Хромосомные аномалии в гематологии - классификация
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Метод определения Культивирование лимфоцитов периферической крови, микроскопия дифференциально окрашенных метафазных хромосом.
КАРИОТИПИРОВАНИЕ ВХОДИТ В СОСТАВ ИССЛЕДОВАНИЙ:
Вне процесса деления клетки хромосомы в её ядре расположены в виде «распакованной» молекулы ДНК, и они трудно доступны для осмотра в световом микроскопе. Для того, чтобы хромосомы и их структура стали хорошо видны используют специальные красители, позволяющие выявлять гетерогенные (неоднородные) участки хромосом и проводить их анализ – определять кариотип. Хромосомы в световом микроскопе на стадии метафазы представляют собой молекулы ДНК, упакованные при помощи особых белков в плотные сверхспирализованные палочковидные структуры. Таким образом, большое число хромосом упаковывается в маленький объём и помещается в относительно небольшом объёме ядра клетки. Расположение хромосом, видимое в микроскопе, фотографируют и из нескольких фотографий собирают систематизированный кариотип - нумерованный набор хромосомных пар гомологичных хромосом. Изображения хромосом при этом ориентируют вертикально, короткими плечами вверх, а их нумерацию производят в порядке убывания размеров. Пару половых хромосом помещают в самом конце изображения набора хромосом.
Современные методы кариотипирования обеспечивают детальное обнаружение хромосомных аберраций (внутрихромосомных и межхромосомных перестроек), нарушения порядка расположения фрагментов хромосом - делеции, дупликации, инверсии, транслокации. Такое исследование кариотипа позволяет диагностировать ряд хромосомных заболеваний, вызванных как грубыми нарушениями кариотипов (нарушение числа хромосом), так и нарушением хромосомной структуры или множественностью клеточных кариотипов в организме.
Нарушения нормального кариотипа у человека возникают на ранних стадиях развития организма. Если это происходит в половых клеток будущих родителей (в процессе гаметогенеза), то кариотип зиготы (см.), образовавшейся при слиянии родительских клеток, также оказывается нарушенным. При дальнейшем делении такой зиготы все клетки эмбриона и развившегося из него организма окажутся с одинаково аномальным кариотипом. Однако, нарушения кариотипа могут возникнуть и на ранних стадиях дробления зиготы. Развившийся из такой зиготы организм содержит несколько линий клеток (клеточных клонов) с разными кариотипами. Такое многообразие кариотипов во всём организме или только в некоторых его органах называют мозаицизмом.
Как правило, нарушения кариотипа у человека сопровождаются различными, в том числе комплексными, пороками развития, и большинство таких аномалий несовместимо с жизнью. Это приводит к самопроизвольным абортам на ранних стадиях беременности. Однако достаточно большое число плодов (~2,5%) с аномальными кариотипами донашивают до окончания беременности.
Ниже приведена таблица, в которой представлены заболевания, обусловленные нарушениями в кариотипе.
Литература
Необходимо сдавать в состоянии сытости, не рекомендуется сдавать данный тест натощак. Следует воздержаться от приёма антибиотиков за месяц до исследования на кариотип.
Не рекомендуется сдавать кровь единовременно с тестами, имеющие строгую подготовку к сдаче биоматериала (биохимический анализ крови, клинический анализ крови, часть тестов на инфекции и т.д.).
Рекомендуется предварительная консультация врача лаборатории ИНВИТРО с обязательным заполнением специальной анкеты .
Литература
- Бесплодие в браке.
- Первичная аменорея.
- Спонтанные выкидыши (два и более).
- Неразвивающиеся беременности.
- Случаи мёртворождения в семье.
- Случаи ранней детской смертности в семье (до 1 года).
- Врождённые пороки развития (особенно множественные пороки) у ребёнка.
- Задержка умственного и/или физического развития ребёнка.
- Нарушение половой дифференцировки у новорождённого.
- Подозрение на хромосомную болезнь или наследственный синдром по клинической симптоматике (например: изменение формы и размеров черепа, аномалии глаз, носа, пальцев, внешних гениталий и пр.).
- Случаи рождения детей с умственной отсталостью, хромосомной аномалией или врождёнными пороками развития в родословной.
- Обследование перед проведением вспомогательных репродуктивных технологий (ЭКО, ИКСИ и др.).
Литература
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Хромосомные аномалии в гематологии - классификация
Хромосомные аномалии в гематологии - классификация
Хромосомные аномалии могут быть числовыми (кариотип с аномальным числом хромосом в результате потери или добавления хромосомы) или структурными, под которыми понимают изменения структуры отдельных хромосом (потеря, перестройка или добавление хромосомных сегментов). Числовые и структурные аномалии могут сосуществовать в одной опухолевой клетке.
Клетка с нормальным комплектом из 46 структурно нормальных хромосом называется диплоидной. Клетки с 46 хромосомами, но с числовыми хромосомными аномалиями (например, потеря одной хромосомы и добавление другой) называются псевдодиплоидными. Аномальное число хромосом называется анеуплоидией, наличие более чем 46 хромосом — гипердиплоидией, менее 46 хромосом — гиподиплоидией.
Потеря одной копии хромосомы приводит к моносомии по этой хромосоме, потеря обеих копий — к нуллисомии, появление добавочной копии хромосомы — к трисомии по этой хромосоме, более редко встречающееся появление двух добавочных копий — к тетрасомии. Добавление и потеря хромосом обозначаются плюсом или минусом. Например, 45,XY,-7 — это кариотип мужской клетки с моносомией по хромосоме 7, а 47,ХХ,+8 — это кариотип женской клетки с трисомией по хромосоме 8.
Наиболее распространены приобретенные трисомии по хромосоме 8, которые встречаются при острых миелоидных лейкозах, миелодиспластических синдромах и бластном кризе хронического миелолейкоза. Другие трисомии при миелопролиферативных заболеваниях включают +4, +6, +9, +11, +13, +19, +21, при остром лимфобластном лейкозе — +4, +6, +10, +14, +17, +18, +20, +21 и +Х.
Числовые хромосомные аномалии особенно часто встречаются при остром лимфобластном лейкозе и имеют прогностическое значение (гипердиплоидия — благоприятное, гиподиплоидия — неблагоприятное). Наиболее распространенная при хроническом лимфолейкозе цитогенетическая аномалия — трисомия 12 связана с неблагоприятным прогнозом. При множественной миеломе различные варианты анеуплоидии выявлены в 90% случаев.
Структурные хромосомные аномалии
В опухолевых клетках больных онкогематологическими заболеваниями можно обнаружить большое разнообразие структурных аномалий, которые определяются точными терминами: делеции, изохромосомы, дицентрические и изодицентрические хромосомы, инверсии, кольцевые хромосомы, транслокации, инсерции, дупликации, дуплицированные мини-хромосомы и маркерные хромосомы.
Хромосомная делеция (del) — потеря хромосомного сегмента. Различают интерстициальные и терминальные делеции. При интерстициальной делеции утрачен внутренний хромосомный сегмент, а смежные с ним дистальный и проксимальный сегменты оказываются соединенными. Интерстициальная делеция del(5)(ql3q33) обозначает потерю участка длинного плеча хромосомы 5 между сегментами ql3 и q33.
При терминальной делеции отсутствует конец хромосомы, например, делеция del(7)(q22) означает утрату хромосомного материала от сегмента q22 длинного плеча хромосомы 7 до ее теломеры включительно. Вероятно, значение хромосомных делеции в развитии онкогематологических заболеваний определяется утратой генов-супрессоров опухолей.

Изохромосома (i) — структурно аномальная хромосома из двух идентичных плеч, ориентированных как зеркальное отражение одна другой. Изохромосомы могут быть моноцентрическими (содержащими одну центромеру) и дицентрическими или изодицентрическими (две центромеры). Например, изохромосома i(17q), которая часто встречается как вторичная цитогенетическая аномалия при бластном кризе хронического миелолейкоза, состоит из двух длинных плеч.
Важное следствие образования i(17q) заключается в потере короткого плеча 17р, в котором содержится ген-супрессор опухолей р53.
Инверсия (inv) — структурное хромосомное изменение, заключающееся в повороте хромосомного сегмента на 180°. Различают перицентрические и парацентрические инверсии. В перицентрической инверсии сегмент с измененной ориентацией содержит центромеру. В парацентрической инверсии инвертированный сегмент находится внутри короткого или длинного плеча хромосомы и не включает центромеру.
Перицентрическая инверсия inv(16)(pl3q22) часто выявляется при М4-варианте острого миелоидного лейкоза, a inv(3)(q21q26), обнаруженная при варианте М7, может служить примером парацентрической инверсии. Молекулярные последствия инверсий заключаются в перемещении генов в несвойственное им положение и изменении их регуляции.
Кольцевая хромосома (r — от англ. ring) — аномальная хромосома, оба плеча которой, короткое и длинное, разорваны, а точки разрывов соединились вместе, образовав замкнутую структуру (кольцо). Кольцевые хромосомы редко встречаются при онкогематологических заболеваниях.
Хромосомная транслокация (t) — обмен генетическим материалом между негомологичными хромосомами. Различают реципрокные и нереципрокные транслокации. При реципрокной транслокации происходит взаимный обмен фрагментами между двумя, реже тремя и более хромосомами, без потери генетического материала, в отличие от нереципрокных транслокаций. При онкогематологических заболеваниях описано большое число транслокаций, во многих случаях идентифицированы и связанные с ними молекулярные изменения и механизмы злокачественной трансформации.
Ассоциация определенных хромосомных транслокаций с отдельными формами злокачественных опухолей хорошо известна при гемобластозах. Транслокации при лейкозах и лимфомах человека либо активируют клеточные протоонкогены, либо приводят к формированию слитных, «химерных» генов, способствующих злокачественной трансформации гемопоэтических клеток. Молекулярно-генетический анализ точек разрывов показывает, что генетические транслокации изменяют структуру или регуляцию генов, имеющих важное значение для роста и/или дифференцировки соответствующего типа клеток.
В связи с этим они могут быть использованы для дифференциальной диагностики миелопролиферативных и лимфопролиферативных заболеваний.
Пример транслокации, активирующей клеточный протоонкоген в результате перемещения его под контроль регуляторного элемента другого гена, находящегося на другой хромосоме, — t(14;18)(q32;q21), закономерно выявляющаяся при фолликулярных неходжкинских лимфомах и имеющая патогенетическое значение. Точки разрывов хромосом находятся в сегментах q32 хромосомы 14 и q21 хромосомы 18; в результате происходит обмен хромосомными фрагментами между хромосомами 14 и 18 с переносом онкогена bcl-2 с хромосомы 18 на хромосому 14.
Это приводит к дисрегуляции и бесконтрольной экспрессии антиапоптозного гена bcl-2, накоплению долгоживущих центроцитов и способствует злокачественной трансформации.
Транслокация t(9;22)(q34;qll) является примером образования химерного гена bcr/abl, который сформирован при слиянии гена bcr из локуса 22qll и гена abl из локуса 9q34. Новый ген экспрессируется с образованием bcr/abl-мРНК и белка, обладающего повышенной тирозинкиназной активностью и способностью индуцировать неограниченную клеточную пролиферацию. Данная хромосомная перестройка выявляется у 95-97% больных хроническим миелолейкозом.
Пример комплексной транслокации с вовлечением трех хромосом — транслокация t(3;9;22)(ql3;q34;qll), которая происходит между локусами 3ql3, 9q34 и 22qll также с образованием химерного гена bcr/abl.
Дицентрическая хромосома (die) — структурно аномальная хромосома с двумя центромерами, которая является результатом реципрокной транслокации и содержит центромеры обеих вовлеченных в транслокацию хромосом. Дицентрическая хромосома dic(7;12)(pll;pll) встречается при остром лимфобластном лейкозе.
Добавление хромосомного материала (add — от англ. addition) — добавление хромосомного материала неизвестного происхождения, которое обозначается знаком плюс. Например, 14q+ означает присутствие дополнительного генетического материала неизвестного происхождения в длинном плече хромосомы 14.
Инсерция (ins — от англ. insertion) — наличие хромосомного сегмента в новом положении в той же самой или другой гомологичной хромосоме (встречается редко). Некоторые инсерции были описаны ранее как транслокации, например ins(3;3)(q26;q21q26) — инсерция сегмента, расположенного между локусами q21 и q26 хромосомы 3, в локус q26 другой хромосомы 3.
Дупликация (dup) — присутствие добавочной копии сегмента хромосомы рядом с первой копией с образованием тандема из двух копий дуплицированного сегмента. Примером может служить вторичная хромосомная аномалия dup(l)(pl2->q31) при остром лимфобластном лейкозе. В отличие от хромосомных дупликаций, молекулярная микродупликация, такая как дупликация части гена ALL1, может быть определена только молекулярными методами.
Дуплицированные мини-хромосомы (dmin) — маркерные хромосомы без центромер, которые обычно являются результатом генной амплификации. Эти мелкие сферические парные структуры, похожие на диплококков, чаще встречаются при солидных опухолях, нежели при гематологических.
Маркерные хромосомы (mar — от англ. marker) — термин используется для описания структурно аномальных хромосом, не имеющих идентификационных признаков. Кариотип может включать один или более маркеров. Присутствие одной маркерной хромосомы в кариотипе обозначается символом +mar, нескольких различных — +marl, +mar2, +mar3 и т. д., нескольких копий одного маркера — +marl x2, +marl хЗ и т. д.
Врожденные и приобретенные хромосомные изменения
Числовые и структурные хромосомные аномалии могут быть врожденными и приобретенными. Врожденные хромосомные аномалии присутствуют во всех или почти во всех клетках организма уже на самых ранних стадиях эмбриогенеза. Приобретенные хромосомные аномалии возникают в соматических клетках и обычно ассоциированы со злокачественной трансформацией. Врожденные хромосомные аномалии связаны с наследственными генетическими синдромами (например, трисомия по хромосоме 21 — с синдромом Дауна) или являются вариантом нормы.
Самая распространенная врожденная хромосомная аномалия, обнаруженная у фенотипически нормальных людей, — перицентрическая инверсия хромосомы 9 inv(9)(pllql3), встречающаяся у 1% популяции. Кариотип с врожденной (конституциональной) аномалией обозначается буквой с (constitutional) — например, кариотип клеток женщины с синдромом Дауна обозначается как 47,ХХ,+21с.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Обзор хромосомных аномалий (Overview of Chromosomal Anomalies)
Хромосомные аномалии являются причиной различных расстройств. Аномалии, которые затрагивают аутосомы (22 парные хромосомы, одинаковые у мужчин и женщин), встречаются чаще, чем вовлекающие половые хромосомы (X и Y).
Хромосомные аномалии подпадают под различные категории, но в широком смысле могут рассматриваться как численные или структурные.
Численные аномалии включают в себя:
Трисомию (дополнительная хромосома)
Моносомию (отсутствие хромосомы)
Структурные аномалии включают в себя:
Транслокации (аномалии, при которых целая хромосома или сегменты хромосом неправильно объединяются с другими хромосомами)
Делеции и дупликации различных частей хромосом;
Терминология
Для описания хромосомных аномалий важны некоторые специфические термины из области генетики:
Анеуплоидия: наиболее распространенная хромосомная аномалия, вызвана дополнительной или недостающей хромосомой.
Кариотип: Полный набор хромосом в клетках человека.
Генотип: Генетическая конституция, определяемая кариотипом.
Диагностика хромосомных аномалий
Хромосомный анализ (кариотипирование)
Дифференциальное окрашивание хромосом
Флуоресцентная гибридизация in situ (FISH)
Хромосомный микроматричный анализ (матричная сравнительная геномная гибридизация)
Для хромосомного анализа обычно используют лимфоциты, за исключением пренатального исследования, при котором используют амниоциты или клетки плацентарных ворсинок хориона (см. Амниоцентез Амниоцентез Генетическая оценка является частью рутинного пренатального наблюдения, в идеале выполняется до зачатия. Объем исследований, включенных в генетическую оценку, зависит от того, как женщина оценивает. Прочитайте дополнительные сведенияНекоторые методы используют для более четкой характеризации хромосом
При классическом бэндинге (например, G [Гимзе]-, Q [флуоресцентном]- и C-бэндинге) краситель используют для окрашивания полос на хромосомах.
При хромосомном анализе высокого разрешения используют специальные культуральные методы, чтобы получить высокий процент клеток в профазе и прометафазе. Хромосомы при этом менее конденсированы, чем при рутинном анализе метафазы, и число выявляемых полос увеличивается, что позволяет проводить более чувствительный кариотипический анализ.
При спектральном кариотипическом анализе (также называется хромосомой живописи) используют методы хромосом-специфичной многоцветной флуоресцентной гибридизации (FISH), которые улучшают выявление определенных дефектов, включая транслокации и инверсии.
Хромосомный микроматричный анализ (ХМА), или матриксная сравнительная геномная гибридизация (aCGH) является одноэтапным методом, позволяющим просканировать весь геном на наличие хромосомных количественных аномалий, включая увеличение (дупликацию) или уменьшение (делецию) хромосом, что может указывать на несбалансированную транслокацию. Микроматричный анализ однонуклеотидного полиморфизма (ОНП) обладает дополнительной способностью определять области гомозиготности, которая может наблюдаться в случаях, когда родители происходят от общих предков (кровное родство), а также тогда, когда имеется однородительская дисомия Однородительская дисомия Некоторые ситуации представляют аберрантное наследование, часто из-за изменения генов или хромосом. Однако, некоторые из этих изменений, такие как мозаичность, очень распространены, другие. Прочитайте дополнительные сведения (например, обе копии хромосом унаследованы от одного родителя вместо того, чтобы 1 копия - от матери и 1 копия - от отца). Важно отметить, что ХMA не обнаруживает сбалансированных перестроек (например, транслокаций, инверсий), которые не связаны с делециями или дупликациями.
Скрининг
В настоящее время доступны методы неинвазивного пренатального скрининга (НИПС). При НИПС свободные от клеток плода ДНК-последовательности, полученные из образца материнской крови, используются, главным образом, для пренатального скрининга трисомии 21 ( синдром Дауна Синдром Дауна (трисомия 21) Синдром Дауна является аномалией 21-й хромосомы, может проявляться нарушением умственного развития, микроцефалией, небольшим ростом и характерным внешним видом. Диагноз предполагают на основании. Прочитайте дополнительные сведения , а также, трисомии 18 Трисомия по 18 хромосоме Трисомия по 18 хромосоме вызывается дополнительной 18 хромосомой и, как правило, связана с умственной отсталостью, малым весом при рождении, а также различными врожденными аномалиями развития. Прочитайте дополнительные сведенияАвторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Хромосомные аномалии в гематологии - классификация
ККМ — клетки костного мозга
КМ — костный мозг
МСК — мезенхимальные стромальные клетки
ОМЛ — острый миелоидный лейкоз
РА — рефрактерная анемия
РАИБ — рефрактерная анемия с избытком бластов
РАКС — рефрактерная анемия с кольцевыми сидеробластами
FISH — флюоресцентная in situ гибридизация

Оценка цитогенетических аномалий в костном мозге (КМ) у больных с миелодиспластическим синдромом (МДС) имеет большое значение для подтверждения клональной природы заболевания, определения прогноза заболевания и выбора тактики лечения больных. Цитогенетические аномалии в клетках костного мозга (ККМ) выявляют у 40—70% пациентов с МДС, и их разнообразие характеризует заболевание. Так, изолированная делеция (5q) выявляется у пациентов с отдельной нозологической формой МДС, 5q-синдромом, характеризующейся своеобразной клинической картиной, хорошим ответом на иммуномодулирующую терапию и благоприятным прогнозом. Изолированная трисомия 8 позволяет отнести пациента к группе промежуточного риска и является прогностическим фактором эффективности иммуносупрессивной терапии. Аномалии 7-й хромосомы (моносомия или делеция q-плеча) определяют крайне неблагоприятный прогноз и диктуют необходимость выбора агрессивной тактики лечения, в том числе выполнения трансплантации аллогенных гемопоэтических стволовых клеток в качестве первой линии терапии. Комплексные хромосомные аномалии (3 и более) могут включать –5/del(5q), –7/del(7q), del(17p) и чаще всего ассоциируются с вторичным МДС и неблагоприятным течением заболевания. Разнообразие выявляемых генетических аномалий при МДС представлено в табл. 1 [1].
