок с болезнью Розаи — Дорфмана. В подтверждение сказанного приводим наше наблюдение.
Больной Г., 3 года (история болезни № 2399), поступил 03.03.2014 г. в хирургическое отделение областной детской клинической больницы (ОДКБ) г. Донецка с жалобами на наличие опухолевидного образования на боковой поверхности шеи слева. Болеет в течение месяца, когда родители заметили асимметрию и деформирующую припухлость шеи. Ребенок получал консервативное лечение по месту жительства без положительного эффекта. Ввиду неэффективности проводимой терапии пациент был консультирован гематологом, а затем направлен в клинику детской хирургии ОДКБ.
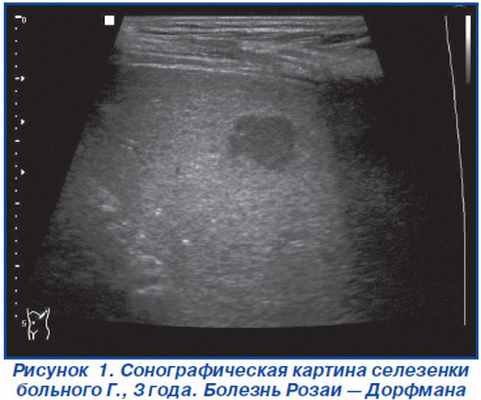
Анамнез жизни без особенностей. Родился путем кесарева сечения. Вес при рождении 3800 г. Привит, растет и развивается по возрасту. Аллергию отрицают. На диспансерном учете не состоит. Наследственность не отягощена. При поступлении самочувствие удовлетворительное. Кожные покровы чистые. В легких дыхание пуэрильное. Деятельность сердца ритмичная. Живот не вздут, мягкий, доступен пальпации во всех отделах. Место болезни: в области шеи слева, в проекции верхней трети кивательной мышцы, определяется бугристое образование неправильной округлой формы, около 4,0 см в диаметре, плотноэластичной консистенции, безболезненное при пальпации, кожа над опухолью не изменена. В клинике ребенок обследован. В общеклиническом анализе крови — без патологии. Обзорная рентгенография органов грудной клетки изменений не выявила. Произведено УЗИ шеи: слева под кивательной мышцей визуализируются увеличенные лимфатические узлы, один из которых размером 31 x 18 x 23 мм, округлой формы, гипоэхогенные, однородные. Ниже него, под кивательной мышцей, визуализируется цепочка мелких (до 8 мм) лимфатических узлов, с четкими ровными контурами. Сосудистый рисунок в узлах сохранен. Окружающие ткани отечные. УЗИ органов брюшной полости: эхопризнаки очаговых изменений селезенки (рис. 1), диффузные изменения печени. Был выставлен клинический диагноз: подозрение на лимфогранулематоз с преимущественным поражением шейных лимфоузлов.
04.03.2014 г. произведено оперативное лечение (хирург — д.м.н., проф. Веселый С.В.). После обработки операционного поля по боковой поверхности шеи произведен разрез кожи длиной 4,0 см. Гемостаз. Рассечена поверхностная фасция, латеральное брюшко кивательной мышцы отведено медиально. При ревизии установлено, что опухоль представлена конгломератом лимфоузлов 4,0 x 4,5 см, в капсуле, белесовато-серого цвета. Конгломерат лимфатических узлов в полном объеме мобилизован из окружающих тканей и удален. Рана дренирована резиновым выпускником через отдельную контрапертуру. Послойное ушивание раны. Туалет. Асептическая повязка. Произведены пункция и аспирация костного мозга из грудины и гребней обеих подвздошных костей, трепан-биопсия обеих подвздошных костей. Диагноз после операции: лимфопролиферативное заболевание, лимфогранулематоз?
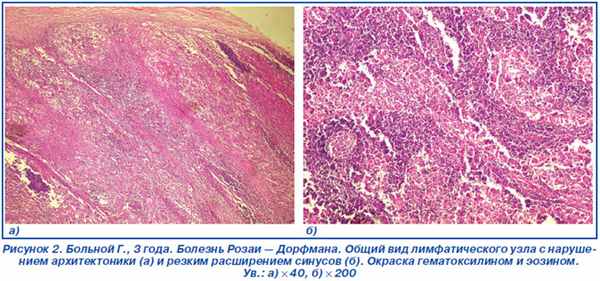
Гистологическое заключение (№ 1619–34). В лимфатических узлах капсула утолщена за счет разрастания плотной волокнистой соединительной ткани, на большом протяжении структура нарушена с резким расширением синусов (рис. 2).
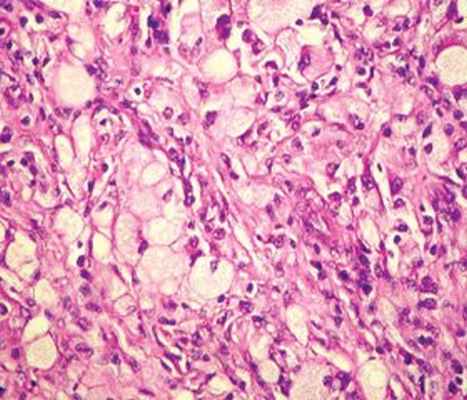
В просвете синусов располагаются лимфоциты, плазматические клетки и большое количество крупных полигональных гистиоцитов с нечеткими контурами, обильной эозинофильной, местами вакуолизированной цитоплазмой. Ядра этих клеток крупные, округлой формы, гипохромные, с четко выраженным, парацентрально расположенным ядрышком и мелкодисперсным хроматином, сконцентрированным преимущественно под ядерной оболочкой (рис. 3а).
Во многих полях зрения выражен эмпериополез с наличием лимфоцитов и плазматических клеток в цитоплазме гистиоцитов (рис. 3б). При иммуногистохимическом исследовании данные клетки экспрессируют S100 (рис. 3г), негативны на CD1a.
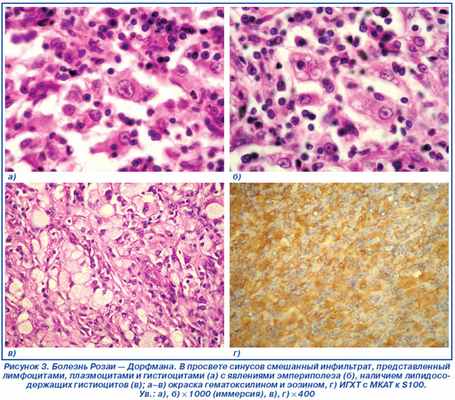
В просвете синусов отмечаются скопления крупных клеток различной формы со светлой пенистой цитоплазмой и округлым светлым ядром, смещенным на периферию клетки (липидосодержащие гистиоциты) (рис. 3в). Заключение: гистологическая картина и иммунофенотип клеток соответствуют синусовому гистиоцитозу с массивной лимфаденопатией (болезнь Розаи — Дорфмана).
Результаты миелограммы: красный росток сокращен, эритропоэз по эритроцитарному типу, среди лимфоцитов встречаются в небольшом количестве атипичные лимфоциты с неправильной формой ядра и бороздчатой структурой хроматина.
Таким образом, синусный гистиоцитоз с массивной лимфаденопатией встречается у детей редко. Заподозрить болезнь Розаи — Дорфмана позволяет возникновение лимфаденопатии, преимущественно цервикальной, с одновременным поражением экстранодальных зон (чаще всего селезенки). Для уточнения диагноза целесообразно использование УЗИ, компьютерной томографии. Окончательный диагноз устанавливается после хирургического удаления (биопсии) конгломерата лимфоузлов, гистологического и гистохимического исследования.
1. Львов А.Н., Волощук И.Н., Варшавский В.А., Горбачева Ю.В., Бобко С.И. Синусный гистиоцитоз (болезнь Розаи — Дорфмана): клиническое наблюдение // Вестник дерматологии и венерологии. — 2011. — № 5. — С. 115–120.
2. Райт Д., Эддис Б., Леонг Э. Морфологическая диагностика патологии лимфатических узлов. — М.: Медлит, 2008. — 176 с.
3. Duval M., Nguyen V.H., Daniel S.J. Rosai-Dorfman disease: An uncommon cause of massive cervical adenopathy in a two-year-old female // Otolaryngol. Head Neck Surg. — 2009. — Vol. 140, № 2. — Р. 274–275.
4. Felipe Barbosa Lima, Pedro Samuel de Valões Barcelos, Ana Paula Nunes Constâncio. Rosai-Dorfman disease with spontaneous resolution: case report of a child // Rev. Bras. Hematol. Hemoter. — 2011. — Vol. 33, № 4. — Р. 312–314.
5. Juskevicius R., Finlay J.L. Rosai-Dorfman disease of the parotid gland, cytologic and histopathologic findings with immunohistochemical correlation // Arch. Pathol. Lab. Med. — 2001. — Vol. 125. — P. 1348–1350.
Читайте также: