Губчатая почка - клиника, диагностика
Добавил пользователь Алексей Ф. Обновлено: 30.01.2026
Дистальноканальцевый ацидоз у детей - причины, клиника, диагностика
В дистальных отделах нефрона происходит секреция выделяющихся в организме кислот. Почкам растущего ребенка приходится секретировать дополнительное количество кислоты, образующейся при росте костной ткани.
В клетках дистальных отделов (как и в клетках проксимальных канальцев) из СО2 и Н2О под действием карбоангидразы образуется Н2СО3, диссоциирующая на НСО3- и Н+.
Н+ переносится через апикальную мембрану благодаря работе Н+-АТФазы и Н+,К+ -АТФазы; последняя секретирует Н+, а в обмен реабсорбирует К+. НСО3 переносится через базолатеральную мембрану с помощью Сl-/НСО3- -обменника.
Теоретически дефект любого из этих белков может вызвать первичный дистальноканальцевый ацидоз. В семьях с аутосомно-доминантным дистальноканальцевым ацидозом выявлена мутация гена Сl-/НСО3-обменника. Кроме того, возможно аутосомно-рецессивное и сцепленное с полом наследование. Дистальноканальцевый ацидоз у детей чаще всего бывает спорадическим, наследственным или вызванным амфотерицином В. В последнем случае секреция протонов не нарушена, но они вновь возвращаются в клетку через образованные амфотерицином В поры.
Причины дистальноканальцевого ацидоза
1. Первичный:
Наследственный (иногда сочетается с глухотой)
Идиопатический
2. Наследственный, сочетающийся с другими заболеваниями:
Синдром Элерса—Данло
Наследственные формы гиперкалыдиурии
Остеопетроз с недостаточностью карбоангидразы II
Наследственный овалоцитоз
Серповидноклеточная анемия
Медуллярная кистозная болезнь
Болезнь Фабри
Болезнь Вильсона
3. Аутоиммунные заболевания:
Синдром Шегрена
Хронический активный гепатит
Первичный билиарный цирроз печени
СКВ
Хронический лимфоцитарный тиреоидит
Гипергаммаглобулинемия
Криоглобулинемия
4. Состояния, при которых развивается нефрокальциноз:
Тиреотоксикоз
Передозировка витамина D
Гиперкальциурия
Губчатая почка
Болезнь Фабри
Болезнь Вильсона
Гиперпаратиреоз
5. Лекарственные средства и токсические вещества:
Амфотерицин В
Литий
Клеи, содержащие толуол или другие органические растворители
Анальгетики (анальгетическая нефропатия)
Цикламат
Циклоспорин
Длительный прием фуросемида при бронхолегочной дисплазии
Соли ванадиевых кислот
6. Интерстициальные заболевания:
Балканская эндемическая нефропатия
Хронический пиелонефрит
Проказа
Обструкция мочевых путей
Отторжение трансплантированной почки
Первичная оксалурия
Как и при проксимальноканальцевом ацидозе, при дистальноканальцевом ацидозе отмечается задержка роста. Лабораторное исследование выявляет гиперхлоремический метаболический ацидоз и гипокалиемию. При выраженной гипокалиемии возникают мышечная слабость, вплоть до паралича, и болезненные мышечные спазмы.
Анионный интервал мочи положителен. Хронический метаболический ацидоз приводитк деминерализации костей и гиперкальциурии. Существуют четкие различительные признаки дистальноканальцевого и проксимальноканальцевого ацидоза. Поскольку окончательное закисление мочи происходит в дистальных канальцах, то при дистальноканальцевом ацидозе рН мочи всегда выше 5,5.
Ацидоз и гипокалиемия усиливают реабсорбцию цитрата в проксимальных канальцах, что приводит к гипоцитратурии. Цитрат - это основной компонент мочи, который препятствует образованию кальциевых камней, поэтому при дистальноканальцевом ацидозе часто встречаются нефрокальциноз и мочекаменная болезнь. При проксимальноканальцевом ацидозе из-за поражения канальцев реабсорбция цитрата нарушена и его содержание в моче нормально или даже повышено.
При дистальноканальцевом ацидозе внутрь назначают основания (смесь калиевой и натриевой солей цитрата или бикарбоната) в дозе, соответствующей ежедневной нормальной секреции протонов в дистальных отделах нефрона. Обычно достаточно 3—5 мэкв/кг/сут основания в три или четыре приема, чтобы нормализовать концентрацию бикарбоната в плазме и предотвратить гипоцитратурию.
Такое лечение позволяет детям не только нормально расти, но и догнать сверстников в росте. Без лечения или при несоблюдении схемы лечения развиваются нарастающий нефрокальциноз и почечная недостаточность. Детям с тяжелой гипокалиемией и ацидозом необходимо до начала лечения основаниями нормализовать уровень калия, чтобы предотвратить усугубление гипокалиемии.
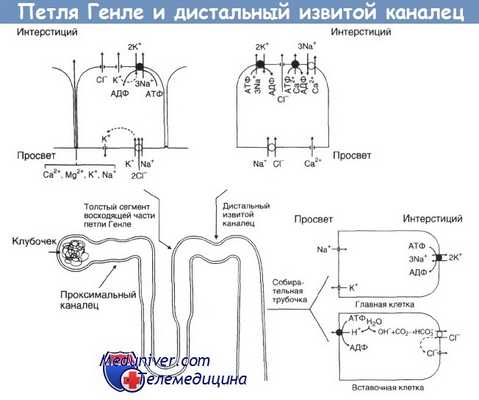
Основные переносчики апикальной и базолатеральной мембраны толстого сегмента восходящей части петли Генле, дистального извитого канальца и коркового отдела собирательной трубочки.
Учебное видео анализа КЩС при респираторном и метаболическом ацидозе
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Амилоидоз почек ( Амилоидная дистрофия почек , Амилоидный нефроз )
Амилоидоз почек – это проявление системного амилоидоза, характеризующееся нарушением белково-углеводного обмена с внеклеточным отложением в почечной ткани амилоида – сложного белково-полисахаридного соединения, приводящего к нарушению функции органа. Патология протекает с развитием нефротического синдрома (протеинурия, отеки, гипо- и диспротеинемия, гиперхолестеринемия) и исходом в хроническую почечную недостаточность. Диагностика включает исследования мочи, крови и копрограммы; проведение УЗИ почек и биопсии. Назначается диета, проводится лекарственная терапия и коррекция первичных нарушений; в тяжелых случаях может потребоваться гемодиализ и трансплантация почки.
МКБ-10
Общие сведения
В практической урологии амилоидоз почек (амилоидная дистрофия, амилоидный нефроз) составляет 1–2,8% от всех заболеваний почек. Амилоидоз почек служит наиболее частым проявлением системного амилоидоза, при котором в межклеточном пространстве различных органов накапливается особое гликопротеидное вещество – амилоид, нарушающее их функции. Наряду с гломерулонефритом является основной причиной развития нефротического синдрома.
Причины
Этиология идиопатического амилоидоза почек в большинстве случаев остается неизвестной; иногда заболевание развивается при множественной миеломе. Кроме почек при первичном амилоидозе могут поражаться язык, кожа, щитовидная железа, печень, легкие, кишечник, селезенка, сердце.
Вторичным амилоидозом поражаются почки, сосуды, лимфоузлы, печень и др. органы. Обычно он ассоциирован с хроническими, длительно протекающими патологиями:
- инфекциями (сифилисом, туберкулезом, малярией);
- гнойно-деструктивными процессами (брохоэктатической болезнью, эмпиемой плевры, остеомиелитом, затяжным септическим эндокардитом);
- системными заболеваниями (ревматоидным артритом, псориатическим артритом, болезнью Бехтерева);
- заболеваниями кишечника (язвенным колитом, болезнью Крона);
- опухолями (лимфогранулематозом, менингиомой, новообразованиями почек) и др.
Причиной диализного варианта заболевания выступает длительное проведение пациенту гемодиализа. Семейный наследственный амилоидоз встречается при периодической болезни, нередко в странах средиземноморского бассейна (в частности, португальский вариант). Старческий амилоидоз рассматривается как признак старения, встречающийся после 80 лет у 80 % людей. Локальные формы патологии могут быть обусловлены опухолями эндокринной системы, болезнью Альцгеймера, сахарным диабетом 2-го типа и др. причинами.
Патогенез
Среди теорий патогенеза амилоидоза почек рассматриваются иммунологическая, мутационная гипотеза и гипотеза локального клеточного синтеза. Заболевание характеризуется внеклеточным отложением в почечной ткани, преимущественно в клубочках, амилоида – особого гликопротеида с высоким содержанием нерастворимого фибриллярного белка.
Классификация
В соответствии с этиопатогенетическими механизмами выделяют пять форм амилоидоза почек: идиопатическую, семейную, приобретенную, старческую, локальную опухолевидную.
- При первичном (идиопатическом) амилоидозе причины и механизмы остаются неизвестными.
- Семейные (наследственные) формы обусловлены генетическим дефектом образования в организме фибриллярных белков.
- Вторичный (приобретенный) амилоидоз развивается при иммунологических нарушениях (хронических инфекциях, ревматических болезнях, злокачественных опухолях и т. д.).
- В основе старческого типа патологии лежит инволютивное изменение обмена белков.
- Природа локального опухолевидного амилоидоза почек неясна.
Идиопатическая, семейная, старческая и локальная опухолевидная формы выделяются в самостоятельные нозологические единицы. Вторичный амилоидоз почек рассматривается как осложнение основного заболевания. В зависимости от типа содержащегося в амилоиде фибриллярного белка различные формы болезни («А») принято обозначать следующими аббревиатурами:
- АА-тип (вторичный, в амилоиде содержится сывороточный a-глобулин);
- АL-тип (идиопатический, в амилоиде - легкие цепочки Ig);
- АTTR-тип (семейный, старческий; в амилоиде – белок транстиретин);
- Аβ2М-тип (диализный; в амилоиде - β2-микроглобулин) и др.
Симптомы амилоидоза почек
При данной патологии сочетаются почечные и внепочечные проявления, обусловливающие полиморфность картины заболевания. В течении болезни выделяют 4 стадии (латентную, протеинурическую, нефротическую, азотемическую), имеющие характерную клиническую симптоматику.
В латентной стадии, несмотря присутствие амилоида, клинические проявления отсутствуют. В этот период преобладает симптоматика первичного заболевания (инфекций, гнойных процессов, ревматических болезней и др.). Латентная стадия может длиться до 3-5 и более лет.
В протеинурическую (альбуминурическую) стадию появляется нарастающая потеря белка с мочой, микрогематурия, лейкоцитцрия, повышение СОЭ. Вследствие склероза и атрофии нефронов, лимфостаза и гиперемии почки увеличиваются, становятся плотными, приобретают матовый серо-розовый цвет.
Нефротическая (отечная) стадия характеризуется склерозом и амилоидозом мозгового слоя почки и, как следствие, развитием нефротического синдрома с типичной тетрадой признаков – массивной протеинурией, гиперхолестеринемией, гипопротеинемией, отеками, резистентными к диуретикам. Может отмечаться артериальная гипертензия, но чаще АД бывает нормальным или пониженным. Нередко отмечается гепато- и спленомегалия.
В азотемической (терминальной, уремической) стадии почки рубцово-сморщенные, плотные уменьшенные в размерах (амилоидная почка). Азотемическая стадия соответствует развитию хронической почечной недостаточности. В отличие от гломерулонефрита сохраняются стойкие отеки.
Осложнения
Амилоидоз почек может осложняться тромбозом почечных вен с анурией и болевым синдромом. Исходом этой стадии часто является гибель пациента от азотемической уремии. Системными проявлениями могут являться головокружение, слабость, одышка, аритмия, анемия и др. В случае присоединения амилоидоза кишечника развивается упорная диарея.
Диагностика
В ранний доклинический период диагностировать амилоидоз почек крайне сложно. В этой стадии приоритет отдается лабораторным методам – исследованию мочи и крови:
- Анализ мочи. В общем анализе мочи рано отмечается протеинурия, которая имеет тенденцию к неуклонному нарастанию, лейкоцитурия (при отсутствии признаков пиелонефрита), микрогематурия, цилиндрурия.
- Исследование крови. Биохимические показатели крови характеризуются гипоальбуминемией, гиперглобулинемией, гиперхолестеринемией, повышением активности щелочной фосфотазы, гипербилирубинемией, электролитным дисбалансом (гипонатриемией и гипокальциемией), повышением содержания фибриногена и b-липопротеидов. В общем анализе крови - лейкоцитоз, анемия, повышение СОЭ.
- Анализ кала. При исследовании копрограммы нередко выявляется выраженная степень стеатореи («жирного стула»), креатореи (значительного содержания в испражнениях мышечных волокон), амилореи (присутствия в кале большого количества крахмала).
- Инструментальная диагностика. В связи с выраженными метаболическими нарушениями на ЭКГ может регистрироваться аритмия и нарушения проводимости; при ЭхоКГ — кардиомиопатия и диастолическая дисфункция. УЗИ брюшной полости выявляет увеличение селезенки и печени. При рентгенографии ЖКТ определяется гипотония пищевода, ослабление перистальтики желудка, ускорение или замедление пассажа бария по кишечнику. На УЗИ почек визуализируется их увеличение (большие жировые почки).
- Биопсия. Достоверным методом диагностики амилоидоза служит биопсия почки. Морфологическое исследование биоптата после окраски конго красным при последующей электронной микроскопии в поляризованном свете выявляет зеленое свечение, характерное для амилоидоза почек. Амилоид может выявляться по ходу сосудов, канальцев, в клубочках. В некоторых случаях производится биопсия слизистой прямой кишки, кожи, десны, печени.
Лечение амилоидоза почек
В терапии вторичного процесса немалую роль играет успешность лечения основного заболевания. В случае полного стойкого излечивания первичной патологии нередко регрессируют и симптомы амилоидоза почек. Пациентам требуется изменение пищевого рациона: необходимо длительное, в течение 1,5-2 лет, употребление сырой печени (по 80-120 мг/сут.), ограничение белка и соли (особенно при почечной и сердечной недостаточности); повышенное потребление углеводов; пищи, богатой витаминами (особенно витамином С) и солями калия.
Патогенетическими препаратами являются производные 4-аминохинолина, десенсибилизирующие средства, колхициновые алкалоиды, комплексообразующие средства и др. Симптоматическая терапия при амилоидозе почек включает назначение диуретиков, гипотензивных препаратов, переливание плазмы, альбумина и т. д. Целесообразность использования кортикостероидов и цитостатиков дискутируется. В терминальной стадии может потребоваться хронический почечный диализ или трансплантация почки.
Прогноз и профилактика
Прогноз во многом определяется течением основного заболевания и скоростью прогрессирования амилоидоза почек. Ухудшает прогноз развитие тромбозов, кровоизлияний, присоединение интеркуррентных инфекций, пожилой возраст. При развитии сердечной или почечной недостаточности выживаемость составляет менее 1 года. Условиями выздоровления служат своевременное обращение к врачу-нефрологу и ранняя диагностика амилоидоза почек, активное лечение и полное устранение основного заболевания. Профилактика требует своевременного лечения любой хронической патологии, которая может обусловить развитие амилоидного нефроза.
1. Клинические рекомендации по диагностикеи лечению системного амилоидоза/ Клиническая фармакология и терапия. - 2020. - 29(1).
2. Амилоидоз: современные методы диагностики и лечения/ Рамеев В.В., Козловская Л.В.// ффективная фармакотерапия. Урология и нефрология. - 2012. - №11.
Губчатая почка ( Медуллярная губчатая почка , Медуллярная спонгиозная почка , Почка с губчатыми пирамидами )
Губчатая почка – это врожденная мультикистозная деформация собирательных почечных канальцев мальпигиевых пирамид, придающая почечной ткани вид мелкопористой губки. При развитии осложнений губчатой почки (нефрокальциноза и пиелонефрита) наблюдаются почечные колики, гематурия, пиурия. Патология выявляется при экскреторной урографии, ретроградной пиелографии. Лечение при наличии клинических проявлений направлено на устранение осложнений; при неэффективности консервативной терапии проводят удаление камней из почки, нефростомию, резекцию почки, нефрэктомию.
Губчатая почка - это врожденная патология, характеризующаяся изменением структуры органа. Обычно имеет двухсторонний характер. Может долгое время протекать бессимптомно и становиться случайной находкой при проведении обследования мочеполовой системы по другому поводу. Функция почки длительное время сохраняется нормальной; ее искажение чаще провоцирует инфекционный процесс, прогрессирование камнеобразования и нарушение проходимости верхних мочевыводящих путей.
Большинство исследований в сфере современной урологии подтверждают, что губчатая почка как аномалия внутриутробного развития по этиологии и патогенезу близка к поликистозной почке. Считается, что возникновение аномалии связано с поздними нарушениями эмбриогенеза, а изменения в собирательных почечных канальцах могут наблюдаться и в постнатальном периоде. Наследственная природа губчатой почки подтверждена, но тип наследования в большинстве случаев - спорадический. Диагностируется патология в основном у людей среднего и старшего возраста, преимущественно у мужчин. В связи с длительным латентным течением заболевания в детском возрасте губчатая почка обнаруживается сравнительно редко.
Патанатомия
В большинстве случаев наблюдается двухсторонняя аномалия медуллярного вещества почек, при этом кистозные изменения различной степени выраженности могут затрагивать некоторые или все почечные сосочки. В отличие от поликистозной, губчатая почка внешне имеет правильную форму, ровные контуры, гладкую поверхность и несколько увеличена в размере по сравнению с возрастной нормой. На разрезе определяются расширения терминальных почечных канальцев с множественными мелкими кистами и полостями в области пирамид.
Размеры кист колеблются в пределах от 1 до 5 мм, увеличиваясь по направлению к центру. Наблюдаются кисты двух видов - боковые веретено- или дивертикулообразные выпячивания в просветы почечных канальцев, выстланные цилиндрическим эпителием, или замкнутые кистозные полости, образованные при слиянии мелких кист, изолированные от почечных канальцев и выстланные плоским эпителием. Кистозные полости могут содержать прозрачную желтоватую жидкость (при отсутствии воспаления), слущившиеся клетки, кальцифицированные конкременты (от песчинок до мелких камней). В результате вымывания мелкие камни из кистозных канальцев могут появиться в чашечках или почечной лоханке.
Почечная ткань в области пирамид, как правило, плотная и фиброзно-измененная, при сопутствующем пиелонефрите – с воспалительной трансформацией. Кальцификация почечной паренхимы (нефрокальциноз) является вторичным нарушением, поскольку стаз мочи в расширенных почечных канальцах и кистозных полостях способствует осаждению солей кальция.
Симптомы губчатой почки
Как правило, в течение длительного времени аномалия клинически не проявляется. Симптоматика возникает обычно в возрастном интервале от 20 до 40 лет при присоединении различных осложнений: камнеобразования в кистозных полостях, инфекций почек и мочевыводящих путей. Основные клинические проявления осложненной губчатой почки – это тупые или острые приступообразные боли в области поясницы, макро- и микрогематурия, пиурия.
Сосочковый нефрокальциноз встречается при губчатой почке более чем в 60% случаев. Почечные колики возникают вследствие миграции мелких камней из кистозных полостей в чашечки и лоханку. Развитие воспаления из-за попадания мелких камней в чашечно-лоханочную систему и нарушения оттока мочи проявляется периодическим повышением температуры тела, нарушением мочеиспускания. Редко, при тяжелом течении мочекаменной болезни и рецидивирующей вторичной инфекции происходит гнойное расплавление и гибель почечной паренхимы, что проявляется признаками почечной недостаточности.
Диагноз губчатой почки устанавливается специалистом-нефрологом по результатам расширенного урологического обследования, в котором основным методом является экскреторная урография. На урограмме видны интенсивно контрастированные, мозаично и веерообразно расположенные гроздевидные кистозные полости и расширения собирательных канальцев. Морфологические изменения затрагивают обычно дистальную медуллярную зону почки, а кортико-медуллярная зона и корковое вещество остаются неизмененными. В кистозных полостях сосочковой зоны рентгенконтрастное вещество задерживается дольше, чем в чашечках, что говорит о стазе в собирательных канальцах. На нефрокальциноз указывают затемненные рентгеноконтрастными средствами сосочковые конкременты.
Реже в диагностике губчатой почки используют ретроградную пиелографию, т. к. на пиелограммах не всегда определяются изменения в расширенных собирательных протоках почки. Рентгенологическое исследование почек целесообразно проводить при сочетании губчатой почки с нефрокальцинозом или нефролитиазом для обнаружения кальцинатов и микролитов, расположенных в дистальных отделах пирамид. В этом случае на обзорном снимке видны тени мелких камней в кистах сосочков, которые частично или полностью совпадают с тенями полостей на экскреторной урограмме.
УЗИ почек не всегда позволяет обнаружить мельчайшие кисты в глубоких слоях почечной ткани. Лабораторные исследования помогают подтвердить наличие гематурии, пиурии, незначительной протеинемии, гиперкальциурии. Дифференциальную диагностику губчатой почки проводят с заболеваниями, при которых имеются поликистозные поражения медуллярной почечной ткани (поликистозом почки, кистозным пиелитом, папиллярным некрозом, хроническим пиелонефритом), а также нефрокальцинозом, туберкулезом, почечнокаменной болезнью.
Лечение губчатой почки
При неосложненной аномалии и ее бессимптомном течении никакого лечения не проводится; показаны профилактические назначения для снижения риска развития осложнений. При клинических проявлениях губчатой почки лечение ориентировано на устранение вторичной инфекции мочевыводящих путей и метаболических нарушений (дальнейшего отложения солей кальция в кистозно-измененных почечных канальцах). При присоединении пиелонефрита назначают обильное питье, диету с низким содержанием Ca, пролонгированную антибактериальную терапию. Для профилактики развития ятрогенной инфекции урологические инструментальные манипуляции больным с губчатой почкой показаны в исключительных случаях.
Нефростомия требуется при осложнении патологии мочекаменной болезнью или пиелонефритом и отсутствии эффективности консервативного лечения. При очаговой кистозной деформации, затрагивающей отдельный сегмент почки, производят ее резекцию. Удаление почки (нефрэктомия) выполняется очень редко и строго при одностороннем поражении почек. Миграция мелких конкрементов, нарушающая отток мочи может быть показанием к хирургическому удалению камней из почек – пиелолитотомии, нефролитотомии, перкутанной нефролитотрипсии, дистанционной нефролитотрипсии.
В неосложненных случаях исход при губчатой почке благоприятный, функции почек сохраняются. При развитии и прогрессировании нефрокальциноза, присоединении инфекции со временем прогноз может ухудшаться. Профилактические мероприятия не разработаны.
Подковообразная почка
Подковообразная почка – это врожденная аномалия, при которой обе почки сращиваются между собой в области нижнего или верхнего полюса с образованием перешейка. Патология часто осложняется пиелонефритом, мочекаменной болезнью, гидронефрозом и другими заболеваниями мочевыделительной системы. В используется УЗИ, урография, пиелография, компьютерная томография почек. Хирургическое лечение показано при развитии в измененной почке урологического заболевания, требующего оперативной тактики.
Подковообразная почка встречается в современной урологии с частотой 10-15% от всех почечных аномалий, в соотношении 1 случай на 500 новорожденных, причем у мальчиков в 2,5 раза чаще, чем у девочек. Порок характеризуется сращением почек в области нижних, реже - верхних полюсов, в результате чего соединенные почки приобретают «U»-образный вид, напоминающий подкову. При этом каждая из почек имеет свой мочеточник, впадающий в мочевой пузырь, и питающие сосуды. В 88,6% подковообразные почки имеют аномальное кровообращение и необычное строение чашечек.
В некоторых случаях почки срастаются между собой медиальными поверхностями (т. н. галетообразная почка), противоположными (верхний с нижним) полюсами (S или L-образная почка), обоими полюсами и срединной поверхностью (дискообразная почка). Патология может сочетаться с другими врожденными пороками – поликистозом почек, гидроцефалией, расщеплением позвоночника, пороками аноректальной системы, аномалиями скелета (расщелиной губы и нёба, полидактилией, косолапостью).
Формирование подковообразной почки является следствием дисэмбриогенеза. У плода развитие почки проходит три последовательных стадии: предпочка (пронефрос), первичная почка (мезонефрос) и вторичная почка. Параллельно с развитием вторичной почки происходит миграция парного органа в область будущего ложа в поясничной области. Окончательное формирование и фиксация почки заканчивается уже после рождения. Аномалия образуется в результате нарушений процессов миграции и ротации почки, обусловленных болезнями матери, инфекциями, воздействием на плод вредных химических или лекарственных веществ.
Подковообразная почка, сросшаяся нижними полюсами, располагается ниже физиологической границы (XI—XII грудных - II поясничного позвонков). Перешеек подковообразной почки может соответствовать уровню IV-V поясничных позвонков и обычно располагается кпереди от аорты, нервных стволов и нижней полой вены. При резких движениях перешеек может давить на нервы и сосуды, вызывая боль. При травмах живота возрастает риск повреждения подковообразной почки; кроме того, данная аномалия предрасполагает в возникновению в почке различного рода урологических заболеваний. Подковообразная почка практически всегда сочетается с дистопией.
Симптомы подковообразной почки
В связи со спецификой топографии, иннервации и кровоснабжения аномалия может сопровождаться характерным болевым симптомокомплексом: болью в области пупка, возникающей при перегибе или разгибании туловища, в пояснице, внизу живота, в эпигастрии после физической нагрузки. Сдавление перешейком почки нервных сплетений корня брыжейки может вызывать запоры, спастические боли в кишечнике, нарушение кишечной перистальтики.
На фоне постоянного боевого синдрома может развиваться эмоциональная неустойчивость, неврастения, истерия. Венозная внутрипочечная гипертензия, обусловленная сдавлением сосудов, иногда сопровождается гематурией. При сдавливании нижней полой вены развивается венозный застой в нижней половине тела: отеки нижних конечностей, варикоз вен нижних конечностей и малого таза, асцит. У женщин возможно нарушение менструального цикла и преждевременные роды. В ряде наблюдений подковообразная почка не сопровождается никакой симптоматикой и выявляется случайно.
Сжатие перешейком начального отдела мочеточника создает препятствие для оттока мочи из лоханок, что приводит к развитию:
- пиелонефрита (19,4%)
- образованию камней почки (23,6%)
- гидронефрозу (41,7%)
- артериальной гипертензии (15,2%)
Существуют сведения, что в перешейке подковообразной почки чаще развивается опухолевая трансформация клеток и рак почки.
План диагностического обследования включает УЗИ, УЗДГ, экскреторную урографию либо ретроградную пиелографию, почечную артериографию, сцинтиграфию, компьютерную томографию. Урограммы позволяют рассмотреть низкое расположение органа, ограниченную подвижность, наслоение теней нижних полюсов почки на контур позвоночного столба, тень перешейка. При ретроградной пиелографии в первые минуты визуализируется четкий силуэт подковообразной почки и ее перешейка, низкое положение почечных лоханок, аномальное расположение чашечек.
При УЗИ почек определяется отсутствие смещаемости почечных контуров, нетипичное расположение почечных лоханок и измененная форма чашечек; при УЗДГ выявляется аномальное кровоснабжение подковообразной почки. Выполнение нефросцинтиграфии фиксирует характерное накопление радионуклидного препарата в виде подковы, огибающей позвоночный столб. Почечная ангиография выполняется для исследования сосудистой архитектоники, определения количества, локализации и наличия дополнительных сосудов, толщины и васкуляризации перешейка, что имеет важное значения при планировании операции.
Лечение подковообразной почки
При отсутствии клинических проявлений лечения подковообразной почки не требуется. Такие пациенты подлежат наблюдению врача-уролога для предотвращения развития вторичных осложнений. При пиелонефрите, осложняющем течение подковообразной почки, назначается соответствующее курсовое патогенетическое лечение. В случае развития болевой симптоматики, гидронефротической трансформации, камнеобразования, опухолей почки показана дифференцированная хирургическая тактика.
При болях и нарушениях уродинамики, обусловленных давлением перешейка, производится его рассечение и разведение концов почки с фиксацией в новом положении. При поражениях одной из половин подковообразной почки и потере ее функций выполняется геминефрэктомия. При выявлении камней в подковообразной почке используют различные методы их удаления, включающие дистанционную литотрипсию, перкутанную нефролитотрипсию, пиелолитотомию, нефролитотомию.
Губчатая почка - клиника, диагностика
Нефронофтиз - типы, диагностика, лечение
Выделяют три формы нефронофтиза — все с аутосомно-рецессивным типом наследования. Нефронофтиз типа II начинается в грудном возрасте, нефронофтиз типа III впервые проявляется у подростков, а начало нефронофтиза типа I (ювенильного нефронофтиза) приходится на препубертатный период. Иногда ювенильный нефронофтиз протекает с внепочечными проявлениями.
Нефронофтиз типа I (ювенильный нефронофтиз)
Для детей с ювенильным нефронофтизом характерны полиурия и полидипсия, анемия и задержка роста. Поскольку заболевание не проявляется типичными признаками гломерулопатии (гематурией, протеинурией, артериальной гипертонией, отеками), почечную недостаточность нередко обнаруживают случайно при обследовании по поводу анемии, полиурии или сонливости. Диагноз нефронофтиза в таких случаях может оказаться полной неожиданностью. Средний возраст развития терминальной почечной недостаточности — 13 лет.
Внепочечные проявления нефронофтиза. Ювенильный нефронофтиз может сопровождаться врожденной глазодвигательной апраксией Когана, при которой у гомозиготных по делециям гена NPHP1 детей младше 2 лет отсутствуют произвольные движения глаз по горизонтали. Вместе с пигментной дегенерацией сетчатки нефронофтиз входит в состав синдрома Сениора—Локен, а вместе с агенезией червя мозжечка и колобомой диска зрительного нерва - в состав синдрома Жубер типа II. Описан также нефронофтиз в сочетании с фиброзом печени и с конусовидными эпифизами фаланг.
Наконец, ювенильный нефронофтиз может быть одним из проявлений синдромов Жена (асфиктическая дисплазия грудной клетки) и Эллисаван Кревельда (хондроэктодермальная дисплазия), синдрома RHYNS (Retinitis pigmentosa, HYpopituitarism, Nephronophthisis, mild Skeletal dysplasia — пигментная дегенерация сетчатки, гипопитуитаризм, нефронофтиз, легкие аномалии развития скелета), а также синдромов Лоренса—Муна и Барде—Бидля.
Диагностика нефронофтиза. Важный метод диагностики нефронофтиза — УЗИ, при котором выявляются сглаженность границы между корковым и мозговым веществом, повышенная эхогенность почечной паренхимы, а у детей старше 9 лет — кисты, расположенные на границе коркового и мозгового вещества. Сцинтиграфия обнаруживает снижение концентрирующей способности почек.
Нефронофтиз типа II
Этот тип нефронофтиза называют инфантильным, так как первые его проявления возникают еще во внутриутробном периоде, сразу после рождения или в первый год жизни ребенка. Недавно при исследовании родословной большой семьи бедуинов было установлено, что ген нефронофтиза типа II (NPHP2) находится на 9-й хромосоме (сегмент q22-q31).
Поскольку морфологическая картина и течение этого заболевания довольно сильно отличаются от двух других форм нефронофтиза, возможно, что в группу нефронофтиза оно попало не совсем обоснованно.
Нефронофтиз типа III
Ген нефронофтиза типа III (NPHP3) идентифицирован при исследовании еще одной большой родословной — на этот раз семьи из Венесуэлы. Этот тип нефронофтиза называют подростковым. Терминальная почечная недостаточность наступает на 6 лет позже, чем у больных ювенильным нефронофтизом, — в среднем в 19 лет. В остальном же ни морфологической картиной, ни клиническими проявлениями нефронофтиз типа III от ювенильного нефронофтиза не отличается.
Генодиагностика нефронофтиза
Идентификация гена NPHP1, ответственного за ювенильный нефронофтиз, позволила использовать для диагностики прямой анализ ДНК. Продукт этого гена нефрокистин содержит SH3-домен, что, по-видимому, указывает на участие нефрокистина в межбелковых взаимодействиях, в том числе во внутриклеточной передаче сигнала в фокальных контактах — участках, в которых клетка соприкасается с внеклеточным матриксом.
Нарушением фокальных контактов могут объясняться характерные разрывы базальной мембраны почечных канальцев, наблюдаемые при нефронофтизе. У 85% детей делеции обнаруживаются на обоих аллелях гена NPHP1. На выявлении этих делеций, а также точечных мутаций гена NPHP1 основывается генодиагностика нефронофтиза. Гомозиготные делеции гена NPHP1 описаны также при врожденной глазодвигательной апраксии Когана и при поздней форме пигментной дегенерации сетчатки.
При глазодвигательной апраксии Когана детям грудного и младшего возраста не удаются горизонтальные движения глаз и, чтобы посмотреть в сторону, они совершают резкие толчкообразные движения головой.
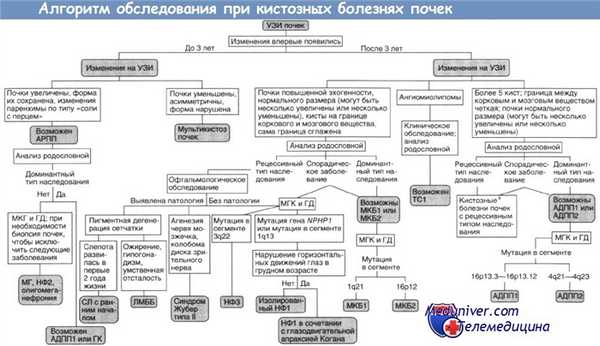
АДПП1, АДПП2 — аутосомно-доминантныи поликистоз почек типов I и II;
АРПП — аутосомно-рецессивный поликистоз почек;
ГД — генодиагностика (при необходимости ГД включают в МГК);
ГК — гломерулокистоз почек;
ЛМББ — синдромы Лоренса—Муна и Барде—Бидля;
МГ — синдром Меккеля—Грубера;
МГК — медико-генетическое консультирование;
МКБ1, МКБ2 — медуллярная кистозная болезнь типов I и II;
НФ1, НФ2, НФЗ — нефронофтиз типов I, II и III;
СЛ — синдром Сениора—Локен;
TC1 — туберозный склероз типа I.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Читайте также:
- Сквозные ранения глазного яблока. Особенности
- Лабораторная диагностика идиопатической тромбоцитопенической пурпуры - анализы
- Снижение уровня андрогенов при гирсутизме. Пероральные контрацептивы и глюкокортикоиды
- Пенетрация глазного яблока: диагностика, лечение
- Потовые железы. Строение и функции потовых желез.