Клиническое течение МДС различно и зависит от варианта заболевания. Рефрактерные цитопении (рефрактерная анемия — РА, рефрактерная тромбоцитопения, рефрактерная нейтропения), как правило, имеют благоприятный прогноз. При увеличении количества бластных клеток в КМ (варианты РА с избытком бластов — РАИБ-1 и РАИБ-2) вероятность трансформации в острый лейкоз увеличивается, и при определении более 20% бластных клеток диагностируют острый миелоидный лейкоз (ОМЛ) с предшествующей миелодисплазией [1—5].
Развитие МДС обусловлено клональным повреждением стволовой кроветворной клетки. Происходит ли это нарушение на уровне истинно стволовой клетки или коммитированных миелоидных предшественников, остается неясным. В последние годы в литературе опубликованы данные о выявлении хромосомных аберраций в популяции гемопоэтических клеток—предшественниц CD34+ у пациентов с МДС и ОМЛ [6—10]. Указывается также на определение патологического клона не только среди миелоидных клеточных линий, но и лимфоидных [11—13], что может быть обусловлено повреждением мультипотентной стволовой клетки.
Кроветворение в КМ обеспечивается постоянным взаимодействием гемопоэтических клеток-предшественниц и клеток стромального микроокружения. Мезенхимальные стромальные клетки (МСК) являются главным компонентом микроокружения и удобной моделью для изучения in vitro. В последнее время опубликованы работы о нарушенной функции стромы при МДС, в частности нарушен механизм миграции гемопоэтических клеток-предшественниц в костномозговые ниши. При сочетанном культивировании гемопоэтических и стромальных предшественников показано, что МСК, полученные от больных с МДС, демонстрируют сниженную способность к поддержанию нормальных гемопоэтических предшественников, у них существенно снижена пластичность по сравнению с таковой у МСК, полученных из КМ здоровых лиц, что может обусловливать неэффективный гемопоэз при МДС [11, 14—18].
Рядом исследовательских групп получены данные, согласно которым в МСК не определяются характерные для МДС хромосомные аберрации, и было сделано предположение, что строма не вовлечена в клональный процесс при данном заболевании и кариотип МСК не изменен [19—21]. Однако в работах E. Flores-Figueroa и соавт. [22, 23], O. Blau и соавт. [24, 25] показано наличие хромосомных нарушений в МСК, в том числе клональных, у отдельных пациентов с МДС. Исследование экспрессии генов с помощью микрочипов демонстрирует наличие генетических аномалий МСК у пациентов с МДС, в частности с 5q-синдромом [26]. Кроме того, на непосредственное участие стромального микроокружения в развитии МДС указывают данные, полученные M. Raaijmakers и соавт. [27], об индуцированном появлении признаков дисплазии в кроветворных клетках мышей при специфическом повреждении стромы.
С учетом несомненной тесной взаимосвязи гемопоэтических и стромальных клеток-предшественниц при развитии МДС представляется важным изучение цитогенетических особенностей этих клеточных популяций у пациентов с МДС.
Материалы и методы
В работе представлены результаты обследования 35 первичных больных, полученные в момент диагностики, в период с июля 2011 г. по май 2012 г.: 29 больных, их которых с МДС (РА) — 4, с рефрактерная цитопенией с мультилинейной дисплазией (РЦМД) — 13, с рефрактерной анемией с кольцевыми сидеробластами (РАКС) — 2, с РАИБ — 7, с 5q-синдром — 3; 6 больных в стадии трансформации в ОМЛ (1 пациент с вторичным ОМЛ) и 7 здоровых доноров КМ. Диагноз устанавливали в соответствии с классификацией ВОЗ (2008) [1]. Соотношение мужчин и женщин 18/17. Медиана возраста составила 60 лет (19—77 лет).
Цитогенетическое исследование ККМ проводили прямым методом и после краткосрочного (16—24 ч) культивирования клеток. Фиксацию и приготовление препаратов хромосом выполняли по общепринятой методике. G-дифференциальную окраску хромосом (G-band) осуществляли по методике M. Seabright [28] в модификации с применением красителя Wright. Цитогенетическое исследование МСК выполняли по методу, описанному Z. Zhang и соавт. [29]. Хромосомный анализ проводили в соответствии с Международной цитогенетической номенклатурой хромосом человека (ISCN 2005) [30]. Анализировали в среднем 20 метафаз. Клональной считали аномалию при обнаружении структурной перестройки и трисомии минимум в 2 метафазах и моносомии минимум в 3. При обнаружении структурной перестройки в одной метафазе перестройку обозначали как неклональную (спонтанную). Единичные неполные митозы не учитывали ввиду возможности потерь хромосом при обработке клеточного осадка. Флюоресцентную in situ гибридизацию (FISH) выполняли на ККМ и изолированных клетках CD34+ с использованием набора ДНК-зондов LSI (5q33—q34) EGR-1 SpectrumOrange/D5S721/D5S23 SpectrumGreen Probe, LSI (7q31) SpectrumOrange/CEP 7 SpectrumGreen Probe, CEP 8 SpectrumOrange DNAProbeKit, LSIAML1/ETO Dual Color Fusion Translocation Probe, CEP X SpectrumOrange/CEPY SpectrumGreen DNA Probe, LSI D20S108 (20q12) SpectrumOrange Probe («Vysis Abbott», США). Гибридизацию осуществляли согласно методике фирмы-производителя. Анализ сигналов проводили под флюоресцентным микроскопом Zeiss-Axioscope с использованием тройного фильтра Orange/Green/Dapi. Анализировали 200 интерфазных ядер.
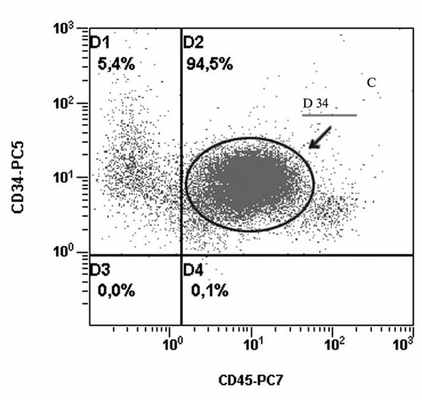
Гемопоэтические клетки—предшественницы СD34+ получали из КМ и периферической крови. Мононуклеарные клетки выделяли, центрифугируя в градиенте плотности фиколла, и метили CD34 MicroBeads human. Изолированную фракцию клеток CD34+ получали, используя MACS колонки для магнитной сепарации, в соответствии с инструкциями фирмы-производителя («Miltenyi Biotec», «Bergisch Gladbach», Германия). Чистота полученной фракции проверена с помощью проточной цитометрии и составила 94,5% (рис. 1). Рисунок 1. Контроль чистоты популяции клеток CD34+ после магнитной сепарации с помощью проточной цитометрии. Выделенную фракцию клеток концентрировали с помощью цитоспина на адгезивном стекле для выполнения FISH-исследования.
Культуру МСК получали из КМ. Фракцию мононуклеаров выделяли из аспирата КМ в градиенте плотности фиколла (1,077 г/см 3 ). Для получения МСК мононуклеары культивировали в пластиковых матрасах с площадью дна 25 и 75 см 2 в концентрации 1—6·10 6 клеток на флакон в среде α-МЕМ («HyClone», США) с 10% эмбриональной телячьей сывороткой («HyClone», США), 2 мМ L-глутамина («BioWest»), 100 ед/мл пенициллина и 50 ед/мл стрептомицина («Ферейн», Россия). После образования конфлюэнтного монослоя из фибробластов клетки снимали 0,05% раствором трипсина и пассировали. В большинстве исследований использовали клетки после 2—3 пассажей.
Результаты
Цитогенетическое исследование ККМ. Характеристика больных и результаты цитогенетического исследования КМ представлены в табл. 2. Нормальный кариотип определен у 19 из 35 больных, у одного из них выявлена конституциональная инверсия inv(9)(p13q21); у 13 (МДС у 11 и ОМЛ у 2) — аномалии кариотипа и у 3 делящиеся клетки не обнаружены. Спектр выделенных аномалий следующий: у 7 больных (МДС у 6 и ОМЛ у 1) выявлены изолированные del(5q), –7, i(14), inv(3); у 1 (МДС) — 2 аномалии — del(5q) и –7, и у 5 (МДС — у 3 и ОМЛ — у 2) комплексный кариотип.
У 5 (14%) из 35 больных методом FISH выявлены скрытые хромосомные аномалии, которые не определены при кариотипировании: у 2 из них (МДС) при цитогенетическом исследовании не получены митозы и методом FISH выявлены хромосомные аномалии: в первом случае — одновременно del(5q) и del(7q) в 84% клеток, во втором — трисомия 8 в 67% клеток. Еще у 2 больных (ОМЛ у 1 и МДС у 1) методом G-band определен нормальный кариотип, однако с помощью метода FISH выявлена del(5q) в обоих случаях. У 1 больного МДС, у которого в кариотипе определена изолированная del(5q), FISH-анализ позволил выявить дополнительно трисомию 21 в 52% клеток (исследование выполнено прицельно, так как имелись анамнестические данные предыдущего цитогенетического анализа у этого пациента, выявившего 2 аномалии). Таким образом, после применения метода FISH дополнительно к исследованию кариотипа число больных с цитогенетическими аномалиями в ККМ увеличилось с 13 до 17 (49%).
FISH-исследование выделенных гемопоэтических клеток—предшественниц CD34+ из КМ и периферической крови. Методом FISH проанализированы гемопоэтические клетки—предшественницы CD34+, полученные из КМ и периферической крови, у 24 из 35 пациентов (у 3 ОМЛ, у 21 МДС). У 10 из 24 пациентов в КМ определены хромосомные аномалии. Мы подтверждали эти аномалии в популяции клеток CD34+ с помощью FISH-исследования.
Цитогенетическое исследование МСК. Кариологический анализ МСК выполнен у 23 из 35 пациентов (МДС у 19 и ОМЛ у 4) и у 7 здоровых доноров КМ (см. табл. 2). У 12 больных из 35 не удалось получить рост МСК в культуре вследствие ограниченной способности стромальных клеток больных с МДС к пролиферации. Нормальный кариотип МСК определен у всех больных ОМЛ и у 6 больных с МДС, у одного из которых в кариотипе МСК определена конституциональная инверсия inv(9). У 2 (11%) из 19 пациентов с МДС выявлены аномалии кариотипа.
У одного из них с конституциональной inv(9) (пациент №12) выявлена неклональная транслокация в одной метафазе: 46ХY,t(2;22)(p10;q11), inv(9)(p13q21)[1]/46ХY, inv(9) (p13q21)[19]. У второго (пациент №21) в кариотипе МСК выявлена клональная перестройка в 7 метафазах из 20: 46ХУ,add(2q)[7]/46ХУ[13]. В КМ у этого больного определен комплексный кариотип (рис. 2). Рисунок 2. Кариотип ККМ и МСК пациента №21 И., 58 лет, диагноз: МДС, РА. а — кариотип ККМ: 45—46,XY,–5,–13,der(19),add(q13?or p13),–20,+2mar,+mar del(13q21)?,+d min. Рисунок 2. Кариотип ККМ и МСК пациента №21 И., 58 лет, диагноз: МДС, РА. б — кариотип МСК: 46,XY,add (2)(q36). У обоих пациентов с инверсией inv(9) в ККМ и МСК ее конституциональный характер был подтвержден результатами исследования кариотипа стимулированной фитогемагглютинином культуры лимфоцитов периферической крови. При кариотипировании МСК от здоровых доноров КМ во всех исследованных образцах получен нормальный кариотип.
В настоящей работе представлен анализ цитогенетических изменений в популяциях гемопоэтических и стромальных клеток-предшественниц у пациентов с различными вариантами МДС, а также в стадии трансформации заболевания в ОМЛ. Интерес к изучению этой области определяется тем, что до сих пор остается неуточненным вопрос об истинном уровне генетического повреждения при МДС, а также участии стромального микроокружения в патогенезе клональной эволюции миелодисплазии.
Определение кариотипа ККМ в настоящее время входит в перечень стандартных исследований, необходимых для верификации диагноза МДС. Выявленные хромосомные аномалии определяют прогноз заболевания и соответственно тактику лечения больных. Современные прогностические шкалы, в частности шкала IPSS-R, помимо количества бластных клеток в КМ, степени цитопении и зависимости от гемотрансфузий, включают данные цитогенетического анализа [31]. В нашем исследовании цитогенетические аномалии в КМ обнаружены у 17 (49%) из 35 обследованных, что коррелирует с данными литературы [1—5]. Стоит еще раз отметить, что не во всех случаях метод стандартного цитогенетического исследования явился достаточным для обнаружения аномалий кариотипа, и у около 10% пациентов не удалось получить делящиеся клетки для анализа. По меньшей мере в 14% случаев (по нашим данным, у 5 из 35 больных) методом FISH могут быть выявлены дополнительные (скрытые) хромосомные аномалии. У всех 5 больных размер выявленных клонов был довольно велик (от 52 до 84%); такие клоны не могут быть незамеченными при хромосомном анализе. По-видимому, все эти клетки находятся в стадии G0, не выходят в митоз и поэтому не обнаруживаются при кариотипировании. У 1 из 5 пациентов с выявленной del(5q) неразделившиеся клетки содержали вторую аномалию — трисомию 21. Можно предположить, что у этого больного наблюдалось 2 клона клеток в КМ — один только с del(5q) и второй одновременно с del(5q) и трисомией 21. Выявление дополнительных хромосомных аномалий у ряда пациентов может иметь принципиальное значение, так как от этого зависят и прогноз заболевания, и терапевтический подход. Так, при определении нормального кариотипа в ККМ может быть выбрана тактика динамического наблюдения с последующими исследованиями КМ, тогда как выявление del(5q) позволяет эффективно применять иммуномодулирующую терапию. Следовательно, в дебюте заболевания всем пациентам целесообразно сначала проводить стандартное цитогенетическое исследование, и в случаях определения нормального кариотипа или отсутствия делящихся клеток необходимо выполнять FISH-исследование с целью определения клинически значимых хромосомных аномалий. Всем пациентам, не дожидаясь получения результатов кариотипирования, проводить FISH-исследование нецелесообразно [32].
В клетках CD34+, выделенных из КМ и периферической крови, определены те же клональные нарушения, что и в ККМ пациентов с аномалиями кариотипа, что подтверждает поражение гемопоэтических предшественников при МДС. По данным литературы, количество клеток—предшественниц CD34+ в КМ больных МДС значительно больше, чем у здоровых лиц, пациентов с апластической анемией и вторичными цитопеническими синдромами. Процент клеток CD34+ неоднороден у пациентов категорий низкого (1,7%) и высокого (10,5%) риска и максимальный у пациентов с ОМЛ (35%), он коррелирует с процентом бластных клеток и немного превышает его [33]. Количество циркулирующих клеток CD34+ у пациентов с МДС также значительно больше, чем у здоровых лиц, и зависит от процентного содержания их в КМ [34]. Наряду с увеличением количества бластных клеток, увеличение количества клеток CD34+ тоже может свидетельствовать о прогрессии заболевания. В нашем исследовании выявлено, что в среднем процент клонально измененных клеток среди гемопоэтических предшественников не отличается от такового в общей популяции ККМ пациентов с аномалиями кариотипа. Однако у 4 из 10 больных отличия выявлены. Согласно нашим наблюдениям размер патологического клона в клетках CD34+ может быть как значительно больше, так и меньше размера клона в общей популяции ККМ. Возможно, выраженность клональных изменений в гемопоэтических клетках-предшественницах может свидетельствовать о динамике развития заболевания и иметь прогностическое значение. Ввиду того что результаты FISH-исследования циркулирующих клеток СD34+ в среднем сопоставимы с результатами исследования ККМ, цитогенетический контроль динамики заболевания в процессе лечения при наличии маркеров для FISH-исследования может осуществляться с использованием изолированной фракции клеток CD34+ или мононуклеарной фракции периферической крови, что поможет снизить кратность пункций КМ у пациентов с МДС. У пациентов с нормальным кариотипом хромосомные перестройки не обнаружены и в популяции клеток CD34+. Таким образом, детальное цитогенетическое исследование (стандартное цитогенетическое исследование и FISH) не выявило у пациентов с нормальным кариотипом в КМ «скрытых» хромосомных аномалий в гемопоэтических предшественниках CD34+.
Почти у 90% пациентов с МДС и у всех пациентов с ОМЛ в нашем исследовании определен нормальный кариотип МСК. Хромосомные аномалии МСК выявлены у 2 (11%) из 19 пациентов с МДС. У обоих выявлены структурные аномалии: у одного — 35% клон add(2q), у другого — с конституциональной хромосомной нестабильностью, inv(9)(p13;q21) — единичный митоз с t(2;22)(p10;q11). Учитывая, что у пациента с клональными изменениями кариотипа МСК в ККМ обнаружен комплексный кариотип, можно предположить, что генетическая нестабильность стромы обусловлена множественными цитогенетическими аномалиями в кроветворных клетках. Численные аномалии кариотипа МСК не выявлены ни в одном случае.
Как упоминалось выше, в литературе есть публикации, согласно которым хромосомные аномалии в МСК у пациентов с МДС не определяются [19, 20]. По-видимому, причина заключается в использовании разных методов цитогенетического анализа, так как авторы с целью определения аномалий кариотипа МСК применяли только FISH-исследование с выявленными в ККМ цитогенетическими маркерами и не исследовали кариотип МСК, тогда как выявленные нами аномалии МСК определяются только при кариотипировании. Таким образом, в МСК у пациентов с МДС, действительно, не определяются характерные для ККМ цитогенетические аномалии, однако возможны отличные от них аберрации. В работах E. Flores-Figueroa и соавт. [22, 23] и Lu-Xi Song и соавт. [35] описаны аномалии кариотипа МСК более чем у 50% обследованных пациентов с МДС, в основном за счет неклональных потерь отдельных хромосом. Однако встречающиеся отдельные неполные митозы нами не учитывались, так как подобные ситуации могут встречаться вследствие технических особенностей обработки клеточного осадка. O. Blau и соавт. [25] описывают клональные структурные аномалии МСК у 16% пациентов с МДС и ОМЛ. Результаты нашего исследования сопоставимы с результатами O. Blau, однако мы не выявили аномалии кариотипа МСК у пациентов в стадии трансформации МДС в ОМЛ.
Заключение
Таким образом, несмотря на небольшое число проанализированных больных, на основании проведенного исследования можно сделать заключение, что в гемопоэтических и мезенхимальных клетках-предшественницах у пациентов с МДС определяются разные цитогенетические аномалии. В клетках CD34+, выделенных из КМ и периферической крови, выявлены те же аномалии, что и в общей популяции ККМ. У 11% пациентов с МДС выявлены аномалии кариотипа МСК, что указывает на генетическую нестабильность стромы при этом заболевании и подверженность возникновению хромосомных поломок. Различия хромосомных аномалий, определяемых в гемопоэтических и мезенхимальных клетках-предшественницах, подтверждают, что клетки стромального микроокружения в отличие от гемопоэтических предшественников не являются частью патологического клона при МДС, однако, вероятно, играют важную роль в патогенезе развития заболевания.
Острые лейкозы
Лейкозы острые — это гетерогенная группа опухолевых заболеваний кроветворной системы, которые начинаются в костном мозге и характеризуются накоплением недифференцированных (бластных) клеток и подавлением нормальных ростков кроветворения. Заболеваемость острыми лейкозами — в среднем 3—5 первичных случаев на 100 000 человек в год. Острые лейкозы распространены повсеместно в разных странах, встречаются во всех возрастных группах, но у детей их удельный вес наибольший среди всех гемобластозов. Мужчины и женщины заболевают с равной частотой.
Классификация
Острые лейкозы классифицируют в зависимости от цитохимических и иммунофенотипических особенностей опухолевых клеток, выделяют:
-острые лимфобластные
-острые нелимфобластные лейкозы.
Современная классификация острых лейкозов предложена группой французских, американских и британских гематологов - FAB-классификация.
Предлейкоз — это доклиническая стадия острого лейкоза, при которой ограниченное количество бластов еще не вызывает явных клинических расстройств (МДС - миелодиспластические синдромы).
Клиника
Клиника развернутой стадии острого лейкоза: характерны нарастающая «беспричинная» слабость, недомогание, повышение Т°. Довольно часто геморрагический синдром: кровоточивость слизистых оболочек (десневые, носовые, маточные, кишечные кровотечения), петехиальная сыпь на коже, в первую очередь голеней, реже наблюдается гематурия и еще реже — кровоизлияние в мозг. Геморрагический синдром - нередко самое раннее проявление острого лейкоза.
При острых миеломнобластных лейкозах характерна гиперплазия десен, из-за которой больные часто впервые обращаются к стоматологу. Весьма характерны язвенно-некротические поражения слизистых оболочек ротовой полости (стоматит), глотки (ангина) и кишечника - некротическая энтеропатия, парапроктит и др.. что объясняется снижением уровня гранулоцитов крови. Начальные симптомы некротической энтеропатии - плеск и урчание при пальпации илеоцекальной зоны, кашицеобразный стул, легкое вздутие живота. В крови в большинстве случаев выявляются бластные клетки - уродливые, с деформированными нуклеолами, двухядерные; обычно много митозов. В костном мозге содержание бластов достигает иногда 95-99%. За счет бластов общий лейкоцитоз в крови иногда очень высок, изредка он в норме; известны и лейкопенические варианты дебюта острого лейкоза. При кариологическом исследовании в бластных клетках обнаруживаются хромосомные аномалии необычное число хромосом, аберрации и т.д.). Анемия при остром лейкозе нередко наступает позднее, чем тромбоцитопения и агранулоцитоз, она объясняется как кровотечениями, так и бластозом в костном мозге. Симптомы анемии весьма типичны для острого лейкоза; обычно малокровие носит нормохромный или гипохромный характер.
Первыми проявлениями острого лейкоза может быть также «беспричинный» субфебрилитет, изолированное увеличение лимфоузла, увеличение селезенки, лейкопения, тромбоцитопения, упорные боли в суставах и др. Увеличение лимфоузлов, печени и селезенки в начале развернутой стадии встречается не всегда, но с течением времени развивается у многих больных острым лейкозом. Болезненность костей выявляется лишь при большой массе лейкозных клеток, т.е. в уже запущенных случаях острого лейкоза. Опыт показывает, что даже при высоком бластозе в крови и костном мозге у пациента может сохраняться достаточный уровень гемоглобина, тромбоцитов и нейтрофилов, и общее состояние остается удовлетворительным (пациент даже отказывается от дальнейшего обследования, утверждая, что он чувствует себя нормально). Обычно это наблюдается при остром лимфобластном лейкозе (изменения в крови выявляются при периодическом осмотре, диспансеризации и т.д.). Задача врача в этих случаях — убедить пациента и его родственников в неотложной необходимости обследования. В других случаях даже небольшой бластоз вызывает выраженную слабость, глубокую тромбоцитопению, анемию и гранулоцитопению. что сразу влечет клинические осложнения. При всех вариантах острого лейкоза наблюдаются те или иные нарушения в свертывающей системе крови, вплоть до ДВС-синдрома, особенно при большом содержании бластов, тем более атипичных промиелоцитов в крови.
ДВС-синдром влечет за собой кровоточивость, нередко летальную.
Острый лимфобластный лейкоз
Острый лимфобластный лейкоз (ОЛЛ) - злокачественная опухоль крови, возникающая в результате мутации на уровне клетки-предшественницы лимфопоэза. Этот тип лейкоза является самым частым онкогематологическим заболеванием у детей (они составляют около 85% всех случаев острых лейкозов). У взрослых острый лимфобластный лейкоз наблюдается в 25% случаев всех острых лейкозов. Учашение заболеваемости острыми лимфобластными лейкозами отмечается у детей в возрасте 2—4 года, затем у лиц в возрасте 50—60 лет.
Классификация: на основе светооптических характеристик лейкозных клеток выделяют три основных варианта ОЛЛ: LI, L2 и L3. У детей чаше наблюдается вариант LI, у взрослых - форма L2.
При иммунофенотипировании различают четыре варианта опухоли:
- пре-В-клеточный вариант,
- ранний пре-В-клеточный,
- Т-клеточный (около 15% всех случаев),
- зрелый В-клеточный (весьма редкий).
После уточнения варианта ОЛЛ, применяют наиболее подходящую лечебную схему и оценивают прогноз для жизни. Так, у детей с ранним пре-В-клеточным вариантом ОЛЛ прогноз достоверно лучше; зрелые В-клеточные острые лимфобластные лейкозы менее благоприятны прогностически, особенно у взрослых. Цитогенетические исследования обнаруживают хромосомные аберрации более чем у половины больных острыми лимфобластными лейкозами. Наиболее серьезный риск-фактор - наличие «филадельфийской» хромосомы. У детей с острым лимфобластным лейкозом эта транслокация встречается в 7% случаев, у взрослых — в 30% случаев. Ухудшает прогноз наличие высокого уровня сывороточной лактатдегидрогеназы в момент выявления ОЛЛ. Один из ведущих риск-факторов — это высокий лейкоцитоз. Отметим, что у больных старше 50 лет ремиссии ОЛЛ удается добиться реже, чем у пациентов более молодого возраста.
Клиническая симптоматика при острых лимфобластных лейкозах: в начале заболевания обычно жалоб нет, или они весьма скудные — умеренная слабость и утомляемость, иногда бледность кожных покровов. В развернутой стадии ОЛЛ может наблюдаться лихорадка, геморрагические явления, малокровие и др. Однако анемизация и геморрагический диатез выражены значительно менее, чем при острых нелимфобластных лейкозах. При острых лимфобластных лейкозах увеличение печени наблюдается не всегда. У молодых больных острыми лимфобластными лейкозами возможна умеренная диффузная лимфоаденопатия. Первыми проявлениями острого лейкоза может быть также «беспричинный» субфебрилитет, бледность кожи и слизистых оболочек, кровоточивость десен, обильные менструации, изолированное увеличение лимфоузла, упорные боли в суставах и др. Почти всегда наблюдается слабость, утомляемость, потливость. Нередко отмечаются рецидивирующие ЛОР-инфекции, герпес. Болезненность костей выявляется лишь при большой массе лейкозных клеток, т.е. в уже запущенных случаях острого лейкоза. Даже при высоком бластозе в крови и костном мозге у пациента может сохраняться достаточный уровень гемоглобина, тромбоцитов и нейтрофилов. и общее состояние остается удовлетворительным.
Диагностика: поскольку специфичных симптомов при острых лейкозах не существует, диагноз устанавливают только после морфологического исследования крови и костного мозга. Появление даже единичных бластных клеток в периферической крови (гемограмма) требует безотлагательного исследования костного мозга (стернальная пункция).
Принципы лечения: госпитализация в гематологический стационар.
Химиотерапия, поспешно начатая в амбулаторных условиях, полностью исключает возможность последующего излечения пациента даже в условиях гематологического центра. Не рекомендуется начинать цитостатическое лечение в амбулаторных условиях, иногда однократное введение цитостатиков или глюкокортикостероидов до неузнаваемости меняет клиническую и гематологическую картину, и в дальнейшем не позволяет поставить правильный диагноз.
Лечение острых лейкозов предполагает:
- индукцию ремиссии,
- консолидацию полученной ремиссии,
- поддерживающую терапию,
- а также профилактику нейролейкемии.
Применяют патогенетическое лечение острого лейкоза с целью достижения полной клинико-гематологической ремиссии. В настоящее время общепринят метод интенсивной комбинированной длительной цитостатической терапии острого лейкоза (тотальная терапия).
Нелимфобластные острые лейкозы
Нелимфобластные острые лейкозы - эти варианты лейкозов возникают в результате мутации одной из миелоидных клеток-предшественников. Преобладают среди лейкозов взрослых.
В группе острых миелоидных лейкозов выделяют следующие формы заболевания:
МО-острый миелобластный лейкоз, при котором бластные клетки имеют минимальные признаки дифференцировки;
M1-2—острый миелобластный лейкоз с наличием клеток разной степени зрелости;
МЗ—острый промиелоцитарный лейкоз;
М4- острый миеломонобластный лейкоз;
М5—острый монобластный лейкоз;
М6—острый эритролейкоз;
М7- острый мегакариобластный лейкоз.
Острый миелобластный (ОМЛ) и миеломонобластный лейкозы (ОММЛ) встречаются чаще всего: среди нелимфобластных острых лейкозов взрослых эти формы составляют ок. 80%. Возрастные пики острого миелобластного и миеломонобластного лейкоза наблюдаются в возрасте до 1-2 лет, затем - в 38 лет (ОМЛ) и в 50 лет (ОММЛ). Клиника: характерно острое начало болезни с высокой лихорадкой, некрозами в горле (при глубокой первичной гранулоцитопении); часто бывают боли в костях, малокровие, геморрагические явления, которые обусловлены не только тромбоцитопенией, но и ДВС-синдромом. Наблюдается кровоточивость слизистых оболочек (десневые, носовые, маточные, кишечные кровотечения), петехиальная сыпь на коже, в первую очередь голеней, реже наблюдается гематурия и еще реже — кровоизлияние в мозг. Геморрагический синдром - нередко самое раннее проявление острого нелимфобластного лейкоза. Увеличение селезенки при остром миелобластном и миеломонобластном лейкозе умеренное. Весьма характерны язвенно-некротические поражения слизистых оболочек ротовой полости (стоматит, гиперпластический гингивит с кровоточивостью), глотки (ангина) и кишечника - некротическая энтеропатия, парапроктит и др., что объясняется снижением уровня гранулоцитов крови. Начальные симптомы некротической энтеропатии: плеск и урчание при пальпации илеоцекальной зоны, кашицеобразный стул, легкое вздутие живота, высокая лихорадка. Прогноз: средняя частота первых ремиссий при остром миелобластном и миеломонобластном лейкозах при современной терапии достигает 60%. Продолжительность первой ремиссии превышает 2 года. Онкологическое выздоровление наблюдается у 10% больных всех возрастов.
Редкие формы острых лейкозов
Макрофагальный острый лейкоз - важнейший и постоянный его клинический признак - высокая асептическая лихорадка, иногда с ознобами. Часто наблюдается гепатоспленомегалия. Бластная инфильтрация печени часто осложняется желтухой.
Мегакариобластный острый лейкоз - клиническая картина сходна с другими острыми лейкозами, в крови и костном мозге наряду с недифференцируемыми бластными клетками присутствуют и мегакариобласты.
Миелодиспластические синдромы (МДС) — гетерогенная группа гемопатий, куда входят разнообразные заболевания неодинаковой природы, сопровождающиеся как значительными изменениями костного мозга, так и его функциональными нарушениями, часто трансформирующиеся в высокобластные острые лейкозы. Разнообразные гемопатии, объединенные экспертами ВОЗ в группу под общим названием МДС, имеют общую причину - повреждение клеток на уровне, близком к стволовой кроветворной клетке. В отличие от других гемобластозов, при МДС клеточный апоптоз сохранен или усилен, что и формирует периферическую би/пан/цитопению на фоне гиперплазии костного мозга.
Преобладающим симптомом является малокровие. Часть гемопатий. входящих в этот «сборник», иногда называют предлейкозом. Современные данные показывают, что предлейкоз - это доклиническая стадия острого лейкоза, при которой ограниченное количество бластов еще не вызывает явных клинических расстройств. Гематологическими признаками формирующегося острого лейкоза являются: нарастающая анемия без ретикулоцитоза; «необъяснимые» гранулоцитопении; «беспричинный» моноцитоз без явной гиперплазии костного мозга в трепанате; хронический нерезко выраженный аутоиммунный гемолиз, немногочисленные очаги гиперплазии в трепанате костного мозга (через несколько лет возможна трансформация в острый лейкоз). Варианты МДС отмечаются в основном у лиц пожилого возраста (старше 60 лет). Характерна слабость, потливость, утомляемость, одышка. Умеренное увеличение печени и селезенки наблюдается только при варианте ХММЛ: лимфоузлы как правило в норме. Лейкозы, развившиеся из миелодиспластических синдромов, отличаются меньшей чувствительностью к терапии, чем первичные острые нелимфобластные лейкозы.
Монобластный острый лейкоз наблюдается весьма редко. Клиническая картина этой формы лейкоза напоминает острый миелобластный лейкоз, однако, в этой форме значительнее выражены интоксикация и гипертермия, чаще наблюдаются некротические изменения слизистой оболочки рта и глотки. Весьма характерна лейкемическая инфильтрация десен, ведущая к гипертрофии сосочков («лейкемический гингивит»). Нередко развивается инфильтрация миндалин и десен, а на поздних этапах прогрессии возникают опухолевые пролифераты во всех внутренних органах, лейкемиды кожи и т.д.
Плазмобластный острый лейкоз отмечается во всех возрастах, в т.ч. у молодых лиц и у детей, характерно острое клиническое течение. Наблюдается увеличение лимфоузлов, селезенки, печени. В сыворотке крови выявляется М-градиент - моноклональный иммуноглобулин (парапротеин).
Промиелоцитарный острый лейкоз - довольно редко встречается - не более 7% всех острых лейкозов. Прежде это заболевание отличалось тяжелейшим прогнозом (массивная кровопотеря или геморрагический инсульт уже в первые недели от момента диагностики), а теперь - это одна из наиболее курабельных форм, при которой наблюдается «онкологическое» излечение на фоне дозревания лейкозных бластов до нормальных форм.
Эритромиелоз острый, эритробластный острый лейкоз, болезнь Ди Гульельмо - редкая форма острого лейкоза. В анамнезе больных острым эритромиелозом часто встречается лучевая или химиотерапия; нередко выявляется как вторичный лейкоз у лиц, пролеченных по поводу лимфогранулематоза, миеломной болезни, эритремии.
Читайте также: