Диагностика хронического идиопатического миелофиброза (ХИМФ) - дифференциация
Добавил пользователь Владимир З. Обновлено: 30.01.2026
Клиника хронического идиопатического миелофиброза (ХИМФ) - спленомегалия, портальная гипертензия, анемия, асцит
Картина крови при хроническом идиопатическом миелофиброзе (ХИМФ) отличается большим разнообразием. Показатели красной крови на момент постановки диагноза чаще нормальны или несколько снижены, но у 15—25 % больных уже имеется анемия. Повышение показателей красной крови, обычно клинически бессимптомное, отмечается у 5—6 % больных.
У большинства больных наблюдается нейтрофильный лейкоцитоз (10—15•10 9 /л), очень характерен выраженный палочкоядерный сдвиг и присутствие единичных мета- и миелоцитов.
У 10—15 % больных наблюдается более выраженный лейкоцитоз (>20•10 9 /л) и большая степень левого сдвига лейкоцитарной формулы крови, циркуляция единичных бластов. Число тромбоцитов нормально у 30—40 % больных и увеличено примерно у 15 %. Отмечается повышение уровня лактатдегидрогеназы, коррелирующее с числом лейкоцитов. Морфологические изменения эритроцитов — частая находка при хроническом идиопатическом миелофиброзе (ХИМФ). Клинические симптомы хронического идиопатического миелофиброза можно разделить на:
1) ассоциированные со значительным увеличением селезенки;
2) обусловленные усиленным клеточным катаболизмом;
3) обусловленные недостаточностью костного мозга.
Под последними имеются в виду анемический и тромбоцитопенический синдромы, хотя в действительности в их развитии участвуют разные патогенетические механизмы.
В зависимости от давности хронического идиопатического миелофиброза (ХИМФ) размеры селезенки широко варьируют. Со временем она достигает гигантских размеров, занимает всю левую и часть правой половины живота, отличается повышенной плотностью и бугристостью. Субъективные расстройства, вызываемые большой селезенкой, — это чувство тяжести, ощущение сдавления (малой вместимости) желудка и кишечника (неустойчивый стул), периодические острые боли, вызываемые инфарктом селезенки и периспленитом.
Классической причиной увеличения селезенки оказывается миелоидная метаплазия селезенки (ММС), но следующей возможной причиной спленомегалии является осложнение портальной гипертонией, а также увеличение депонирующей и секвестрирующей функций селезенки, т. е. рабочая гипертрофия органа.

Гепатомегалия и портальная гипертензия при хроническом идиопатическом миелофиброзе
Более чем у половины больных при установлении диагноза определяют гепатомегалию. Изолированное увеличение печени изредка возможно, как и преобладание гепатомегалии над спленомегалией. Значительное увеличение печени наблюдается обычно у больных, перенесших спленэктомию (СЭ), а частота прогрессирующей гепатомегалии после СЭ составляет 26—22 %.
Функциональные нарушения печени редки, чаще наблюдаются в терминальной стадии заболевания.
Развитие синдрома портальной гипертонии сопровождается значительным увеличением селезенки, не обусловленным ее участием в кроветворении, варикозным расширением вен пищевода, а затем периферическими отеками и асцитом. По данным Silverstein, портальная гипертония осложняет течение хронического идиопатического миелофиброза в 6—8 %, из них в 70 % она является результатом гиперкинетического тока крови и в 30 % — внутрипеченочных блоков. Их причины — миелоидная метаплазия, которая локализуется в синусоидах печени, вызываемый ею реактивный фиброз, а в отдельных случаях — постнекротический цирроз печени, обусловленный перенесенным гепатитом.
Среди вариантов портальной гипертонии, частота которой другими авторами оценивается в 10—15 %, выделяют пресинусоидальный тромботический блок, синусоидальную обструкцию, вызываемую миелоидной метаплазией печени в сочетании с гиперкинетическим током крови, постсинусоидальный, очевидно, тромботический блок, аналогичный синдрому Бадда — Киари.
Считается, что при функциональном внутрипеченочном портальном блоке анатомических изменений в печени нет, но есть и такая точка зрения, что для развития портальной гипертонии одного гиперкинетического тока крови недостаточно и структурные нарушения в печени должны присутствовать.
Уровень блока току крови в портальной системе в настоящее время определяется неинвазивным методом — ультразвуковой допплерографией.
При подпеченочной портальной гипертензии (ПГ) (тромбоз селезеночной или воротной вены) имеются большие размеры селезенки и отсутствие или незначительность увеличения печени. При пункции селезенки кровь поступает в шприц под большим давлением, в пунктате селезенки элементов миелоидной метаплазии обычно мало, за исключением случаев с большой продолжительностью заболевания и смешанным генезом спленомегалии.
При осложнении тромбозом надпеченочных вен клиническая картина выглядит значительно более драматично: болевой синдром имеет различную выраженность, но может и отсутствовать; формируется массивный, резистентный к лечению асцит, нередко желтуха, признаки печеночной недостаточности (клинические и лабораторные) тяжелое общее истощение; возможны периодические кровотечения из расширенных вен пищевода и желудка (кровавая рвота и мелена). Печень обычно значительно увеличена, тогда как селезенка — умеренно. Течение синдрома Бадда — Киари может быть острым, подострым и хроническим.
Неизвестно почему, но у всех шести наблюдаемых нами больных это осложнение возникло на раннем этапе, до постановки гематологического диагноза, и у женщин молодого возраста. Изменения в анализах крови были не столь однозначными, чтобы на их основании поставить гематологический диагноз и уточнить, какой именно. С подобной ситуацией сталкиваются и другие авторы. Расширение возможностей углубленного обследования больных с помощью мегакариоцитарной и эритроидной культур и цитогенетического анализа позволило прийти к заключению, что у 2/3 больных этот синдром является осложнением ХМПЗ.
Склонность к тромбозам в системе воротной, надпеченочных и чревных вен в целом отмечена преимущественно у больных эссенциальной тромбоцитемией (ЭТ) с моноклональным ге-мопоэзом, у носителей гена PRV-1, у больных со спонтанным ростом эритроидной культуры. Пока неизвестно, в какой степени эти патогенетические особенности распространяются на больных хроническим идиопатическим миелофиброзом (ХИМФ).
В клиническом отношении значение проблемы портальной гипертензии у больных хроническим идиопатическим миелофиброзом весьма существенно. Ее своевременная диагностика позволяет принять решение в пользу назначения спленэк-томии у больных с под- и внутрипеченочной портальной гипертензией и назначить адекватное консервативное или хирургическое (наложение шунтов) лечение при осложнении тромбозом надпеченочных вен.
Чаще всего большие размеры селезенки врачи относят за счет основного заболевания и проводят довольно агрессивную терапию с целью ее сокращения, что в случаях осложнения портальной гипертензией обречено на неудачу и может привести к серьезным проблемам.
В одном нашем наблюдении больную лечили по месту жительства миелосаном в течение 6 мес с целью сокращения размеров селезенки. В анализах крови наблюдались только небольшой тромбоцитоз и нейтрофилез. Заболевание расценивалось как сублейкемический миелоз. Селезенка была увеличена до уровня пупка. К концу этого лечения развился тяжелый геморрагический тромбоцитопенический синдром, в течение 3 мес больная находилась в критическом состоянии. Еще через 3 мес она была подвергнута спленэктомии. На операции выявлен цирроз печени без миелоидной метаплазии селезенки (ММС). Диагноз пересмотрен в пользу эссенциальной тромбоцитемии (ЭТ).
В течение последующих 5 лет больная находилась в хорошем состоянии и принимала гидроксимочевину в небольшой дозе, которая контролировала тромбоцитоз. Затем внезапно развился тромбоз в системе мезентериальных сосудов, распознанный с опозданием. Во время операции удалена значительная часть тонкого кишечника.
Случай демонстрирует часто имеющую место неточность гематологического диагноза в группе ХМПЗ, нераспознанную внутрипеченочную портальную гипертензию, в данном случае, видимо, обусловленную сопутствующим заболеванием, неадекватную цитостатическую терапию, осложнившуюся гипоплазией кроветворения и геморрагическим синдромом, а также развитие тромбоза мезентериальных сосудов при контролируемом тромбоцитозе.
Причиной развития асцита может оказаться не только портальная гипертензия, но и имплантация очагов кроветворения на брюшине и сальнике. В таких случаях в асцитической жидкости обнаруживают мегакариоциты и гранулоциты. Плевральный и абдоминальный выпот часто носит геморрагический характер. Эта и другие атипичные локализации миелоидной метаплазии: в лимфатических узлах со сдавлением спинного мозга, тонком кишечнике, средостении, почках, легких, других висцеральных органах — относятся к числу раритетов. Имеются данные об увеличении периферических лимфатических узлов у 32 % больных, но, по нашим наблюдениям, это значительно более редкий феномен.
К симптомам, обусловленным клеточным гиперкатаболизмом, относятся потеря массы тела и повышение температуры тела, гиперурикемия. Она может быть бессимптомной или протекать с признаками подагрической полиартралгии, подагры, мочекаменной болезни, осложняться хроническим пиелонефритом, обтурацией мочеточников, хронической почечной недостаточностью. У отдельных больных интенсивность камнеобразования в почках необычайно велика. Развитию урикемии способствует проведение массивной цитостатической терапии .
Хотя повышение температуры тела может быть результатом клеточного гиперкатаболизма, это справедливо по отношению к умеренному субфебрилитету, а значительные подъемы температуры тела обычно обусловлены инфекцией, особенно мочевыводящих путей, или латентно протекающим МДС-синдромом, который может проявиться как типичный, развернутый острый лейкоз через ряд месяцев и даже лет.

В случаях, протекающих с количественной и качественной патологией тромбоцитов, возможны сосудистые осложнения: тромботические микроциркуляторные расстройства, тромбозы артерий и вен, геморрагический синдром, ДВС-синдром.
Внутренние кровотечения обычно обусловлены разрывом вен пищевода при осложнении портальной гипертонией. Присущая этому заболеванию, как и другим ХМПЗ, качественная дефектность тромбоцитов объясняет появление экхимозов на коже при сравнительно умеренной тромбоцитопении. Несостоятельность гемостаза особенно четко проявляет себя при спленэктомии.
Анемический синдром нередко выходит на передний план, особенно в поздних стадиях заболевания. Его причины разнообразны. Среди них могут быть:
• недостаточное образование эритроцитов;
• гиперволемия;
• усиление депонирования и секвестрации клеток крови в увеличенной селезенке (гиперспле-низм);
• аутоиммунный гемолиз эритроцитов;
• ускоренный гемолиз эритроцитов в результате синдрома пароксизмальной ночной гемоглобинурии или ферментных дефектов (дефицит Г-6-ФДГ и др.);
• дефицит железа и фолиевой кислоты.
Количественная недостаточность эритропоэза определяется замещением кроветворного костного мозга миелофиброзом и остеомиелосклерозом с возможным присутствием жировой ткани. Компенсаторный эритропоэз в трубчатых костях со временем также редуцируется, а компенсаторные возможности селезеночного эритропоэза ограничены его частой неэффективностью и одновременным усилением депонирования и деструкции клеток крови в большой селезенке.
Гемодилюционная анемия является результатом гиперволемии, обусловленной спленомегалией. Она хорошо переносится больными и является по существу только лабораторным феноменом.
Дефекты мембраны эритроцитов, сходные с наблюдаемыми при пароксизмальной ночной гемоглобинурии (ПНГ), описаны при хроническом идиопатическом миелофиброзе многими авторами. Их последствием является синдром гемолитической анемии. Повышенному гемолизу эритроцитов способствует и усиление перекисного окисления липидов клеточных мембран эритроцитов.
Дефицит фолиевой кислоты, приводящий к появлению макроцитарной анемии с кольцами Кебота, базофильной пунктацией эритроцитов, тельцами Жолли, наблюдается в поздней стадии заболевания. Его объясняют повышенным расходом фолиевой кислоты при усиленном гемопоэзе.
К количественной недостаточности эритропоэза приводит и его подавление при прогрессирующей гиперплазии лейкоцитарного ростка, хронической и острой. Возможно развитие сидеробластной анемии без- и с малопроцентной бластемией, которая является предстадией острого лейкоза. Описаны случаи парциальной красноклеточной аплазии, завершившиеся острым лейкозом. Отметим, что обычно к анемии приводит сочетание нескольких причинных факторов. Удельный вес каждого из них подлежит уточнению, что особенно необходимо при решении вопроса о назначении спленэктомии. Это же относится и к тромбоцитопеническому синдрому. Причинами его развития являются:
• усиление депонирования и деструкции тромбоцитов в увеличенной селезенке (и печени);
• вторичный аутоиммунный гемолиз тромбоцитов;
• нарушение образования тромбоцитов в результате редукции числа мегакариоцитов или их качественной дефектности;
• сочетание этих процессов;
• ДВС-синдром (тромбоцитопения потребления).
Среди известных клинических проявлений заболевания могут иметь место и аутоимунные симптомы, такие как дерматиты и кожные васкулиты, опосредованные активацией Т-лимфоцитов.
При рентгенографическом исследовании костного скелета обнаруживаются признаки уплотнения структуры плоских костей, особенно позвонков, иногда эбурнеация трубчатых костей с сужением их просвета, иногда определяют очаговый остеолиз.
Эволюция хронического идиопатического миелофиброза характеризуется постепенным нарастанием лейкоцитоза при исходно различном числе лейкоцитов: нормальном, сниженном и повышенном. Развитие острого лейкоза наблюдается как в случаях прогрессирующего лейкоцитоза (более 30•109/л), так и лейкопении (3•109/л). Большинство больных не доживает до развития типичного острого лейкоза. Картины рефрактерной анемии, тромбоцитопении, панцитопении или, наоборот, нарастающего лейкоцитоза и левого сдвига лейкоцитарной формулы, выход из-под контроля размеров селезенки, появление упорной асептической лихорадки являются показателями терминальной фазы заболевания.
Терминальное состояние больных может определяться и висцеральными осложнениями: сердечной, печеночной и почечной недостаточностью, к которым имеются соответствующие патофизиологические предпосылки. Жизнь больных может оборвать острое кровотечение из расширенных вен пищевода при осложнении портальной гипертонией. Гематологические и соматические терминальные состояния нередко сочетаются с тяжелой общей дистрофией и компрессионными осложнениями.
Течение заболевания обычно хроническое, медленно прогрессирующее. Длительное время оно может протекать почти полностью бессимптомно, и только случайное исследование анализа крови выявляет небольшие отклонения или осмотр позволяет обнаружить спленомегалию. При доброкачественном, медленно протекающем варианте хронического идиопатического миелофиброза (ХИМФ) наблюдаются «мягкая» степень развития миелофиброза или только клеточная пролиферация, отсутствие анемии, нормальное или незначительно повышенное число лейкоцитов и тромбоцитов, отсутствие циркулирующих бластов и незначительное число CD334-позитивных клеток в периферической крови. У подобных больных прогрессия заболевания может длительное время отсутствовать.
Возможны варианты более злокачественного течения, которые терминологически обозначаются как варианты с ускоренным развитием терминальной фазы. Выделяют и вариант с подострым течением, при котором морфологические изменения костного мозга не отличаются от обычного хронического идиопатического миелофиброза, но при этом нет значительной спленомегалии, и довольно быстро развиваются анемия и другие проявления недостаточности кроветворения.
К атипичным хронически протекающим вариантам хронического идиопатического миелофиброза можно отнести и так называемые гибридные формы, имеющие признаки двух ХМПЗ:
• истинной полицитемии и хронического идиопатического миелофиброза — упорная плетора, но раннее и значительное увеличение селезенки за счет миелоидной метаплазии, лейкоцитозные формы, устойчивость к цитостатической терапии, эволюция в острый лейкоз через длительный период зрелоклеточного лейкоцитоза, частые цитогенетические аномалии;
• хронического идиопатического миелофиброза и эссенциальная тромбоцитемия. С первым заболеванием их сближает выраженный миелофиброз, со вторым — небольшая величина селезенки, значительный тромбоцитоз, отсутствие лейкоэритробластической картины периферической крови, характерной для иМФ.
Атипичными в определенном смысле являются и случаи хронического идиопатического миелофиброза с существенным увеличением печени, а не селезенки, а также осложненные портальной гипертонией, синдромом Бадда — Киари, при которых проявления гематологического заболевания по анализу крови могут быть минимальными, и только культуральные и цитогенетические исследования выявляют их принадлежность к ХМПЗ.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Диагностика хронического идиопатического миелофиброза (ХИМФ) - дифференциация
Диагностика хронического идиопатического миелофиброза (ХИМФ) до настоящего времени основана на критериях, предложеных PVSG, США. Ими являются:
• миелофиброз, занимающий более 1/3 среза гистологического препарата костного мозга подвздошной кости;
• лейкоэритробластическая картина периферической крови;
• спленомегалия;
• отсутствие увеличения массы циркулирующих эритроцитов (МЦЭ);
• отсутствие Ph+-хромосомы в клетках костного мозга.
Первые три критерия прямо подтверждают диагноз, а два последующих — это критерии исключения других заболеваний.
Под лейкоэритробластической картиной периферической крови понимается небольшой или умеренный лейкоцитоз, увеличение содержания палочко-ядерных форм, небольшой левый сдвиг в лейкоцитарной формуле, присутствие единичных эритронормобластов и каплевидных (tear-drop) пойкилоцитов.
Эти критерии имеют ряд недостатков, к тому же они не рассчитаны на диагностику ранней стадии хронического идиопатического миелофиброза, при которой изменения в анализах периферической крови могут отсутствовать.
Морфологический критерий диагноза хронического идиопатического миелофиброза — присутствие миелофиброза — с учетом наличия клеточнопролиферативной, т. е. дофиброзной стадии заболевания, имеет относительное значение. Эти факторы и некоторые другие объясняют потребность в переработке критериев диагностики хронического идиопатического миелофиброза (ХИМФ), и такая попытка была сделана Итальянской согласительной комиссией.
Важными для установления диагноза авторы считают 2 основных и 6 дополнительных (факультативных) критериев.

К основным критериям хронического идиопатического миелофиброза отнесены:
1) присутствие миелофиброза в гистоморфологическом препарате костного мозга;
2) отсутствие Ph+-хромосомы или BCR-ABL в миелоидных клетках.
Факультативные критерии хронического идиопатического миелофиброза:
1) спленомегалия;
2) анизопойкилоцитоз с каплевидной формой эритроцитов различной степени выраженности;
3) присутствие в периферической крови миелоидных клеток и эритронормобластов;
4) присутствие в срезах костного мозга кластеров мегакариоцитов;
5) аномалия величины, формы и структуры мегакариоцитов;
6) миелоидная метаплазия селезенки.
Для постановки диагноза необходимо присутствие 2 основных критериев и 4 из 6 факультативных.
Существует много оттенков в состоянии гемопоэза, степени и расположении миелофиброза, остеосклероза и жировой ткани, на основании которых можно углубить и расширить патоморфологическую классификацию хронического идиопатического миелофиброза и его стадийность, однако основой морфологической диагностики являются гиперплазия и морфологическая анаплазия мегакариоцитов и их кластерное расположение в сочетании с миелофиброзом.
При морфологической диагностике хронического идиопатического миелофиброза большое значение имеют гиперплазия сосудов стромы и их структурные изменения, которые критерии итальянских авторов не учитывают. Вероятно, это не последний вариант диагностических критериев хронического идиопатического миелофиброза. Когда поиск молекулярного маркера заболевания завершится успехом, подходы к диагностике данного заболевания упростятся.
В задачи диагностики должны входить и определение клинической стадии, вариантов и фаз течения заболевания, разработка которых происходит в настоящее время. В терминологической расшифровке нуждается и все понятие терминальной стадии заболевания.
Поскольку и другие ХМПЗ могут завершиться миелофиброзом с миелоидной метаплазией (МММ), возникает необходимость в дифференциальной диагностике хронического идиопатического миелофиброза от постполицитемического и завершающего эссенциальной тромбоцитемией (ЭТ) миелофиброза, хотя на этой стадии первичность или вторичность миелофиброза с миелоидной метаплазией большого практического значения, во всяком случае для его лечения, не имеет.

При дифференциальной диагностике хронического идиопатического миелофиброза и постэритремического миелофиброза решающее значение имеет анамнез заболевания: если обнаружению миелофиброза предшествовал длительный срок заболевания истинной полицитемией (ИП), то это постполицитемический (постэритремический) миелофиброз. Аналогичным образом диагностируют миелофиброз, завершивший длительное течение эссенциальной тромбоцитемией (ЭТ).
В дифференциальной диагностике на ранних стадиях обоих заболеваний следует учитывать различие причин спленомегалии: при хроническом идиопатическом миелофиброзе (ХИМФ) — это миелоидная метаплазия, при эссенциальной тромбоцитемии (ЭТ) — депонирование тромбоцитов. Каждому из этих заболеваний присущи свои патоморфологические диагностические критерии.
Некоторые прежние диагностические принципы, например исключение диагноза хронического идиопатического миелофиброза (ХИМФ) при увеличении показателей красной крови, даже если все остальные параметры заболевания тождественны хроническому идиопатическому миелофиброзу (ХИМФ), вероятно, устарели.
Теперь, когда мы больше знаем, почему это может быть при хроническом идиопатическом миелофиброзе (ХИМФ) (носительство больными хроническим идиопатическим миелофиброзом гена PRV-1 или наличие спонтанного роста эритроидных колоний), исключать диагноз хронического идиопатического миелофиброза на этом основании при соответствии всех остальных его проявлений критериям его диагностики, включая и патоморфологический, было бы неправильным.
У ряда больных хроническим идиопатическим миелофиброзом имеется и симптом кожного зуда, связанный с приемом водных процедур, который прежде считался патогномоничным для истинной полицитемии (ИП). Большее значение следует придавать патоморфологическим разграничительным критериям отдельных хронических миелопролиферативных заболеваний (ХМПЗ).
Симптомокомплекс, сходный с хроническим идиопатическим миелофиброзом, может наблюдаться в редких случаях хронического миелолейкоза, включая длительное существование умеренного зрелоклеточного нейтрофильного лейкоцитоза, поэтому исследование Ph-хромосомы обоснованно включено в диагностические критерии хронического идиопатического миелофиброза. Гистоморфологические различия между хроническим миелолейкозом и хроническим идиопатическим миелофиброзом также существенны, но имеют второстепенное значение.
Проблема дифференциальной диагностики хронического идиопатического миелофиброза со злокачественным миелофиброзом, миелодиспластическими синдромами, когда миелодиспластический синдром (МДС) сопровождается реактивным миелофиброзом, так называемым острым панмиелозом, разрешается с помощью оценки клинических, гематологических и патоморфологических данных.
Большинству этих заболеваний присущ симптом тяжелой анемии с начала заболевания, лихорадочный синдром, острое или подострое течение, что отличает их от хронического идиопатического миелофиброза.
В дифференциальной диагностике хронического идиопатического миелофиброза с раковым остеосклерозом и миелокарциноматозом имеют значение:
• наличие на данный момент или в анамнезе раковых заболеваний (отметим, что между временем их диагностики, кажущимся вполне успешным лечением и обнаружением ракового остеосклероза может пройти много лет);
• клиническая картина миелокарциноматоза, для которой характерны лихорадка и мучительные оссалгии, не свойственные хроническому идиопатическому миелофиброзу, за исключением периода развития острого лейкоза. В то же время раковый остеосклероз может и не сопровождаться болями в костях;
• наличие признаков метастазирования в другие органы (лимфатические узлы, легкие, печень);
• обнаружение раковых клеток в цитологическом или гистологическом препарате костного мозга. Селезенка может быть увеличена в обоих случаях, но при миелокарциноматозе и раковом остеосклерозе обычно незначительно;
• обнаружение на рентгенограммах очагов деструкции в костях, характерных для миелокарциноматоза;
• диффузный характер остеосклероза при его раковом происхождении. Этот признак непостоянный. Изредка возникает необходимость в проведении дифференциального диагноза между хроническим идиопатическим миелофиброзом (ХИМФ), осложненным портальной гипертонией или протекающим со значительным увеличением печени, и самостоятельными заболеваниями печени.
В классических случаях детальный анализ периферической крови разрешает диагностическую проблему: лейкоцитоз, тромбоцитоз, эритрокариоцитоз, нейтрофилез не характерны для самостоятельных заболеваний печени. Трудности возникают при отсутствии этих гематологических изменений при хроническом идиопатическом миелофиброзе, что вполне возможно.
В связи с этим рекомендуется во всех неясных случаях провести полное обследование больных, включающее трепанобиопсию подвздошной кости, пункцию или биопсию печени при отсутствии противопоказаний, культуральные исследования, которые позволят верифицировать гематологический диагноз или его отвергнуть.
Хронический идиопатический миелофиброз (ХИМФ) - определение, эпидемиология, патогенез
Хронический идиопатический миелофиброз (ХИМФ) относится к группе клоновых хронических миелопролиферативных заболеваний (ХМПЗ). Уникальной особенностью ХИМФ является раннее и значительное развитие фиброза костного мозга и миелоидной метаплазии селезенки (ММС).
Существует много синонимов для обозначения данного заболевания: сублейкемический миелоз, алейкемический миелоз, остеомиелопоэтическая дисплазия, агногенная или просто миелоидная метаплазия, миелосклероз с миелоидной метаплазией, хронический гранулоцитарно-мегакариоцитарный миелоз и многие другие. В классификации ВОЗ 2001 г. данное заболевание обозначено как «хронический идиопатический миелофиброз» и отнесено в группу хронических миелопролиферативных заболеваний.
Хронический идиопатический миелофиброз (ХИМФ) — сравнительно редкая патология. Заболеваемость составляет 0,3—1,5 на 100 000 населения в год, а средний возраст на момент заболевания — 60 лет. По данным французских авторов, в год регистрируется от 0,3 до 0,7 новых случаев заболевания.
Мужчины болеют несколько чаще женщин. В 90 % заболевание диагностируется после 40 лет, однако известны случаи заболевания хроническим идиопатическим миелофиброзом в молодом и даже детском возрасте. Отмечается более доброкачественное течение заболевания у детей.

Патогенез хронического идиопатического миелофиброза (ХИМФ)
Хронический идиопатический миелофиброз является клоновым миелопролиферативным заболеванием с первичным поражением стволовой кроветворной клетки, что доказывается обнаружением одного типа изоэнзимов глюкозо-6-фосфатдегидрогеназы в лейкоцитах, эритроцитах и мегакариоцитах больных миелофиброзом женщин-мулаток, а также анализом рестрикции Х-хромосомы у этих пациенток.
Имеются отдельные данные о том, что к патологическому клону при хроническом идиопатическом миелофиброзе относятся также В- и Т-лимфоциты, что является лишним подтверждением стволового уровня поражения гемопоэза при данном заболевании.
Патогенез хронического идиопатического миелофиброза (ХИМФ) нельзя считать полностью расшифрованным. Заболевание характеризуется пролиферацией гранулоцитов и мегакариоцитов с образованием экстрамедуллярных очагов кроветворения, главным образом в селезенке, т. е. развитием миелоидной метаплазии и фиброза костного мозга.
Характер и степень миелофиброза настолько отличает данное заболевание от других хронических миелопролиферативных заболеваний (ХМПЗ), что составляет основу его нозологического обозначения как хронический идиопатический миелофиброз. Между тем миелофиброз при этом заболевании вторичен, реактивен. Реактивный характер миелофиброза доказывается наличием двух типов изоэнзимов глюкозо-6-фосфатдегидрогеназы в фибробластах костного мозга у больных с одним типом фермента в кроветворных клетках: поликлональность фибробластов у больных хроническим идиопатическим миелофиброзом (ХИМФ) свидетельствует о реактивном характере миелофиброза.
Раннее образование экстрамедуллярных очагов гемопоэза, возможно, обусловлено повышенной при этом заболевании циркуляцией в крови полипотентных CD34-позитивных и линейно-рестриктированных предшественников гемопоэза — CFU-GEMM, BFU-E, CFU-GM, GFU-MK. В периферической крови больных ХИМФ количество циркулирующих гемопоэтических предшественников (CD34-позитивных клеток) почти в 2000 раз превышает норму. Высокая экспрессия стволовыми клетками при ХИМФ рецептора фактора стволовых клеток (c-Kit), по-видимому, обусловливает преимущества в пролиферации патологического клона.

Специфические хромосомные аберрации при хроническом идиопатическом миелофиброзе (ХИМФ) не описаны, но те же изменения, которые характерны для других заболеваний этой группы, встречаются у 45—60 % больных. В 90 % обнаруженные изменения представлены 13q-, 20q-, +8, +9, 12р-, +lq. В 46 % обнаруживаются только 13q- и 20q-. В одной из работ цитогетический анализ 14 больных выявил 13q- у 3 больных, 20q- у 1, у остальных десяти больных изменений не было. Эти и многие другие исследования подтверждают факт высокой частоты 13q- аберрации при хроническом идиопатическом миелофиброзе, что позволяет некоторым авторам высказать предположение о патогенетической связи между развитием миелофиброза и делецией 13q-.
Механизм этой связи может заключаться в том, что делегированный участок приходится на район хромосомы 13, в котором локализован какой-то пока не идентифицированный ген, кодирующий один из факторов, ингибирующих развитие опухоли. Другие авторы, однако, не считают, что 13q- и 20q- могут быть связаны с инактивацией какого-то гена. Данные, полученные авторами, которые использовали метод геномной гибридизации, позволил им предположить, что к патогенезу заболевания, скорее всего, имеет отношение изменение хромосомы 9.
В исследовании популяции CD34-позитивных клеток частота цитогенетических аномалий составила 80 %, причем в подавляющем большинстве обнаруживалась 13q-делеция. Когда исследовались зрелые лейкоциты, такие же нарушения кариотипа обнаруживались только в 34,8 % случаев. Часть CD34+-клеток характеризовалась также экспрессией CD38, антигена, экспрессированного на самых ранних гемопоэтических клетках, что подтверждает происхождение хронического идиопатического миелофиброза из самых примитивных гемопоэтических предшественников.
Об их пролиферативном преимуществе свидетельствовало нарастание числа этих клеток в культуре на 7-й и 14-й дни. Ни селезеночные фибробласты, ни В-лимфоциты не имели цитогенетических нарушений.
Несмотря на отсутствие диагностических хромосомных аберраций при хроническом идиопатическом миелофиброзе, значение цитогенетических исследований в прогнозе заболевания несомненно.

Анализ течения заболевания у 165 больных показал, что наличие 20q- и 13q- не влияет на прогноз, в то время как +8 и 12р- часто сочетаются с короткой продолжительностью заболевания и повышенной частотой развития острого лейкоза.
При хроническом идиопатическом миелофиброзе повышено содержание эритропоэти-на в крови, в то же время возможен спонтанный рост эритроидных колоний, с высокой частотой обнаруживается носительство гена PRV-1, повышено содержание тромбопоэтина, и степень этого повышения коррелирует с выраженностью миело-фиброза.
Установлено повышение чувствительности клеток-предшественниц при хроническом идиопатическом миелофиброзе к воздействию различных цитокинов и увеличение экспрессии рецепторов к соответствующим цитокинам: ИЛ-3, GM-CSF, эритропоэтину и стволовому фактору роста (SCF).
В то же время уровень экспрессии тромбопоэтинового рецептора Mpl на мегакариоцитах и тромбоцитах снижен, как это наблюдается при эссенциальной тромбоцитемии (ЭТ) и истинной полицитемии (ИП), из чего следует, что механизм гиперплазии мегакариоцитов и их повышенного колониеобразования в культуре не связан непосредственно с тромбопоэтином. В этом отношении представляет интерес установление повышенной экспрессии фактора транскрипции — GATA-1 — в мегакариоцитах и CD34-позитивных клетках по сравнению со здоровыми лицами. Этот фактор участвует в генерации из стволовой клетки мегакариоцитарных предшественников и бипотенциальных эритроидно-мегакариоцитарных колоний.
Обнаружено, что фибробласты селезенки при хроническом идиопатическом миелофиброзе экспрессируют определенный набор молекул адгезии, отличный от такового у здоровых, и что рост CD34-позитивных предшественников при этом заболевании зависит от их взаимодействия с фибробластами селезенки, что снижает их зависимость от внешних воздействий и способствует экспансии патологического клона в органе и прогрессированию ММС.
Развитие миелофиброза происходит под влиянием фиброгенных цитокинов с участием ряда других факторов и белков.
Многочисленными исследованиями показано, что при хроническом идиопатическом миелофиброзе (ХИМФ) увеличена концентрация как в крови, так и внутри клеток многих цитокинов, участвующих в формировании фиброза и в неоангиогенезе — ИЛ-1, ростового фактора, выделяемого тромбоцитами (platelet-derived growth factor — PDGF, трансформирующего ростового фактора b — TGF-b), основного фактора роста фибробластов (basic fibroblast growth factor- bFGF), фактора роста эндотелия сосудов (vascular endothelial growth factor — VEGF). TGF-b усиливает синтез коллагена и фибронектина, способствует деградации компонентов экстрамедуллярного матрикса, участвующего в образовании фиброзной ткани. Высвобождение фиброгенных цитокинов происходит непосредственно в костном мозге.
Уровень кальмодулина — белка, связывающего ионы кальция, участвующего в их транспорте и тем самым вносящего вклад в формирование фиброза, в моче больных хроническим идиопатическим миелофиброзом (ХИМФ) превышает уровень кальмодулина у здоровых в 3 раза.
Цитокины, повышение уровня которых обнаружено при хроническом идиопатическом миелофиброзе, продуцируются мегакариоцитами, тромбоцитами и моноцитами.
Основные цитокины, участвующие в формировании фиброза (PDGF, bFGF и TGF-b), содержатся в а-гранулах мегакариоцитов и высвобождаются под действием ИЛ-1.
Установлено, что пептид, носящий название субстанции Р и участвующий в передаче сигнала нейронами, содержится в повышенном количестве в крови больных хроническим идиопатическим миелофиброзом. Этот пептид индуцирует продукцию ИЛ-1. Другим источником повышенного уровня ИЛ-1 являются моноциты. При хроническом идиопатическом миелофиброзе они активированы, содержат увеличенное по сравнению с нормой количество TGF-b и даже в отсутствие стимуляции продуцируют ИЛ-1.
С помощью моноклональных антител получены дополнительные доказательства локализации TGF-b в мононуклеарах периферической крови, хотя этот фактор долго считался маркером мегакариоцитов.
Одновременно с фиброзирующим костный мозг действием TGF-b совместно с гранулоцитарно-макрофагальным и макрофагальным факторами роста (GM-CSF и M-SCF) стимулирует рост гранулоцитарно-макрофагальных предшественников, что способствует увеличенной при хроническом идиопатическом миелофиброзе миелопролиферации. Таким образом, при данном заболевании существует аутокринный механизм стимуляции как миелопролиферации, так и миелофиброза. Активированные стромальные клетки также синтезируют цитокины и компоненты экстрацеллюлярного матрикса. Эти факторы поддерживают клональную гемопоэтическую пролиферацию, ответ которой на регуляторные факторы роста при этом заболевании нарушен.
Неоангиогенез поддерживает как фиброзирование костного мозга, так и миелопролиферацию. Экспансия гемопоэтических клеток происходит не только вследствие стимуляции их роста, но и ослабления отрицательных регуляторных сигналов.
Между степенью развития миелофиброза и экспансией гемопоэтических предшественников в периферическую кровь и селезенку имеется прямая связь, однако ее нельзя объяснить простым механическим вытеснением гемопоэтических предшественников фиброзированным костным мозгом. Эта возможность выглядела бы вполне логично, если бы не было нарушений в рецепторном аппарате гемопоэтических клеток по отношению к цитокинам и экспансии кроветворения уже на клеточно-пролиферативной, дофиброзной стадии заболевания. Вероятны более сложные клеточно-клеточные взаимоотношения между гемопоэтическими предшественниками и фибробластами на уровне костного мозга и селезенки.
В заключение следует осветить еще одну сторону этой проблемы. Роль тромбопоэтина и его рецептора в патогенезе миелофиброза в эксперименте на мышах выглядит в высшей степени убедительно: мыши, инфицированные протоонкогеном V-Mpl, дают развитие синдрома миелопролиферации и миелофиброза, аналогичного человеческому. В развитии тромбопоэтининдуцированного миелофиброза у мышей ведущая роль принадлежит TGF-b.
У мышей, подвергшихся чрезмерному воздействию тромбопоэтина, а также у мышей — носителей мутантного гена фактора транскрипции GATA-1, в связи с чем у них имеется редуцированная экспрессия этого фактора транскрипции, принимающего участие в конечной дифференцировке мегакариоцитов и эритроцитов, а также при трансплантации мыши костно-мозговых клеток, которые генно-инженерным путем изменены так, что они гиперэкспрессируют ген тромбопоэтина, также развивается синдром, полностью идентичный хронический идиопатический миелофиброз (ХИМФ), — гиперпролиферация мегакариоцитов, миелофиброз и экстрамедуллярный гемопоэз с постепенным увеличением селезенки.
После трансплантации нормального костного мозга наступает реверсия миелофиброза, без трансплантации болезнь прогрессирует и часто заканчивается острым лейкозом. В то же время показано, что значительная часть мегакариоцитов больных хроническим идиопатическим миелофиброзом плохо или совсем не реагирует на антитела против GATA-1, что совместно с обнаруженным нарушением транскрипционных факторов Scl и FOG-1 в CD34+-клетках больных хроническим идиопатическим миелофиброзом свидетельствует, скорее всего, о множественных молекулярных нарушениях, имеющих значение в патогенезе данного заболевания у человека.
Миелофиброз ( Агногенная миелоидная метаплазия , Сублейкемический миелоз )
Миелофиброз – это хроническое гематологическое заболевание, характеризующееся опухолевой пролиферацией гемопоэтических стволовых клеток и фиброзом костного мозга. Основные клинические проявления включают симптомы опухолевой интоксикации и анемического синдрома (прогрессирующую слабость, бледность кожи и слизистых оболочек, потерю веса), а также увеличение селезенки (спленомегалию). Диагноз устанавливается на основании молекулярно-генетических исследований, изучения гистологической картины костного мозга. Лечение проводится с помощью химиотерапевтических препаратов. Хирургические методы лечения подразумевают трансплантацию костного мозга и удаление селезенки.
МКБ-10
Общие сведения
Миелофиброз (агногенная миелоидная метаплазия, сублейкемический миелоз) – злокачественное заболевание, при котором происходит постепенное замещение костного мозга опухолевыми стволовыми клетками и разрастающейся соединительной тканью. Впервые эту патологию описал немецкий врач Г. Хойк в 1879 году. А в 1951 году американским гематологом Уильямом Дамешеком миелофиброз был выделен в самостоятельную нозологическую единицу. При неблагоприятном течении миелофиброз способен трансформироваться в более тяжелую болезнь ‒ острый лейкоз. Распространенность миелофиброза составляет от 0,3 до 0,7 случаев на 100 тыс. населения. Пик заболеваемости приходится на возраст от 50 до 70 лет, но встречаются и молодые пациенты. Чаще страдают мужчины.
Причины миелофиброза
Существует первичный и вторичный сублейкемический миелоз. Точная причина первичного миелофиброза до сих пор не установлена. Наибольшей популярностью среди специалистов в области гематологии пользуется теория влияния генетической мутации. У большинства пациентов выявляются мутации гена тирозинкиназы (JAK2V617F), кальретикулина (CALR), тромбопоэтина (MPL), регулирующих экспрессию белков JAK-STAT сигнального пути. Гены локализуются в локусе хромосомы del3p24.
В качестве этиологического фактора изучается действие большой дозы радиоактивного излучения. Также рассматривается роль персистирующих вирусных инфекций (вируса простого герпеса, Эпштейна-Барра, цитомегаловируса), длительного приема оральных контрацептивов, миелосупрессивных лекарственных препаратов, контакта с различными органическими и неорганическими соединениями (бензолом, мышьяком). Вторичный миелофиброз развивается как исход других хронических миелопролиферативных заболеваний – истинной полицитемии, эссенциальной тромбоцитемии, хронического миелолейкоза.
Патогенез
В результате повышенной экспрессии сигнальных белков в одной из стволовых костномозговых клеток запускается активная пролиферация (опухолевая трансформация). Этот процесс сопровождается вторичным воспалением с выделением цитокинов и факторов роста. Факторы роста фибробластов и эндотелия сосудов индуцируют выработку стромальными клетками костного мозга большого количества коллагена и разрастание соединительной ткани (собственно фиброз). Постепенно нормальная ткань костного мозга замещается опухолью и соединительной тканью.
При массивном поражении опухолью костного мозга клетки крови, не достигнув стадии полного созревания, попадают в системный кровоток. Это приводит к образованию очагов экстрамедуллярного (внекостномозгового) кроветворения, главным образом в печени и селезенке. Распад опухоли ведет к высвобождению мочевой кислоты, которая откладывается в тканях суставов и почечных канальцах.
Симптомы миелофиброза
Длительное время пациент чувствует себя удовлетворительно. Через несколько лет от начала заболевания постепенно появляется опухолевая интоксикация в виде общей слабости, повышения температуры до субфебрильных цифр, потливости, усиливающейся по ночам. У больного снижается аппетит, он стремительно теряет в весе. Присоединяется анемический синдром (бледность кожных покровов, головокружение, учащение сердцебиения). Характерны носовые, десневые кровотечения, геморрагические высыпания на коже. Возникают боли в суставах, кожный зуд, боли в костях.
Пациент ощущает тяжесть и боли в левом подреберье вследствие выраженного увеличения селезенки. На фоне спленомегалии развивается синдром гиперспленизма, который заключается в массивном разрушении клеток крови (в основном эритроцитов) в синусоидах селезенки. В этом случае встречаются признаки гемолиза (желтушность кожи, слизистых оболочек, потемнение мочи).
Редкие симптомы связаны с необычной локализацией очагов экстрамедуллярного кроветворения – в легких (кашель, затруднение дыхания, кровохарканье), желудочно-кишечном тракте (боли в животе, кровавая диарея). При расположении очагов в центральной и периферической нервной системе наблюдаются эпилептические судороги, нарушения чувствительности, слабость движений в конечностях, вплоть до полного паралича.
Осложнения
При миелофиброзе часто образуются тромбы, которые приводят к острому нарушению мозгового кровообращения, инфаркту миокарда, тромбоэмболии легочной артерии. Стойкое снижение уровня лейкоцитов нередко сопряжено с различными инфекциями, приобретающими тяжелое течение. Наиболее неблагоприятным осложнением считается трансформация миелофиброза в миелолейкоз (бластный криз), трудно поддающийся терапии. К нетипичным осложнениям следует отнести патологические переломы из-за деструкции трубчатых костей и портальную гипертензию, причиной которой служит длительная обструкция микротромбами внутрипеченочных вен.
Диагностика
Курацией пациентов с миелофиброзом занимаются врачи-гематологи. При общем осмотре обращает на себя внимание изменение цвета кожных покровов, слизистых (бледность или желтушность), спленомегалия при пальпации и перкуссии селезенки, иногда достигающей гигантских размеров (до лобкового симфиза). Дополнительные методы диагностики включают:
- Общие лабораторные исследования. В начале заболевания в общем анализе крови выявляется увеличение эритроцитов, тромбоцитов, лейкоцитов, со временем сменяющееся на низкие показатели. Часто в периферической крови присутствуют незрелые формы эритроцитов, лейкоцитов (миелоциты, промиелоциты). В биохимическом анализе крови наблюдаются повышенные концентрации лактатдегидрогеназы (ЛДГ), ионизированного кальция. Отмечаются изменения коагулограммы – ускорение свертывания крови, уменьшение активированного частичного тромбопластинового времени, торможение процессов фибринолиза. В анализе мочи обнаруживаются уробилин, гемоглобин, ураты (соли мочевой кислоты).
- Исследование костного мозга. Образец костного мозга получают с помощью трепанобиопсии. Гистологическая картина зависит от фазы заболевания. Для ранней (префибротической фазы) характерны гиперплазия всех ростков кроветворения (гранулоцитарного, мегакариоцитарного, эритроидного) с незрелостью клеток. В позднюю (фибротическую) фазу определяется большое количество коллагеновых и ретикулярных волокон (фиброз), замещающих гемопоэтическую ткань, выраженная клеточная атипия. Высокий уровень бластных клеток (более 20%) свидетельствует о трансформации миелофиброза в острый лейкоз.
- Молекулярно-генетические тесты. Диагностика мутации генов JAK2V617F, CALR, MPL осуществляется методом FISH. Для идентификации аллельной нагрузки мутации проводится полимеразная цепная реакция real-time. Также выполняется HLA-типирование для решения вопроса о возможности трансплантации костного мозга.
- Цитогенетические и цитохимические анализы. При цитогенетическом исследовании (кариотипировании) клеток костного мозга находят аномалии 1, 3, 6 хромосом (транслокация, трисомия, комплексные нарушения). При анализе химического состава (цитохимии) нейтрофилов активность щелочной фосфатазы оказывается в 3 раза выше нормы.
Для достоверной постановки диагноза гематологическим сообществом разработаны специальные критерии. Большие критерии включают повышенную клеточность костного мозга с ретикулярным и коллагеновым фиброзом, наличие мутаций генов JAK2V617F, MPL, CALR. К малым критериям относятся анемия, спленомегалия, лейкоэритробластоз (присутствие в крови незрелых форм лейкоцитов, эритроцитов), а также повышение лактатдегидрогеназы. Диагноз считается подтвержденным, если имеются 2 больших критерия или 1 большой и 3 малых критерия.
Миелофиброз следует дифференцировать в первую очередь с гематологическими заболеваниями, такими как аутоиммунные гемолитические анемии, гемобластозы (лейкозы, лимфомы). Сочетание спленомегалии с симптомами интоксикации (слабостью, субфебрилитетом, ночной потливостью) требует исключения туберкулеза, подострого инфекционного эндокардита.
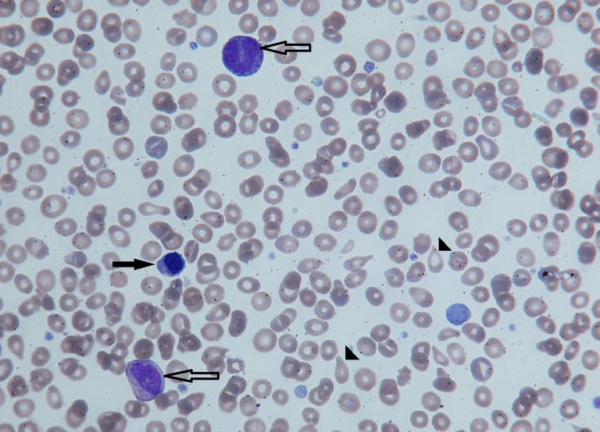
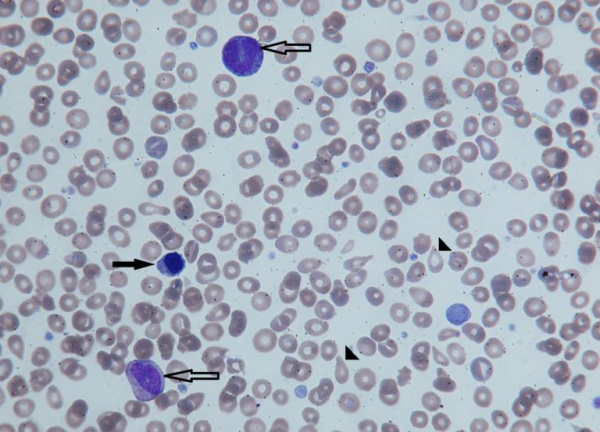
Незрелые формы эритроцитов (черная стрелка) и гранулоцитов (контурная стрелка) в периферической крови
Лечение миелофиброза
После постановки диагноза пациент должен быть госпитализирован в гематологический стационар. Для принятия решения о выборе тактики лечения необходимо определить степень риска, а именно - вероятность бластной трансформации и ориентировочную продолжительность жизни. С этой целью была создана «Международная шкала оценки риска и прогноза» (DIPSS). Она учитывает возраст пациента, количество форменных элементов крови, выраженность симптомов опухолевой интоксикации. Каждый признак соответствует одному баллу. Различают низкий, первый и второй промежуточный, высокий риски, при которых проводится дифференцированная терапия:
Прогноз и профилактика
Миелофиброз – это тяжелое заболевание с неблагоприятным прогнозом. С момента постановки диагноза средняя продолжительность жизни составляет около 5 лет. При манифестации в более молодом возрасте миелофиброз имеет менее агрессивное течение, что сопряжено с лучшим ответом на терапию и большей выживаемостью больных. Эффективных методов профилактики не разработано ввиду неизвестности этиологического фактора. Предупреждение развития вторичного миелофиброза заключается в своевременной диагностике и лечении патологий, на фоне которых он возникает - истинной полицитемии и эссенциальной тромбоцитемии.
2. Патофизиологические основы лечения сублейкемического миелоза. Патофизиология крови. Экстремальные состояния/ Под ред. А.И. Воробьева и Н.А. Горбуновой - 2004.
3. Критерии диагностики и современные методы лечения первичного миелофиброза/ Абдулкадыров К. М., Шуваев В. А., Мартынкевич И. С.// Вестник гематологии. - 2013 - №9(3).
4. Клинические рекомендации по диагностике и терапии Ph-негативных миелопролиферативных заболеваний (истинная полицитемия, эссенциальная тромбоцитопения, первичный миелофиброз. - 2014.
Хронический лимфолейкоз
Хронический лимфолейкоз – это онкологическое заболевание, сопровождающееся накоплением атипичных зрелых В-лимфоцитов в периферической крови, печени, селезенке, лимфоузлах и костном мозге. На начальных стадиях проявляется лимфоцитозом и генерализованной лимфоаденопатией. При прогрессировании хронического лимфолейкоза наблюдаются гепатомегалия и спленомегалия, а также анемия и тромбоцитопения, проявляющиеся слабостью, утомляемостью, петехиальными кровоизлияниями и повышенной кровоточивостью. Отмечаются частые инфекции, обусловленные снижением иммунитета. Диагноз устанавливается на основании лабораторных исследований. Лечение – химиотерапия, пересадка костного мозга.
Хронический лимфолейкоз – заболевание из группы неходжкинских лимфом. Сопровождается увеличением количества морфологически зрелых, но неполноценных В-лимфоцитов. Хронический лимфолейкоз является самой распространенной формой гемобластозов, составляет треть всех лейкозов, диагностируемых в США и странах Европы. Мужчины страдают чаще женщин. Пик заболеваемости приходится на возраст 50-70 лет, в этом периоде выявляется около 70% от общего количества хронических лимфолейкозов.
Пациенты молодого возраста страдают редко, до 40 лет первые симптом болезни возникают всего у 10% больных. В последние годы специалисты отмечают некоторое «омоложение» патологии. Клиническое течение хронического лимфолейкоза очень вариативно, возможно как продолжительное отсутствие прогрессирования, так и крайне агрессивный вариант с летальным исходом в течение 2-3 лет после постановки диагноза. Существует ряд факторов, позволяющих прогнозировать течение заболевания. Лечение осуществляют специалисты в области онкологии и гематологии.
Причины
Причины возникновения окончательно не выяснены. Хронический лимфолейкоз считается единственным лейкозом с неподтвержденной связью между развитием заболевания и неблагоприятными факторами внешней среды (ионизирующим излучением, контактом с канцерогенными веществами). Специалисты считают, что основным фактором, способствующим развитию хронического лимфолейкоза, является наследственная предрасположенность. Типичные хромосомные мутации, вызывающие повреждения онкогенов на начальной стадии болезни, пока не выявлены, однако исследования подтверждают мутагенную природу заболевания.
Клиническая картина хронического лимфолейкоза обусловлена лимфоцитозом. Причиной лимфоцитоза становится появление большого количества морфологически зрелых, но иммунологически дефектных В-лимфоцитов, неспособных к обеспечению гуморального иммунитета. Ранее считали, что аномальные В-лимфоциты при хроническом лимфолейкозе являются долго живущими клетками и редко подвергаются делению. В последующем эта теория была опровергнута.
Исследования показали, что В-лимфоциты быстро размножаются. Ежедневно в организме больного образуется 0,1-1% от общего количества атипичных клеток. У разных больных поражаются различные клоны клеток, поэтому хронический лимфолейкоз можно рассматривать как группу близкородственных заболеваний с общим этиопатогенезом и сходной клинической симптоматикой.
При изучении клеток выявляется большое разнообразие. В материале могут преобладать широкоплазменные либо узкоплазменные клетки с молодыми либо сморщенными ядрами, почти бесцветной либо ярко окрашенной зернистой цитоплазмой. Пролиферация аномальных клеток происходит в псевдофолликулах – скоплениях лейкозных клеток, располагающихся в лимфоузлах и костном мозге.
Причинами цитопении при хроническом лимфолейкозе являются аутоиммунное разрушение форменных элементов крови и подавление пролиферации стволовых клеток, обусловленное повышением уровня Т-лимфоцитов в селезенке и периферической крови. Кроме того, при наличии киллерных свойств разрушение кровяных клеток могут вызывать атипичные В-лимфоциты.
Классификация
С учетом симптомов, морфологических признаков, скорости прогрессирования и реакции на терапию различают следующие формы болезни:
- Хронический лимфолейкоз с доброкачественным течением. Состояние больного долго остается удовлетворительным. Отмечается медленное увеличение количества лейкоцитов в крови. С момента постановки диагноза до стабильного увеличения лимфоузлов может пройти несколько лет или даже десятилетий. Больные сохраняют трудоспособность и привычный образ жизни.
- Классическая (прогрессирующая) форма хронического лимфолейкоза. Лейкоцитоз нарастает в течение месяцев, а не лет. Отмечается параллельное увеличение лимфоузлов.
- Опухолевая форма хронического лимфолейкоза. Отличительной особенностью этой формы является нерезко выраженный лейкоцитоз при выраженном увеличении лимфоузлов.
- Костномозговая форма хронического лимфолейкоза. Выявляется прогрессирующая цитопения при отсутствии увеличения лимфатических узлов, печени и селезенки.
- Хронический лимфолейкоз с увеличением селезенки.
- Хронический лимфолейкоз с парапротеинемией. Отмечаются симптомы одной из вышеперечисленных форм заболевания в сочетании с моноклональной G- или M-гаммапатией.
- Прелимфоцитарная форма хронического лимфолейкоза. Отличительной особенностью этой формы является наличие лимфоцитов, содержащих нуклеолы, в мазках крови и костного мозга, образцах ткани селезенки и лимфоузлов.
- Волосатоклеточный лейкоз. Выявляются цитопения и спленомегалия при отсутствии увеличения лимфоузлов. При микроскопическом исследовании обнаруживаются лимфоциты с характерным «моложавым» ядром и «неровной» цитоплазмой с обрывами, фестончатыми краями и ростками в виде волосков либо ворсинок.
- Т-клеточная форма хронического лимфолейкоза. Наблюдается в 5% случаев. Сопровождается лейкемической инфильтрацией дермы. Обычно быстро прогрессирует.
Выделяют три стадии клинических стадии хронического лимфолейкоза: начальную, развернутых клинических проявлений и терминальную.
Симптомы хронического лимфолейкоза
На начальной стадии патология протекает бессимптомно и может выявляться только по анализам крови. В течение нескольких месяцев или лет у больного хроническим лимфолейкозом выявляется лимфоцитоз 40-50%. Количество лейкоцитов приближено к верхней границе нормы. В обычном состоянии периферические и висцеральные лимфоузлы не увеличены. В период инфекционных заболеваний лимфатические узлы могут временно увеличиваться, а после выздоровления снова уменьшаться. Первым признаком прогрессирования хронического лимфолейкоза становится стабильное увеличение лимфоузлов, нередко – в сочетании с гепатомегалией и спленомегалией.
Вначале поражаются шейные и подмышечные лимфоузлы, затем – узлы в области средостения и брюшной полости, потом – в паховой области. При пальпации выявляются подвижные безболезненные плотноэластические образования, не спаянные с кожей и близлежащими тканями. Диаметр узлов при хроническом лимфолейкозе может колебаться от 0,5 до 5 и более сантиметров. Крупные периферические лимфоузлы могут выбухать с образованием видимого косметического дефекта. При значительном увеличении печени, селезенки и висцеральных лимфоузлов может наблюдаться сдавление внутренних органов, сопровождающееся различными функциональными нарушениями.
Пациенты с хроническим лимфолейкозом жалуются на слабость, беспричинную утомляемость и снижение трудоспособности. По анализам крови отмечается увеличение лимфоцитоза до 80-90%. Количество эритроцитов и тромбоцитов обычно остается в пределах нормы, у некоторых больных выявляется незначительная тромбоцитопения. На поздних стадиях хронического лимфолейкоза отмечаются снижение веса, ночные поты и повышение температуры до субфебрильных цифр. Характерны расстройства иммунитета. Больные часто страдают простудными заболеваниями, циститом и уретритом. Наблюдается склонность к нагноению ран и частое образование гнойников в подкожной жировой клетчатке.
Причиной летального исхода при хроническом лимфолейкозе часто становятся тяжелые инфекционные заболевания. Возможны воспаления легких, сопровождающиеся спаданием легочной ткани и грубыми нарушениями вентиляции. У некоторых больных развивается экссудативный плеврит, который может осложняться разрывом или сдавлением грудного лимфатического протока. Еще одним частым проявлением развернутого хронического лимфолейкоза является опоясывающий лишай, который в тяжелых случаях становится генерализованным, захватывая всю поверхность кожи, а иногда и слизистые оболочки. Аналогичные поражения могут наблюдаться при герпесе и ветряной оспе.
В числе возможных осложнений хронического лимфолейкоза – инфильтрация преддверно-улиткового нерва, сопровождающаяся расстройствами слуха и шумом в ушах. В терминальной стадии хронического лимфолейкоза может наблюдаться инфильтрация мозговых оболочек, мозгового вещества и нервных корешков. По анализам крови выявляются тромбоцитопения, гемолитическая анемия и гранулоцитопения.
Возможна трансформация хронического лимфолейкоза в синдром Рихтера – диффузную лимфому, проявляющуюся быстрым ростом лимфоузлов и формированием очагов за пределами лимфатической системы. До развития лимфомы доживает около 5% пациентов. В остальных случаях смерть наступает от инфекционных осложнений, кровотечений, анемии и кахексии. У некоторых больных хроническим лимфолейкозом развивается тяжелая почечная недостаточность, обусловленная инфильтрацией почечной паренхимы.
В половине случаев патологию обнаруживают случайно, при обследовании по поводу других заболеваний или при проведении планового осмотра. При постановке диагноза учитывают жалобы, анамнез, данные объективного осмотра, результаты анализов крови и иммунофенотипирования. Диагностическим критерием хронического лимфолейкоза является увеличение количества лейкоцитов в анализе крови до 5×109/л в сочетании с характерными изменениями иммунофенотипа лимфоцитов. При микроскопическом исследовании мазка крови выявляются малые В-лимфоциты и тени Гумпрехта, возможно – в сочетании с атипичными или крупными лимфоцитами. При иммунофенотипировании подтверждается наличие клеток с абберантным иммунофенотипом и клональность.
Определение стадии хронического лимфолейкоза осуществляют на основании клинических проявлений заболевания и результатов объективного осмотра периферических лимфоузлов. Для составления плана лечения и оценки прогноза при хроническом лимфолейкозе проводят цитогенетические исследования. При подозрении на синдром Рихтера назначают биопсию. Для определения причин цитопении выполняют стернальную пункцию костного мозга с последующим микроскопическим исследованием пунктата.
Лечение хронического лимфолейкоза
На начальных стадиях хронического лимфолейкоза применяют выжидательную тактику. Пациентам назначают обследование каждые 3-6 месяцев. При отсутствии признаков прогрессирования ограничиваются наблюдением. Показанием к проведению активного лечения является увеличение количества лейкоцитов вдвое и более в течение полугода. Основным методом лечения хронического лимфолейкоза является химиотерапия. Наиболее эффективной комбинацией лекарственных препаратов обычно становится сочетание ритуксимаба, циклофосфамида и флударабина.
При упорном течении хронического лимфолейкоза назначают большие дозы кортикостероидов, осуществляют пересадку костного мозга. У больных пожилого возраста с тяжелой соматической патологией использование интенсивной химиотерапии и пересадка костного мозга могут быть затруднены. В подобных случаях проводят монохимиотерапию хлорамбуцилом или применяют данный препарат в сочетании с ритуксимабом.
Прогноз
Хронический лимфолейкоз рассматривается как практически неизлечимое длительно текущее заболевание с относительно удовлетворительным прогнозом. В 15% случаев наблюдается агрессивное течение с быстрым нарастанием лейкоцитоза и прогрессированием клинической симптоматики. Летальный исход при этой форме хронического лимфолейкоза наступает в течение 2-3 лет. В остальных случаях отмечается медленное прогрессирование, средняя продолжительность жизни с момента постановки диагноза колеблется от 5 до 10 лет. При доброкачественном течении срок жизни может составлять несколько десятилетий. После прохождения курса лечения улучшение наблюдается у 40-70% больных хроническим лимфолейкозом, однако полные ремиссии выявляются редко.
Читайте также: