Гломерулокистоз почек - клиника, диагностика
Добавил пользователь Cypher Обновлено: 20.01.2026
Аутосомно-доминантный поликистоз почек - типы, диагностика, лечение
В США и в Европе аутосомно-доминантный поликистоз почек типа I встречается чаще всех других наследственных болезней с аутосомно-доминантным наследованием.
Распространенность этого заболевания составляет 1 на 1000 живых новорожденных, и 10—15% взрослых, находящихся на постоянном диализе, страдают именно аутосомно-доминантным поликистозом. Терминальная почечная недостаточность наступает в возрасте 50—60 лет. Ген PKD1, ответственный за аутосомно-доминантный поликистоз почек, находится на 16-й хромосоме (сегмент 16р13.1) и кодирует недавно открытый белок поликистин 1, в состав которого входят крупный трансмембранный и сложный внеклеточный домены.
Полагают, что поликистин 1 связывается с продуктом гена PKD2 — поликистином 2.
Клиническая картина аутосомно-доминантного поликистоза почек
Аутосомно-доминантный поликистоз почек обычно проявляется лишь в зрелом возрасте. Однако иногда тяжелые клинические проявления, в том числе характерные аномалии лица (лицо Поттер), наблюдаются уже у новорожденного. У детей старшего возраста и у взрослых на аутосомно-доминантный поликистоз могут указывать боль в пояснице, гематурия, частые инфекции мочевых путей, артериальная гипертония.
В брюшной полости пальпируются объемные образования, нередки грыжи брюшной стенки. У взрослых могут возникать аневризмы внутричерепных артерий и дивертикулез кишечника.
УЗИ почек. У детей с отягощенным семейным анамнезом даже единичная киста при нормальном размере почек может с большой вероятностью оказаться первым проявлением аутосомно-доминантного поликистоза. Помимо почек могут страдать и другие органы, однако до пубертатного периода кисты печени, поджелудочной железы и яичников выявляются редко.
Генодиагностика аутосомно-доминантного поликистоза почек. Несмотря на аутосомно-доминантный тип наследования, длительное течение заболевания с постепенным (за несколько десятилетий) формированием кист предполагает, что в клетках некоторых кист происходят соматические мутации, приводящие к потере гетерозиготности по гену PKD1. Мутации этого гена можно выявить с помощью прямого анализа ДНК.
Лечение аутосомно-доминантного поликистоза почек
Детей из группы риска аутосомно-доминантного поликистоза почек ежегодно обследуют на гематурию, артериальную гипертонию и наличие объемных образований в брюшной полости. Активное лечение артериальной гипертонии и инфекций мочевых путей позволяет замедлить развитие терминальной почечной недостаточности. Для выявления аневризм внутричерепных артерий используют КТ и МРТ. Эти исследования проводят лишь при наличии соответствующей симптоматики или аневризм у родственников больного ребенка.
Аутосомно-доминантный поликистоз почек типа I у грудных детей
Изредка аутосомно-доминантный поликистоз почек проявляется уже в грудном возрасте. Ген PKD1 у таких детей несет крупные делеции, которые могут захватывать и соседний с ним ген TSC2, ответственный за ту-берозный склероз типа II, — так называемый синдром соседних генов. У детей с туберозным склерозом при этом обнаруживаются крупные кисты в обеих почках и первые признаки аутосомно-доминантного по-ликистоза выявляются еще до года.
Аутосомно доминантный поликистоз почек типа II
Это заболевание редко встречается у детей, протекает легче и позднее приводит к терминальной почечной недостаточности — в остальном же его клиническая картина почти неотличима от аутосомно-доминантного поликистоза почек типа I. Ген, ответственный за аутосомно-доминантный поликистоз почек II, идентифицирован, так что отличить один тип поликистоза от другого можно с помощью генодиагностики.
Генодиагностика особенно важна, когда больному аутосомно-доминантным поликистозом планируют провести трансплантацию почки и донором выступает один из членов семьи. Поскольку заболевание проявляется лишь в зрелом возрасте, необходимо убедиться, что предполагаемый донор не несет мутантного аллеля.
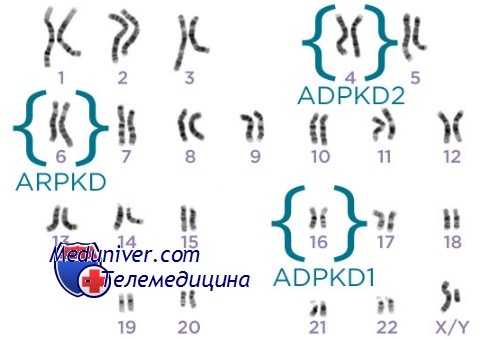
АДПП1, АДПП2 — аутосомно-доминантныи поликистоз почек типов I и II;
АРПП — аутосомно-рецессивный поликистоз почек;
ГД — генодиагностика (при необходимости ГД включают в МГК);
ГК — гломерулокистоз почек;
ЛМББ — синдромы Лоренса—Муна и Барде—Бидля;
МГ — синдром Меккеля—Грубера;
МГК — медико-генетическое консультирование;
МКБ1, МКБ2 — медуллярная кистозная болезнь типов I и II;
НФ1, НФ2, НФЗ — нефронофтиз типов I, II и III;
СЛ — синдром Сениора—Локен;
TC1 — туберозный склероз типа I.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Гломерулокистоз почек - клиника, диагностика
Нефронофтиз - типы, диагностика, лечение
Выделяют три формы нефронофтиза — все с аутосомно-рецессивным типом наследования. Нефронофтиз типа II начинается в грудном возрасте, нефронофтиз типа III впервые проявляется у подростков, а начало нефронофтиза типа I (ювенильного нефронофтиза) приходится на препубертатный период. Иногда ювенильный нефронофтиз протекает с внепочечными проявлениями.
Нефронофтиз типа I (ювенильный нефронофтиз)
Для детей с ювенильным нефронофтизом характерны полиурия и полидипсия, анемия и задержка роста. Поскольку заболевание не проявляется типичными признаками гломерулопатии (гематурией, протеинурией, артериальной гипертонией, отеками), почечную недостаточность нередко обнаруживают случайно при обследовании по поводу анемии, полиурии или сонливости. Диагноз нефронофтиза в таких случаях может оказаться полной неожиданностью. Средний возраст развития терминальной почечной недостаточности — 13 лет.
Внепочечные проявления нефронофтиза. Ювенильный нефронофтиз может сопровождаться врожденной глазодвигательной апраксией Когана, при которой у гомозиготных по делециям гена NPHP1 детей младше 2 лет отсутствуют произвольные движения глаз по горизонтали. Вместе с пигментной дегенерацией сетчатки нефронофтиз входит в состав синдрома Сениора—Локен, а вместе с агенезией червя мозжечка и колобомой диска зрительного нерва - в состав синдрома Жубер типа II. Описан также нефронофтиз в сочетании с фиброзом печени и с конусовидными эпифизами фаланг.
Наконец, ювенильный нефронофтиз может быть одним из проявлений синдромов Жена (асфиктическая дисплазия грудной клетки) и Эллисаван Кревельда (хондроэктодермальная дисплазия), синдрома RHYNS (Retinitis pigmentosa, HYpopituitarism, Nephronophthisis, mild Skeletal dysplasia — пигментная дегенерация сетчатки, гипопитуитаризм, нефронофтиз, легкие аномалии развития скелета), а также синдромов Лоренса—Муна и Барде—Бидля.
Диагностика нефронофтиза. Важный метод диагностики нефронофтиза — УЗИ, при котором выявляются сглаженность границы между корковым и мозговым веществом, повышенная эхогенность почечной паренхимы, а у детей старше 9 лет — кисты, расположенные на границе коркового и мозгового вещества. Сцинтиграфия обнаруживает снижение концентрирующей способности почек.
Нефронофтиз типа II
Этот тип нефронофтиза называют инфантильным, так как первые его проявления возникают еще во внутриутробном периоде, сразу после рождения или в первый год жизни ребенка. Недавно при исследовании родословной большой семьи бедуинов было установлено, что ген нефронофтиза типа II (NPHP2) находится на 9-й хромосоме (сегмент q22-q31).
Поскольку морфологическая картина и течение этого заболевания довольно сильно отличаются от двух других форм нефронофтиза, возможно, что в группу нефронофтиза оно попало не совсем обоснованно.
Нефронофтиз типа III
Ген нефронофтиза типа III (NPHP3) идентифицирован при исследовании еще одной большой родословной — на этот раз семьи из Венесуэлы. Этот тип нефронофтиза называют подростковым. Терминальная почечная недостаточность наступает на 6 лет позже, чем у больных ювенильным нефронофтизом, — в среднем в 19 лет. В остальном же ни морфологической картиной, ни клиническими проявлениями нефронофтиз типа III от ювенильного нефронофтиза не отличается.
Генодиагностика нефронофтиза
Идентификация гена NPHP1, ответственного за ювенильный нефронофтиз, позволила использовать для диагностики прямой анализ ДНК. Продукт этого гена нефрокистин содержит SH3-домен, что, по-видимому, указывает на участие нефрокистина в межбелковых взаимодействиях, в том числе во внутриклеточной передаче сигнала в фокальных контактах — участках, в которых клетка соприкасается с внеклеточным матриксом.
Нарушением фокальных контактов могут объясняться характерные разрывы базальной мембраны почечных канальцев, наблюдаемые при нефронофтизе. У 85% детей делеции обнаруживаются на обоих аллелях гена NPHP1. На выявлении этих делеций, а также точечных мутаций гена NPHP1 основывается генодиагностика нефронофтиза. Гомозиготные делеции гена NPHP1 описаны также при врожденной глазодвигательной апраксии Когана и при поздней форме пигментной дегенерации сетчатки.
При глазодвигательной апраксии Когана детям грудного и младшего возраста не удаются горизонтальные движения глаз и, чтобы посмотреть в сторону, они совершают резкие толчкообразные движения головой.
Гломеруоцитоз почек гипопластического типа
Гломеруоцитоз почек гипопластического типа – генетическое заболевание, проявляющееся недоразвитием мочевыделительной системы в сочетании с поражением печени, половых органов и поджелудочной железы. Сопровождается поликистозом почек, протеинурией, сахарным диабетом и пороками развития других органов. Диагностика заболевания производится на основании данных ультразвукового исследования почек (иногда их нарушения выявляются уже внутриутробно), клинических анализов и молекулярно-генетических техник. Лечение гломеруоцитоза почек гипопластического типа только симптоматическое, в некоторых случаях возможна трансплантация почек.
Общие сведения
Гломеруоцитоз почек гипопластического типа (синдром диабета и почечных кист) – редкая форма гипопластической дисплазии почек, характеризующаяся выраженными расстройствами со стороны мочевыделительной, эндокринной, половой и других систем. Некоторое время после открытия гломеруоцитоза полагали, что первичным нарушением при данном заболевании является сахарный диабет, а поражение мочевыделительной системы рассматривали как следствие диабетической нефропатии. Впоследствии специалистам удалось доказать, что гломеруоцитоз почек гипопластического типа следует определять как особую полиогранную патологию, практически все проявления которой обусловлены именно генетическими нарушениями. Врачи-генетики установили генетическую разнородность этого состояния и выяснили, что у больных обычно выявляется аутосомно-доминантный механизм наследования. Заболевание одинаково часто поражает мужчин и женщин. Распространенность гломеруоцитоза почек гипопластического типа в популяции пока определить не удалось из-за редкой встречаемости патологии.
Причины гломеруоцитоза почек гипопластического типа
Гломеруоцитоз почек гипопластического типа сопровождается поражением мочевыделительной системы, возникающим еще в пренатальном периоде. В настоящее время определено пять генов, мутации которых способны вызывать дефекты развития почек и других органов на фоне инсулинозависимого сахарного диабета. Этиология и патогенез патологического состояния лучше всего изучены на примере гена HNF1B, локализующегося на 17-й хромосоме. Этот ген кодирует последовательность одноименного белка, являющегося одним из так называемых транскрипционных факторов – протеином, который контролирует работу и экспрессию других генов. Наиболее активно образование белка HNF1B происходит в почках, органах репродуктивной системы, поджелудочной железе и печени, поэтому гломеруоцитоз почек гипопластического типа характеризуется поражением перечисленных анатомических образований.
Ген HNF1B имеет достаточно сложную структуру (9 экзонов). На сегодняшний день описано более 70-ти типов мутаций, способных вызвать клинические проявления синдрома почечных кист с сахарным диабетом. Несмотря на то, что продуцируемый геном белок является фактором транскрипции, его функции в различных органах весьма разнообразны. В частности, белок HNF1B контролирует образование почечных трубочек и общее развитие почек, отвечает за формирование органов репродуктивной системы и определяет структуру островков Лангерганса в поджелудочной железе. Именно поэтому дефекты в структуре этого гена (и, соответственно, белка) приводят к многочисленным порокам развития. Другие гены, способные вызывать развитие гломеруоцитоза почек гипопластического типа, пока изучены недостаточно, известно только, что они расположены на разных хромосомах. Предполагается, что все эти гены также являются теми или иными транскрипционными факторами, контролирующими формирование различных структур почек и других органов.
Симптомы гломеруоцитоза гипопластического типа
Гломеруоцитоз почек гипопластического типа характеризуется разнообразными нарушениями, некоторые из которых проявляются сразу после рождения ребенка. У девочек наблюдаются пороки развития половых органов (например, зияние половой щели у доношенного ребенка), у мальчиков диагностируются крипторхизм или эпидидимальные кисты. Начиная с младенческого возраста, у больных регистрируются признаки нарушения функции почек: изменение диуреза (олиго- или полиурия), боли в пояснице, мутная моча и осадок в моче. Могут выявляться общие симптомы патологии выделительной системы: запах аммиака изо рта, неприятный запах пота и кожных покровов. Кроме того, возможны желтуха, обусловленная поражением печени, замедленный набор массы тела, вызванный нарушениями в работе желудочно-кишечного тракта, и ряд других симптомов.
Инсулинозависимый сахарный диабет при гломеруоцитозе почек гипопластического типа возникает до 25 лет. У ряда больных симптомы диабета появляются еще в детские годы. Кроме раннего развития, заболевание обычно ничем не отличается от диабета взрослых, поэтому его относят в группу «диабетов взрослых у детей» (MODY). Тип этого эндокринного нарушения, обусловленного синдромом сахарного диабета и почечных кист, относят к 5-му типу MODY. Клиническая картина заболевания включает в себя похудение, полиурию (не всегда, зависит от функциональной активности почек) и полидипсию. При отсутствии инъекций инсулина развиваются помутнение сознания и кетоацидотическая кома. Последняя намного быстрее возникает при наличии гломеруоцитоза почек гипопластического типа, сопровождающегося резким замедлением экскреции кетоновых тел из организма.
В отдаленной перспективе проявления синдрома сахарного диабета и почечных кист представляют собой сочетание эндокринных, выделительных и обменных расстройств. Из-за нарушений работы почек замедляется выведение мочевой кислоты, что приводит к развитию подагры, которая характеризуется болью и воспалением суставов и иногда сопровождается мочекаменной болезнью. Возникают нарушения, характерные для больных сахарным диабетом, в том числе – ангиопатии сетчатки и трофические расстройства в области нижних конечностей. Страдает репродуктивная система, из-за пороков ее развития у мужчин и женщин нередко диагностируется бесплодие. У девушек также наблюдаются частые нарушения менструального цикла и аменорея, у мужчин на фоне не излеченного крипторхизма могут развиваться злокачественные опухоли яичек.
Диагностика гломеруоцитоза почек гипопластического типа
Поскольку заболевание, обусловленное мутацией гена HNF1B, сопровождается множественными поражениями органов и систем, для диагностики данной патологии используют большое количество методик. Программа обследования включает в себя различные типы ультразвуковых исследований, консультации медицинских специалистов разного профиля (уролога, эндокринолога, гинеколога и пр.), анализы мочи и крови (как общий, так и биохимический) и методы молекулярной генетики. Именно на основании результатов всех этих диагностических методик можно надежно определить гломеруоцитоз почек гипопластического типа. Могут также потребоваться дополнительные исследования: анализ на уровень гликозилированного гемоглобина, глюкозотолерантный тест и другие.
Первые проявления заболевания можно выявить при помощи ультразвуковой диагностики. Патология почек у плода нередко определяется уже при проведении скрининговых УЗИ у беременных. Чаще всего в почках образуются кисты, иногда множественные (поликистоз почек), сами органы уменьшены в размерах и могут срастаться между собой (такое явление носит название подковообразной почки). При гломеруоцитозе почек гипопластического типа также может определяться аномальное расположение сосудов органов и их разветвление. Нередко обнаруживается необычная локализация мочеточника. При УЗИ малого таза у женщин с гломеруоцитозом почек гипопластического типа часто выявляются пороки развития репродуктивной системы (двурогая или рудиментарная матка). УЗИ печени свидетельствует об аномальном строении органа (изменении его формы и размеров).
В анализах мочи обнаруживают протеинурию, гиалиновые и другие цилиндры. Микрогематурия и глюкозурия проявляются по мере развития сахарного диабета. Из-за выраженной протеинурии в биохимическом анализе крови может выявляться снижение общего белка и увеличение концентрации липопротеидов. Повышение уровня мочевой кислоты в плазме крови (гиперурикемия) также является постоянным проявлением гломеруоцитоза почек гипопластического типа. По мере нарастания сахарного диабета повышается концентрация глюкозы с развитием гипергликемии и глюкозурии. Дополнительный анализ на уровень гликозилированного гемоглобина показывает его увеличение – это служит доказательством длительной гипергликемии у больного.
При проведении консультаций узких специалистов выявляются многочисленные нарушения. Гинеколог часто обнаруживает атрезию влагалища, уролог диагностирует эпидидимальные кисты и нередко крипторхизм. Все вышеперечисленные данные дают основание для проведения генетической диагностики, которая осуществляется методом прямого секвенирования последовательности гена HNF1B. Иногда при клинически выраженном гломеруоцитозе почек гипопластического типа это исследование не подтверждает наличия генетических дефектов в HNF1B, что не исключает мутаций в других генах, способных стать причиной развития заболевания. Дифференциальную диагностику гломеруоцитоза проводят с другими почечными дисплазиями и сахарным диабетом.
Лечение гломеруоцитоза почек гипопластического типа
Специфического лечение гломеруоцитоза почек гипопластического типа не разработано, применяют симптоматическую и поддерживающую терапию. Необходим постоянный контроль функциональной активности почек, поэтому больные должны состоять на диспансерном учете и регулярно проходить обследование у нефролога и эндокринолога. Для снижения нагрузки на мочевыделительную систему назначают сбалансированный рацион, советуют соблюдать правильный водный режим и ограничить потребление соли. Больным рекомендуют уменьшить количество мяса и фруктозы в рационе, поскольку компоненты этих пищевых продуктов в процессе метаболизма способны превращаться в мочевую кислоту, усугубляя проявления гиперурикемии и подагры. При симптомах острой или хронической почечной недостаточности проводят курс гемодиализа.
План лечения сахарного диабета составляют с учетом отсутствия реакции на препараты сульфонилмочевины. Необходимы регулярные инъекции инсулина. Следует придерживаться низкоуглеводной диеты и рассчитывать количество сахара в рационе. Симптоматическое лечение гломеруоцитоза почек гипопластического типа также производят при поражении печени, поджелудочной железы и органов репродуктивной системы. Хирургическое вмешательство может потребоваться для коррекции крипторхизма и других аномалий развития. Решение о трансплантации почки при ее выраженном поражении принимается с учетом общего состояния пациента и степени нарушений со стороны других органов.
Прогноз и профилактика
Прогноз гломеруоцитоза почек гипопластического типа, как правило, неблагоприятный, что обусловлено ранним появлением симптомов (многие из которых являются врожденными), их тяжестью, широким спектром поражений различных органов и систем. При тяжелом течении заболевания летальный исход может наступить еще в детские годы из-за сочетания выделительных расстройств и сахарного диабета. При адекватной симптоматической терапии, пожизненном соблюдении правил составления рациона и водного режима гломеруоцитоз почек гипопластического типа иногда имеет доброкачественное течение, некоторым больным удается дожить до старости. Профилактика заболевания сводится к пренатальной генетической диагностике, которая особенно показана при наличии данного состояния у близких родственников.
Термин «гломерулокистоз почек» объединяет группу наследственных и спорадических заболеваний с характерной морфологической картиной. В корковом веществе почек находят множество мелких кист. Все они берут начало из капсул Боумена или начальных отделов проксимальных извитых канальцев, при этом петли Генле, дистальные извитые канальцы и собирательные трубочки остаются нетронутыми.
Этим гломерулокистоз отличается от других кистозных болезней почек, при которых кисты формируются главным образом из почечных канальцев. Подобная морфологическая картина встречается и при аутосомно-рецессивном поликистозе почек, а также при некоторых редких наследственных синдромах (например, ротопальцелицевом синдроме типа I). Тем не менее семейную гипопластическую гломерулокистозную болезнь почек рассматривают как самостоятельную форму гломерулокистоза с аутосомно-доминантным типом наследования.
Клиническая картина гломерулокистоза почек
При аутосомно-доминантной семейной гипопластической гломерулокистозной болезни почек признаки ХПН появляются рано, но прогрессирует она обычно медленно, и терминальная почечная недостаточность наступает не скоро. При других гломерулокистозах объемные образования в брюшной полости и почечную недостаточность выявляют уже у грудных детей. Кисты могут обнаруживаться и в печени. АД часто повышено.
Иногда диагноз ставят в зрелом возрасте — больным с артериальной гипертонией, болью в пояснице и гематурией. Функция почек в той или иной степени нарушена.
УЗИ почек при гломерулокистозе. Отличить семейную гипопластическую гломерулокистозную болезнь почек от других гломерулокистозов помогает УЗИ почек. При семейной гипопластической гломерулокистозной болезни почки уменьшены, видны аномалии чашечно-лоханочной системы и пирамид мозгового вещества, почечные сосочки отсутствуют. При прочих гломерулокистозах обе почки увеличены, но форма их не нарушена, эхогенность коркового и мозгового вещества повышена, четкой границы между ними нет, в корковом веществе видны мелкие кисты.
Гломерулонефрит
Гломерулонефрит – это заболевание почек иммунновоспалительного характера. Поражает преимущественно почечные клубочки. В меньшей степени в процесс вовлекаются интерстициальная ткань и канальцы почек. Гломерулонефрит протекает, как самостоятельное заболевание или развивается при некоторых системных патологиях. Клиническая картина складывается из мочевого, отечного и гипертонического синдромов. Диагностическую ценность имеют данные анализов мочи, проб Зимницкого и Реберга, УЗИ почек и УЗДГ почечных сосудов. Лечение включает препараты для коррекции иммунитета, противовоспалительные и симптоматические средства.
Гломерулонефрит – поражение почек иммунновоспалительного характера. В большинстве случаев развитие гломерулонефрита обусловлено чрезмерной иммунной реакцией организма на антигены инфекционной природы. Существует также аутоиммунная форма гломерулоронефрита, при которой поражение почек возникает в результате разрушительного воздействия аутоантител (антител к клеткам собственного организма).
Гломерулонефрит занимает второе место среди приобретенных заболеваний почек у детей после инфекций мочевыводящих путей. По статистическим данным современной урологии, патология является самой частой причиной ранней инвалидизации пациентов вследствие развития хронической почечной недостаточности. Развитие острого гломерулонефрита возможно в любом возрасте, но, как правило, заболевание возникает у больных в возрасте до 40 лет.
Причины гломерулонефрита
Причиной болезни обычно является острая или хроническая стрептококковая инфекция (ангина, пневмония, тонзиллит, скарлатина, стрептодермия). Заболевание может развиться, как следствие кори, ветряной оспы или ОРВИ. Вероятность возникновения патологии увеличивается при длительном пребывании на холоде в условиях повышенной влажности («окопный» нефрит), поскольку сочетание этих внешних факторов изменяет течение иммунологических реакций и вызывает нарушение кровоснабжения почек.
Существуют данные, свидетельствующие о связи гломерулонефрита с заболеваниями, вызываемыми некоторыми вирусами, Toxoplasma gondii, Neisseria meningitidis, Streptococcus pneumoniae и Staphylococcus aureus. В подавляющем большинстве случаев поражение почек развивается через 1-3 недели после стрептококковой инфекции, причем, результаты исследований чаще всего подтверждают, что гломерулонефрит был вызван «нефритогенными» штаммами b-гемолитического стрептококка группы А.
При возникновении в детском коллективе инфекции, вызванной нефритогенными штаммами стрептококка, симптомы острого гломерулонефрита отмечаются у 3-15% инфицированных детей. При проведении лабораторных исследований изменения в моче выявляются у 50% окружающих больного детей и взрослых, что свидетельствует о торпидном (бессимптомном или малосимптомном) течении гломерулонефрита.
После скарлатины острый процесс развивается у 3-5% детей, получавших лечение в домашних условиях и у 1% больных, пролеченных в условиях стационара. К развитию гломерулонефрита может привести ОРВИ у ребенка, который страдает хроническим тонзиллитом или является носителем кожного нефритогенного стрептококка.
Патогенез
Комплексы антиген-антитело откладываются в капиллярах почечных клубочков, ухудшая кровообращение, вследствие чего нарушается процесс выработки первичной мочи, происходит задержка в организме воды, соли и продуктов обмена, снижается уровень противогипертензивных факторов. Все это приводит к артериальной гипертензии и развитию почечной недостаточности.
Классификация
Гломерулонефрит может протекать остро или хронически. Выделяют два основных варианта течения острого процесса:
- Типичный (циклический). Характерно бурное начало и значительная выраженность клинических симптомов;
- Латентный (ациклический). Стертая форма, характеризующаяся постепенным началом и слабой выраженностью симптомов. Представляет значительную опасность вследствие позднего диагностирования и тенденции к переходу в хронический гломерулонефрит.
При хроническом гломерулонефрите возможны следующие варианты течения:
- Нефротический. Преобладают мочевые симптомы.
- Гипертонический. Отмечается повышение артериального давления, мочевой синдром выражен слабо.
- Смешанный. Представляет собой сочетание гипертонического и нефротического синдромов.
- Латентный. Довольно распространенная форма, характеризующаяся отсутствием отеков и артериальной гипертензии при слабо выраженном нефротическом синдроме.
- Гематурический. Отмечается наличие эритроцитов в моче, остальные симптомы отсутствуют или слабо выражены.
Симптомы гломерулонефрита
Симптомы острого диффузного процесса появляются спустя одну-три недели после инфекционного заболевания, обычно вызванного стрептококками (ангина, пиодермия, тонзиллит). Для острого гломерулонефрита характерны три основные группы симптомов: мочевой (олигурия, микро- или макрогематурия), отечный, гипертонический. Острый гломерулонефрит у детей, как правило, развивается бурно, течет циклически и обычно заканчивается выздоровлением. При возникновении острого гломерулонефрита у взрослых чаще наблюдается стертая форма, для которой характерны изменения мочи, отсутствие общих симптомов и тенденция к переходу в хроническую форму.
Начинается заболевание с повышения температуры (возможна значительная гипертермия), познабливания, общей слабости, тошноты, снижения аппетита, головной боли и боли в поясничной области. Больной становится бледным, его веки отекают. При остром гломерулонефрите наблюдается уменьшение диуреза в первые 3-5 суток от начала заболевания. Затем количество выделяемой мочи увеличивается, но снижается ее относительная плотность. Еще один постоянный и обязательный признак гломерулонефрита – гематурия (наличие крови в моче). В 83-85% случаев развивается микрогематурия. В 13-15% возможно развитие макрогематурии, для которой характерна моча цвета «мясных помоев», иногда – черная или темно-коричневая.
Одним из наиболее специфичных симптомов являются отеки лица, выраженные по утрам и уменьшающиеся в течение дня. Следует отметить, что задержка 2-3 литров жидкости в мышцах и подкожной жировой клетчатке возможна и без развития видимых отеков. У полных детей дошкольного возраста единственным признаком отеков иногда становится некоторое уплотнение подкожной клетчатки.
У 60% больных развивается гипертония, которая при тяжелой форме заболевания может длиться до нескольких недель. В 80-85% случаев острый гломерулонефрит вызывает у детей поражение сердечно-сосудистой системы. Возможны нарушения функции центральной нервной системы и увеличение печени. При благоприятном течении, своевременном диагностировании и начале лечения основные симптомы (отеки, артериальная гипертензия) исчезают в течение 2-3 недель. Полное выздоровление отмечается через 2-2,5 месяца.
Для всех форм хронического гломерулонефрита характерно рецидивирующее течение. Клинические симптомы обострения напоминают или полностью повторяют первый эпизод острого процесса. Вероятность рецидива увеличивается в весеннее-осенний период и наступает спустя 1-2 суток после воздействия раздражителя, в роли которого обычно выступает стрептококковая инфекция.
Осложнения
Острый диффузный гломерулонефрит может приводить к развитию острой почечной недостаточности, острой сердечной недостаточности, острой почечной гипертензивной энцефалопатии, внутримозгового кровоизлияния, преходящей потере зрения. Фактором, увеличивающим вероятность перехода острой формы в хроническую, является гипопластическая дисплазия почки, при которой почечная ткань развивается с отставанием от хронологического возраста ребенка.
Для хронического диффузного процесса, характеризующегося прогрессирующим течением и резистентностью к активной иммунодепрессивной терапии, исходом становится вторично-сморщенная почка. Гломерулонефрит занимает одно из ведущих мест среди заболеваний почек, приводящих к развитию почечной недостаточности у детей и ранней инвалидизации больных.
Диагностика
Постановка диагноза производится на основании анамнеза (недавно перенесенное инфекционное заболевание), клинических проявлений (отеки, артериальная гипертензия) и данных лабораторных исследований. По результатам анализов характерны следующие изменения:
- микро- или макрогематурия. При макрогематурии моча становится черной, темно-коричневой, или приобретает цвет «мясных помоев». При микрогематурии изменения цвета мочи не наблюдается. В первые дни заболевания в моче содержатся преимущественно свежие эритроциты, затем – выщелочные.
- умеренная (обычно в пределах 3-6%) альбуминурия в течение 2-3 недель;
- зернистые и гиалиновые цилиндры при микрогематурии, эритроцитарные – при макрогематурии по результатам микроскопии мочевого осадка;
- никтурия, снижение диуреза при проведении пробы Зимницкого. Сохранность концентрационной способности почек подтверждается высокой относительной плотностью мочи;
- снижение фильтрационной способности почек по результатам исследования клиренса эндогенного креатинина;
По результатам общего анализа крови при остром гломерулонефрите выявляется лейкоцитоз и повышение СОЭ. Биохимический анализ крови подтверждает увеличение содержания мочевины, холестерина и креатинина, повышение титра АСТ и АСЛ-О. Характерна острая азотемия (повышение содержания остаточного азота). Проводится УЗИ почек и УЗДГ сосудов почек. Если данные лабораторных исследований и УЗИ сомнительны, для подтверждения диагноза производится биопсия почки и последующее морфологическое исследование полученного материала.
Лечение гломерулонефрита
Лечение патологии осуществляется в условиях стационара. Назначается диета №7, постельный режим. Больным назначается антибактериальная терапия (ампициллин+оксациллин, пенициллин, эритромицин), проводится коррекция иммунитета негормональными (циклофосфамид, азатиоприн) и гормональными (преднизолон) препаратами. В комплекс лечебных мероприятий входит противовоспалительное лечение (диклофенак) и симптоматическая терапия, направленная на уменьшение отеков и нормализацию артериального давления.
В последующем рекомендуется санаторно-курортное лечение. После перенесенного острого гломерулонефрита больные в течение двух лет находятся под наблюдением врача-нефролога. При лечении хронического процесса в период обострения проводится комплекс мероприятий, аналогичных терапии острого гломерулонефрита. Схема лечения в период ремиссии определяется, исходя из наличия и выраженности симптомов.
Читайте также: