Миотоническая дистрофия: причины, признаки, фенотип
Добавил пользователь Валентин П. Обновлено: 30.01.2026
Миотоническая дистрофия. Причины и признаки миотонической дистрофии.
Миотоническая дистрофия — это самый распространенный тип мышечной дистрофии у взрослых. Заболевание обычно не ограничивается поражением скелетных мышц, а является, мультисистемным, с изменениями в поджелудочной железе, гонадах, щитовидной железе, миокарде и головном мозге. Заболевание передается по наследству аутосомно-доминантным путем. Дефектный ген локализован в хромосоме 19 (19q 13.2—13.3), и в норме кодирует миотонин-протеинкиназу — фермент, встречающийся в различных тканях и отвечающий за процесс фосфорилирования белков. Генерализацию заболевания объясняют широкой представленностью фермента в клетках. На молекулярном уровне дефект гена характеризуется экспансией триплетов (амплификацией повторения триплетов) гуанин-цитозинтимин.
Частота триплетных повторов варьирует и напрямую связана с тяжестью заболевания и обратно пропорциональна возрасту начала заболевания.
Поражение мышц. Характерной особенностью является слабость мышц лица. Лицо становится удлиненным и худым с ранним облысением в области лба. Также наблюдается птоз, который, однако, не столь выражен, как при миастении gravis или при синдроме Kearns-Sayre. Обычно отмечается атрофия височных и жевательных мышц. Слабость грудино-ключично-сосцевидной мышцы, как правило, более выраженная, чем в мышцах плеча и задней группе мышц шеи. В конечностях преимущественно страдают задние группы мышц, причем они поражаются значительно позже, чем вышеперечисленная мускулатура.
Проксимальная мускулатура конечностей вовлекается в процесс в самую последнюю очередь. Таким образом, в отличие от больных другими видами дистрофий, пациенты с миотонической дистрофией длительное время сохраняют способность к самостоятельному передвижению. Миотония — это удлинение периода расслабления мышцы после сокращения. Довольно часто больные даже не жалуются на миотонию. Изредка их беспокоит скованность мышц. Выявить миотонию можно перкуссией возвышения большого пальца или языка. Больные не могут быстро разжать руку после рукопожатия. При истинной миотонии повторное сокращение мышц способствует уменьшению миотонии.
Клинический диагноз подтверждается наличием миотонических разрядов на ЭМГ. Методы молекулярной диагностики (ПЦР) позволяют выявить экспансию триплетных повторов.
Общие признаки миотонической дистрофии. Кроме неврологических симптомов у многих больных отмечаются признаки системного поражения, которые даже могут быть ведущими в клинической картине. В этих случаях диагностика миотонической дистрофии затруднена. Поздняя диагностика очень опасна, т. к. пациентам необходима профилактическая и лечебная поддержка. Признаки системного поражения включают катаракту, тубулярную атрофию яичек у мужчин, блокаду сердечного проведений, нарушения ритма сердца, которые могут привести к внезапной смерти.
Тяжелые аритмии часто наблюдаются в фенотипически легких случаях, при этом кардиомиопатия у этих больных не выражена. Даже при легком поражении мышц отмечаются выраженные запоры и холелитиаз. При вовлечении в процесс диафрагмы может возникнуть гиповентиляция. Часто больные жалуются на чрезмерную сонливость. Из-за нарушения функции сердечно-сосудистой и легочных систем большую опасность для больных представляют хирургические вмешательства под наркозом. Препараты группы деполяризующих миорелаксантов могут значительно ухудшить состояние пациента, опиаты и барбитураты могут способствовать развитию дыхательной недостаточности.
К сожалению, часто бывает так, что диагноз миотонической дистрофии ставится только тогда, когда у больного наблюдаются осложнения со стороны сердечно-сосудистой системы после проведения общей анестезии. Нередко у больных миотонической дистрофией отмечаются легкое снижение интеллекта, апатия и сонливость. Врожденная миотоническая дистрофия — это более тяжелая форма дистрофии, чем описанная выше. Она манифестирует при рождении. У новорожденных наблюдается двусторонняя слабость лицевой мускулатуры, гипотония, задержка умственного развития, угнетение дыхания. Во время беременности часто отмечаются многоводие и вялые движения плода. Носителями патологического гена являются только женщины.
- Вернуться в оглавление раздела "Неврология."
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Миотония
Миотония — наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. Характерные признаки миотонии — миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании. Врожденная миотония сопровождается мышечной гипертрофией, дистрофическая миотония, напротив, сопровождается мышечными атрофиями. Диагностика миотонии осуществляется при помощи ЭМГ, ЭНГ и исследования вызванных потенциалов. До настоящего времени радикальная медикаментозная терапия миотонии не разработана. Пациентам проводится симптоматическое и метаболическое лечение, массаж, ЛФК, электростимуляция.
Общие сведения
Миотония — наследственное заболевание, относящееся к каналопатиям (заболевания, связанные с патологией ионных каналов). Проявляется замедленным расслаблением мышц. Характерные признаки миотонии — миотонические разряды, выявляемые игольчатой ЭМГ, и миотонические феномены, которые выявляются при клиническом обследовании.
Этиология и патогенез миотонии
Среди двух типов дистрофической миотонии тип 1, ген которого картирован в локусе 19q13, встречается наиболее часто (98%). Как и все типы дистрофической миотонии он передается по наследству по аутосомно-доминантному типу. Основной этиологический фактор дистрофической миотонии 1 типа — увеличением количества тринуклеотидных повторов CTG (до нескольких тысяч). Миотонин-протеинкиназа, кодируемая геном DMPK, присутствует не только в скелетных, но и миокарде, а также ЦНС. Этим и объясняются основные клинические проявления дистрофической миотонии.
Врожденная парамиотония Эйленбурга, связанная с патологией натриевых каналов, передается по аутосомно-доминантному типу. Ген SCN4A картирован в локусе 17q23.1-q25.3. Парамиотонические проявления развиваются в связи с повышенной возбудимостью мембраны мышечных волокон и нарушением функционирования сократительных элементов мышцы.
В отличие от большинства нейромиотоний, являющихся приобретенными заболеваниями, идиопатическая нейромиотония (синдром Исаакса) передается по наследству и встречается наиболее часто. Эффективность введения кураре для купирования мышечных спазмов говорит о неврогенной природе нейромиотонии, однако окончательно патогенез заболевания неясен и сегодня. С выявлением повышенного титра антител к потенциал-зависимым калиевым каналам нейромиотонию стали считать аутоиммунным заболеванием. Об аутоиммунном характере заболевания говорит также наблюдаемая в ряде случаев эффективность плазмафереза.
Классификация миотонии
Наиболее распространенные формы миотонии:
- дистрофическая (двух типов)
- хондродистрофическая
- конгенитальная аутосомно-доминантная
- конгенитальная аутосомно-рецессивная
- парамиотония Эйленбурга
- нейромиотония
Клиническая картина миотонии
Симптом «кулака» — основной клинический тест на выявление миотонии: пациент не может быстро разжать кулак, для этого ему нужно время и определенные усилия. При повторных попытках такой миотонический феномен угасает за исключением миотонии Эйленбурга, когда скованность, наоборот, усиливается с каждой повторной попыткой. Скованность также наблюдается при разжимании сжатых челюстей, быстро открыть зажмуренные глаза, быстро встать со стула. На игольчатой электромиографии выявляют один из самых характерных для миотонии феноменов — миотонические разряды, сопровождающиеся звуком «пикирующего бомбардировщика», возникающие при введении и перемещении игольчатого электрода.
Отличительной клинической особенностью врожденной миотонии является гипертрофия отдельных мышечных групп, которая создает впечатление об атлетическом телосложении пациента. В большинстве случаев мышечная сила сохранена, но иногда бывает снижена в дистальных мышцах рук. Дистрофическая миотония — мультисистемное заболевание. В большинстве случаев неврологические симптомы сочетаются с сердечной патологией (гипертрофия левого желудочка, аритмия), церебральными симптомами (гиперсомния, сниженный уровень интеллекта), эндокринными расстройствами (нарушение менструального цикла у женщин; гипогонадизм и импотенция у мужчин). Для парамиотонии типична т. н. «холодовая миотония» — возникновение мышечного спазма и пареза на холоде; такие приступы могут длиться от нескольких минут до нескольких часов. Клиническими проявлениями нейромиотоний являются мышечная скованность, спазмы (безболезненные) и постоянная мышечная активность на ЭМГ.
Диагностика миотонии
Дебют дистрофической миотонии приходится, как правило, на юношеский либо взрослый возраст, степень прогрессирования заболевания зависит от генетического дефекта, поэтому тщательный сбор семейного анамнеза имеет большое значение в диагностировании дистрофической миотонии. Миотония Беккера дебютирует в 5-12 лет и характеризуется медленным течением, а миотония Томсена может дебютировать как в детском, так и в зрелом возрасте и протекает, как правило, тяжело и с осложнениями. Физикальное обследование при дистрофической миотонии выявляет атрофию мышц и снижение их силы. Для дистрофической миотонии типа 1 характерна мышечная слабость в дистальных отделах конечностей, для дистрофической миотонии типа 2 — в проксимальных.
С помощью лабораторных исследований при нейромиотонии выявляют антитела к потенциал-зависимым калиевым каналам, а дистрофическая миотония отличается незначительным повышением активности КФК в крови.
Основными инструментальными методом диагностики миотонии является игольчатая электромиография (ЭМГ), на которой определяют миотонические разряды — патогномоничный признак миотонии. Проводится исследование при помощи вызванных потенциалов и электронейрография. При парамиотонии на ЭМГ регистрируют нормальные ПДЕ и редкие миотонические разряды. Для врожденной миотонии типично сохранение параметров ДЕ в пределах нормы, для дистрофической миотонии — сочетание невропатических и миопатических черт. Для диагностики парамиотонии проводят холодовую пробу: незначительное охлаждение вызывает миотонические разряды, при дальнейшем охлаждении наступает «биоэлектрическое молчание» (исчезают как миотонические феномены, так и ПДЕ). ДНК-диагностика дистрофической миотонии основана на выявлении повышенного количества CTG-повторов в гене DMPK.
Дифференциальный диагноз
Как правило, дифференцировать врожденную миотонию от дистрофической миотонии неврологу удается по клиническим признакам. Однако в ряде случаев врожденной миотонии определяют легкую слабость дистальных мышц рук и слабую активность при ЭМГ — признаки, типичные для дистрофической миотонии. Постоянная мышечная активность — клинический признак нейромиотонии — входит в состав синдрома "ригидного человека" (stiff-man syndrome), однако в отличие от нейромиотонии мышечная активность при синдроме "ригидного человека" снижается после введения диазепама, а также во время сна.
Лечение миотонии
Целью лечения нейромиотонии является устранение постоянной мышечной активности и достижение возможной ремиссии, целью лечения миотонии — снижение выраженности миотонических проявлений. Немедикаментозное лечение миотонии состоит из диеты с ограничением солей калия, ЛФК, массажа, электромиостимуляции, а также предупреждения переохлаждений, так как при холоде усиливаются все миотонические реакции. Радикального медикаментозного лечения миотонии не существует, поэтому в целях уменьшении выраженности миотонических проявлений применяют фенитоин, а для снижения уровня калия — диуретики. В некоторых случаях удается достичь ремиссии с помощью иммуносупрессивной терапии: внутривенное введение иммуноглобулина человека, преднизолон, циклофосфамид.
Прогноз при миотонии
Прогноз для жизни при миотонии в целом благоприятный за исключением редких случаев дистрофической миотонии типа 1, когда возможно наступление внезапной сердечной смерти по причине кардиальной патологии. Прогноз для трудоспособности пациентов с врожденными миопатиями также благоприятен (при рациональном трудоустройстве).
Дистрофическая миотония Россолимо-Штейнерта-Куршмана
Дистрофическая миотония Россолимо-Штейнерта-Куршмана — наследственное медленно прогрессирующее заболевание, в основе которого лежит дефект миотонин-протеинкиназы, приводящий к развитию миотонии в сочетании с дистрофическими изменениями мышечной ткани. Заболевание проявляется миотоническими спазмами, атрофическими изменениями мышц шеи, лица и дистальных отделов конечностей, снижением интеллекта, аритмиями и эндокринной патологией. Диагностика дистрофической миотонии основывается на клинических данных, результатах генеалогического анализа и исследования ДНК. Лечение симптоматическое, направленное против симптомов миотонии (фенитоин, прокаинамид, хинин, мочегонные) и мышечной дистрофии (анаболические стероиды, АТФ).
МКБ-10
Дистрофическая миотония Россолимо-Штейнерта-Куршмана является наследственным заболеванием и передается от родителей к детям по аутосомно-доминантному типу. Классическая форма этого заболевания развивается преимущественно в возрастном периоде от 10 до 20 лет. В более редких случаях встречается врожденная дистрофическая миотония Россолимо-Штейнерта-Куршмана, клинические симптомы которой проявляются сразу же после рождения.
Морфологически при миотонии Россолимо-Штейнерта-Куршмана отмечается сочетание гипертрофических изменений одних мышечных волокон с атрофией других, замещение части мышечных волокон жировой и соединительной тканью. Изучение образцов мышечной ткани под электронным микроскопом показывает деструкцию миофибрилл и изменение размера митохондрий.
Причины
Последние исследования генетического набора больных дистрофической миотонией показали, что основу заболевания составляет дефект в гене DMPK, находящемся в 19-й хромосоме и отвечающем за синтез миотонин-протеинкиназы. У больных дистрофической миотонией выявляется значительное увеличение тринуклеотидных CTG-повторов в основной части гена DMPK. При этом именно от количества повторов зависит форма и тяжесть миотонии.
В норме число тринуклеотидных повторов варьирует от 5 до 37. Увеличение повторов до 50-80 приводит к появлению мягкой формы миотонии Россолимо-Штейнерта-Куршмана. Если количество тринуклеотидных повторов находится в промежутке от 100 до 500, развивается поздняя форма заболевания. Врожденные формы дистрофической миотонии возникают при повышении числа CTG-повторов от 500 до 2000. Исследования показали, что увеличение тринуклеотидных повторов происходит в основном в женских гаметах в процессе мейоза. В связи с этим при передаче заболевания от матери у ребенка возникает более тяжелая форма миотонии или ее врожденный вариант.
Симптомы классической формы
В классическом варианте миотония Россолимо-Штейнерта-Куршмана начинает проявляться после первых 5 лет жизни и может манифестировать до 35-летнего возраста. Но наиболее часто клинические проявления заболевания возникают в возрастном диапазоне от 10 до 20 лет. Они представляют собой сочетание типичных симптомов миотонии с признаками миопатии, поражением сердечно-сосудистой системы и ЦНС, эндокринными нарушениями и катарактой.
Из миотонических проявлений для миотонии Россолимо-Штейнерта-Куршмана характерны миотонические спазмы, наиболее выраженные в жевательных мышцах и мышцах-сгибателях кисти. Наблюдаются также механические реакции миотонического типа, выявляемые при ударе неврологическим молоточком. Отличительной особенностью миотонии Россолимо-Штейнерта-Куршмана является наличие атрофических изменений в различных группах мышц. При этом течение заболевания характеризуется постепенным угасанием симптомов миотонии на фоне прогрессирующей мышечной дистрофии.
Чаще всего при миотонии Россолимо-Штейнерта-Куршмана поражаются мышцы дистальных отделов конечностей, мимическая мускулатура, грудино-ключично-сосцевидные и височные мышцы. Поражение мимических мышц проявляется характерным маскообразным печальным выражением лица больных дистрофической миотонией. Атрофические изменения мышц глотки и гортани приводят к развитию миопатического пареза гортани с нарушением голоса и затруднением глотания. Миопатические изменения могут возникать в дыхательной мускулатуре. Наряду с миотоническими спазмами они приводят к ухудшению легочной вентиляции, появлению приступов апноэ во сне, возникновению застойной или аспирационной пневмонии.
Нарушения сердечно-сосудистой системы наблюдаются примерно в половине случаев дистрофической миотонии. К ним относятся аритмии, связанные с нарушением проводимости, и гипертрофия левого желудочка. Наиболее распространена блокада ножек пучка Гиса. Из признаков поражения ЦНС чаще всего наблюдается гиперсомния и снижение интеллектуальных способностей, доходящее до легкой степени дебильности.
Эндокринные расстройства при миотонии Россолимо-Штейнерта-Куршмана затрагивают в основном половую сферу. У мужчин они проявляются снижением либидо, крипторхизмом, импотенцией, гипогонадизмом, у женщин — гирсутизмом, нарушениями менструального цикла (олигоменореей, дисменореей) и ранним климаксом. Типичным является изменение структуры волос в сочетании с алопецией. У мужчин отмечается выпадение волос на висках и в области лба, у женщин — диффузное или очаговое облысение.
Симптомы врожденной формы
Первые признаки врожденной формы миотонии Россолимо-Штейнерта-Куршмана могут проявляться еще в период внутриутробного развития. Как правило, они выражаются в значительном снижении двигательной активности плода, которое диагностируется акушером-гинекологом по данным акушерского УЗИ в III триместре беременности.
После рождения ребенка преобладают симптомы миопатии. Отмечается диффузная гипотония мышц, более выраженная в мимической, жевательной и глазодвигательной мускулатуре, а также в мышечных группах дистальных отделов конечностей. Характерны затруднения вскармливания и дыхательные расстройства. Миотоническая симптоматика начинает проявляться несколько позже. Врожденная дистрофическая миотония сопровождается задержкой моторного развития и олигофренией. Типично быстрое прогрессирование симптомов заболевания, часто приводящее к смертельному исходу еще в раннем детстве.
Диагностика
Типичное сочетание миотонии с признаками дистрофических изменений мышечной ткани, умственной отсталостью, нарушениями со стороны сердечно-сосудистой и эндокринной систем позволяет неврологу предположить миотонию Россолимо-Штейнерта-Куршмана. Подтверждением диагноза являются результаты генеалогического анализа, свидетельствующие об аутосомно-доминантном типе наследования заболевания, и данные ДНК-анализа. Дополнительно проводится электромиография, электронейрография, исследования половых гормонов, ЭКГ. К диагностике пациентов с миотонией Россолимо-Штейнерта-Куршмана могут дополнительно привлекаться генетики, кардиологи, эндокринологи, гинекологи, андрологи.
При диагностике дистрофической миотонии ее необходимо дифференцировать ее от других видов миотонии. Так, наличие мышечных атрофий позволяет отличить миотонию Россолимо-Штейнерта-Куршмана от миотонии Томсена, для которой типична мышечная гипертрофия. От миотонии Беккера заболевание отличается ранним поражением мышц лица и доминантным типом наследования. Кроме того, следует проводить дифференциальный диагноз миотонии Россолимо-Штейнерта-Куршман с миопатиями, БАС и амиотрофией Шарко-Мари-Тута.
Лечение миотонии Россолимо-Штейнерта-Куршмана
Радикальной терапии миотонии Россолимо-Штейнерта-Куршмана пока не существует. Пациентам, имеющим это заболевание, показана диета со сниженным содержанием калия. Им также следует избегать переохлаждения, которое провоцирует миотонические спазмы. Уменьшению миотонических проявлений способствует прием хинина, прокаинамида, фенитоина в сочетании с ацетазоламидом. Показаны анаболические стероиды ( нандролона деканоат, метиландростендиол, метандростенол), небольшие дозы АТФ, витамины группы В.
Миотонический синдром у детей
Миотонический синдром у детей — это нервно-мышечная патология, для которой характерна затрудненная релаксация мускулатуры после ее напряжения. Встречается при наследственных генетических дефектах либо как проявление неврологических, эндокринных, метаболических, аутоиммунных заболеваний. Синдром характеризуется нарушениями двигательной активности, асимметрией мышц, задержкой психомоторного развития. Для диагностики необходим неврологический осмотр, инструментальные методы (ЭМГ, МРТ) и генетическое обследование. Лечение миотонии симптоматическое с применением физиотерапии, ЛФК и массажа, психолого-педагогической коррекции.
Частота встречаемости наследственных форм миотонического синдрома (МС) варьирует от 14 до 23 случаев на 100 тыс. детского населения. В отношении приобретенных вариантов статистических данных нет, поскольку в таких случаях патология не регистрируется как самостоятельная нозологическая единица и входит в структуру других неврологических болезней. Актуальность проблемы в педиатрической практике заключается в прогрессирующем течении заболевания, невозможности назначить этиотропное лечение и значительном ухудшении качества жизни детей при наследственных видах патологии.
В детском возрасте манифестируют врожденные формы, которые передаются преимущественно по аутосомно-доминантному, реже — аутосомно-рецессивному типу (синдром Шварца-Джампела, в части случаев — болезнь Томпсона). Самая частая форма наследственного МС — миотоническая дистрофия Россолимо-Штейнерта-Баттена-Куршмана, которая составляет до 97% всех генетических миотоний и связана с мутацией гена DMPK на 19-й хромосоме. Реже это заболевание обусловлено генетическими дефектами в 3-й (3q21) и 15-й (15q21-q24) хромосомах.
Вторичным миотоническим синдромом могут сопровождаться:
- родовые травмы с церебральными повреждениями;
- тяжелые формы перинатальной энцефалопатии;
- органические поражения ЦНС (опухоли, последствия перенесенных энцефалитов, ЧМТ);
- врожденные и воспалительные миопатии;
- миоплегии (болезнь Гамсторпа, периодический паралич);
- дефицитные состояния (рахит);
- эндокринные и метаболические расстройства (микседема, гипофункция паращитовидных желез, гиперкалиемия);
- аутоиммунные нарушения (приобретенная нейромиотония);
- длительное лечение клофибратом, калийсберегающими диуретиками, препаратами калия.
Патогенез
В механизме развития заболевания ведущим является нарушение процесса расслабления мышцы в результате метаболического или ионного дисбаланса. При этом изменяются свойства мембран миоцитов, происходит их усиленная деполяризация и повышение возбудимости. При совершении определенного действия с участием мускулатуры миофибриллы долго не могут расслабиться и вернуться в исходное положение, вследствие чего и возникает феномен миотонической задержки.
Лучше всего изучен патогенез дистрофической миотонии 1 типа, связанной с нарушениями на уровне экспрессии нуклеотидного повтора CTG. У детей изменяется концентрация миотонин-протеинкиназы DMPK, которая присутствует в скелетной мускулатуре, миокарде и ЦНС. В редких случаях клинические проявления обусловлены дисфункцией ионных каналов клеточных мембран, которая отмечается при аутоиммунных нарушениях.
Симптомы
Главный признак миотонического синдрома — скованность мышечных групп в конце движения. Это проверяется с помощью симптома «кулака»: после сжатия пальцев ребенок несколько секунд не может разжать кулак, и для этого ему нужно прилагать много усилий. Синдром характеризуется затруднениями жевания, неспособностью быстро открыть глаза после зажмуривания. Для наследственных форм МС у детей типично усиление симптоматики при целенаправленных движениях, в холодное время года.
При врожденных формах миотонического синдрома возможно асимметричное развитие мускулатуры. Если патология проявляется с первых месяцев жизни, родители замечают, что ребенок не способен удерживать голову, слабо сосет грудь, у него хуже сформированы рефлексы. Для грудничков с миотоническим синдромом характерно запоздалое становление моторики. В раннем детстве наблюдается повышенная утомляемость малыша, отсутствие интереса к активным играм, отставание в физическом развитии.
На поздних стадиях заболевания у детей происходит атрофия мышц, поэтому типичные признаки миотонического синдрома ослабевают, а потом исчезают. В клинической картине преобладает выраженная мышечная слабость, особенно в дистальных мышцах конечностей. Это обуславливает изменение походки, приводит к появлению вялых парезов и параличей. Поражение лицевых мышц проявляется постоянно «печальным» выражением лица и бедностью мимики.
Осложнения
Врожденные миотонические синдромы относят к мультисистемным поражениям, поэтому у таких детей зачастую диагностируют сопутствующие сердечно-сосудистые, неврологические и эндокринные нарушения. Особую опасность представляет нарушение иннервации дыхательной мускулатуры, из-за чего у пациентов нарастать дыхательная недостаточность вплоть до асфиксии. Церебральные симптомы, как правило, сочетаются с когнитивными нарушениями.
Лечение наследственных разновидностей миотонического синдрома затруднено, поэтому нередки летальные исходы в молодом возрасте. До 80% смертей вызваны вторичной пневмонией на фоне аспирации пищи и нарушений глотания, жизнеугрожающими аритмиями, которые развиваются вследствие дегенерации проводящей системы сердца. У детей может возникать глубокая умственная отсталость, приводящая к инвалидности.
При физикальном осмотре детский невролог проверяет симптом «кулака», оценивает сохранность рефлексов, мышечную силу. На основании полного физикального осмотра специалисту удается поставить предварительный диагноз. Чтобы подтвердить наличие и выяснить причины миотонического синдрома, назначаются следующие диагностические исследования:
- Электромиография. Патогномоничный признак миотонии на ЭМГ — появление высокоамплитудных разрядов со звуковым феноменом при использовании игольчатых электродов. При прогрессировании болезни присутствуют симптомы миопатии: снижение амплитуды потенциалов, полифазные двигательные единицы.
- Биопсия мышц. На ранних стадиях в биоптате определяются неспецифические миопатические изменения: центрально расположенные ядра, уменьшение длины волокон, признаки денервации — наличие пикнотических ядерных глыбок и маленьких угловых мускульных волокон.
- Магнитно-резонансная томография. По результатам МРТ конечностей врач оценивает степень развития мускулатуры и замещения ее жировой тканью. Учитывая сопутствующее поражение ЦНС, рекомендована МРТ головного мозга для обнаружения структурных изменений белого вещества и желудочков.
- Генетическая диагностика. Исследование генома для выявления типичной мутации — «золотой стандарт» для постановки диагноза врожденных вариантов миотонического синдрома у детей. По показаниям можно проводить тестирование в антенатальном периоде, если в семьях родителей были случаи миотонии.
Лечение миотонического синдрома у детей
Консервативная терапия
Этиопатогенетическое лечение миотонического синдрома пока не разработано, поэтому в детской неврологии ограничиваются симптоматическими средствами. Медикаменты подбираются с учетом конкретной ситуации и выраженности патологических изменений. Для замедления прогрессирования проводится лечение противосудорожными средствами и миорелаксантами, для подавления аутоиммунных реакций назначают глюкокортикоиды.
Решающее значение в терапевтической схеме отводится упражнениям ЛФК, которые укрепляют мышечный корсет, помогают сбалансировать развитие разных групп мышц, что улучшает качество жизни больных детей. Показано физиотерапевтическое лечение в виде электрофореза с кальцием, электростимуляции мышц, бальнеотерапии. Для уменьшения миотонического синдрома полезен массаж.
Реабилитация
Учитывая серьезные двигательные нарушения, лечение дополняется регулярными занятиями в реабилитационных центрах с использованием тренажеров и индивидуальных методик. Нарушения артикуляции у детей корректируются занятиями с логопедом. При задержке умственного развития ребенку может потребоваться помощь олигофренопедагога либо обучение в специализированной школе.
Прогноз и профилактика
Если вовремя начать лечение, при приобретенных формах удается восстановить мышечную активность. Наследственный миотонический синдром отличаются сомнительным прогнозом. Средняя продолжительность жизни пациентов составляет 35 лет при раннем дебюте болезни. Специфические меры профилактики миотонического синдрома не разработаны. Семьям с отягощенным анамнезом требуется медико-генетическое консультирование.
1. Клинический случай дистрофической миотонии 1-го типа/ Н.В. Ноздрюхина, А.А. Струценко, Е.Н. Кабаева// Трудный пациент. — 2019.
2. Миотоническая дистрофия. Современное представление и собственное наблюдение/ Т.И. Стеценко// Современная педиатрия. — 2014.
Миотоническая дистрофия: причины, признаки, фенотип
Миотоническая дистрофия (dystrophia myotonica, болезнь Штейна) у ребенка
Миотония означает нарушение расслабления скелетной мышцы после ее напряжения и медленный, тонический ответ на механическую или электрическую стимуляцию. Электромиографически миотония характеризуется повторяющимися ответами двигательных единиц, что создает классический рисунок «пикирующего бомбардировщика» (подъем — спад). Миотония вызывается нестабильностью клеточной мембраны мышц и имеет чисто мышечное происхождение.
Она может возникать при некоторых генетических заболеваниях и синдромах, включая миотоническую дистрофию, наиболее часто встречающееся генетическое расстройство, так же как и при «каналопатиях» или расстройствах ионных каналов.
Миотоническая дистрофия (dystrophia myotonica, болезнь Штейна) — относительно часто встречающееся заболевание с частотой 13,5 на 100000 живых новорожденных. Оно передается по аутосомно-доми-нантному механизму и характеризуется сочетанием миотонии с дистрофическими процессами в мышцах. Миотоническая дистрофия — мультисистемное заболевание, поражающее скелетную и гладкую мышечную ткань, а также глаза, сердце, эндокринную систему и центральную нервную систему.
Клинические проявления составляют континуум от легких до тяжелых форм и разделяются на три частично перекрывающих друг друга фенотипа: легкий, классический и врожденный.
а) Клинические проявления классической формы. Клиническая картина миотонической дистрофии вариабельна. У пациентов с легкой миотонической дистрофией 1 типа (ДМ1) могут наблюдаться только катаракта, легкая миотония или сахарный диабет. Они могут жить полноценной активной жизнью, длительность жизни нормальная или немного снижена. При классической миотонической дистрофии заболевание обычно начинается в возрасте двадцати или тридцати лет, реже — после 40 лет. Однако классическая ДМ1 может проявляться и в детском возрасте, когда наблюдаются невыраженные симптомы, такие, как миотоническое лицо и миотония.
Миотония выявляется при постукивании по мышце, например, по тенару или языку, большой палец при этом находится в положении отведения, на языке на несколько секунд после перкуссии остается ямочка. Миотония расслабления выявляется при пожимании руки пациента: пациент отпускает захват, разжимая сгибатели пальцев сгибанием запястья.
Наблюдается характерное распределение мышечной атрофии и слабости. Атрофия начинается с мышц лица — особенно жевательных и височных мышц, что делает лицо пациента длинным и тонким, с пустыми височными ямками,— и грудинноключичнососцевидных мышц. Затем поражается плечевой пояс, и, что характерно, также истончаются плечелучевые мышцы и передние мышцы голени. У некоторых пациентов отмечается незначительная мышечная слабость, даже при наличии видимой атрофии, но если она развивается до достижения возраста 20 лет, высока вероятность ее прогрессирования и развития тяжелой мышечной слабости дистальных отделов в среднем возрасте.
Редко, после нескольких десятилетий течения болезни, миотоническая дистрофия приковывает пациента к инвалидному креслу. У больных с развитой стадией заболевания могут развиться поражения дыхательной мускулатуры и выраженная аспирация (Thornton, 1999).

Миотоническая дистрофия (болезнь Штейнера). Неонатальная форма (Слева).
Обратите внимание на эквиноварусные стопы. Ребенок с легкой формой заболевания, начавшейся в неонатальном периоде (справа).
Обратите внимание на выгнутую «шатровую» верхнюю губу, открытый рот, небольшой птоз и pectum excavatum.
Нарушения интеллекта у пациентов со взрослой формой миотонической дистрофии встречаются редко; но могут наблюдаться нарушения личности в виде замкнутости, обсессивно-компульсивных и пассивно-агрессивных черт (Delaporte, 1998).
Гиперсомния и апноэ во сне относятся к другим характерным, хотя и поздним проявлениям заболевания (Rubinsztein et al., 1998). Продолжительность жизни может быть снижена из-за пневмонии и дыхательной недостаточности или сердечно-сосудистых осложнений, например аритмии (de Die-Smulders et al., 1998).
б) Клинические проявления врожденной формы. Врожденная форма болезни имеет совершенно другую клиническую картину (Hageman А.Т. et al, 1993). Заболевание начинается в пренатальном периоде; примерно в половине случаев развивается многоводие; роды обычно в тазовом предлежании, младенец может быть маленьким для своего срока гестации. Часто встречаются множественные врожденные контрактуры. Гипотония, мышечная слабость и лицевая диплегия с выгнутой кверху «шатровой» губой — наиболее заметные признаки. Из-за поражения межреберных мышц и диафрагмы примерно в половине случаев развивается дыхательная недостаточность, которая обычно рано приводит к смерти.
Состояние выживших младенцев постепенно улучшается, обычно они обретают способность ходить, хотя позже у них развивается прогрессирующая миопатия в классической форме. Миотония никогда не развивается до достижения возраста 3-4 лет, даже по данным ЭМГ, часто и значительно позже. У менее тяжело пораженных пациентов, переживающих неонатальный период, в большинстве случаев присутствуют психические нарушения, которые обычно связаны с расширением желудочков и церебральная атрофия (Regev et al., 1987).
Тяжелые и относительно легкие формы врожденной миотонической дистрофии могут наблюдаться у кровных родственников.
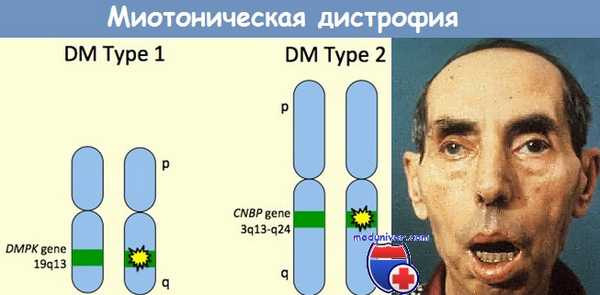
в) Диагностика и лечение. Существует два основных генетических варианта миотонической дистрофии — ДМ1 и ДМ2. ДМ1 вызывается увеличением тринуклеотидной последовательности ЦТГ гена миотонин-протеинкиназы, DMPK. Длина цепочки ЦТГ, превышающая 35 последовательностей, является аномальной. Молекулярное генетическое тестирование выявляет мутации почти у 100% больных, тестирование используется в клинике для диагностики, в том числе и пренатальной. Как и при других заболеваниях, вызываемых удлинением ДНК, наблюдаются влияние пола и постепенное ухудшение при передаче по наследству.
В последующих поколениях длина повторяемого участка обычно увеличивается, особенно при передаче по материнской линии. Существует стойкая корреляция между длиной участка, возрастом дебюта заболевания и тяжестью клинической картины, самые длинные участки вызывают врожденные формы болезни (Jaspert et al., 1995; Takahashi et al., 1996). Классическая ДМ1 вызывается при количестве повторов в 100-1000, врожденная миотоническя дистрофия связана с числом повторов >2000. В подавляющем большинстве случаев врожденная ДМ1 наследуется от матери. Увеличение числа повторов ЦТГ вызывает аномалию процессинга транскриптов РНК, нарушает альтернативный сплайсинг и уровень экспрессии других генов, что отражается различными проявлениями болезни (например, поражение хлоридных каналов вызывает миотонию, аномалии инсулиновых рецепторов приводят к диабету) (Ranum и Day, 2004).
Миотоническая дистрофия 2 типа (ДМ2, ранее проксимальная миотоническая миопатия) гораздо более редкое заболевание, встречающееся чаще всего у взрослых. Она также наследуется по аутосомно-доминантному типу и вызывается увеличением количества последовательностей ЦЦТГ интрона 1 гена ZNF9, кодирующего протеин «цинкового пальца» 9 (Thornton et al., 1994; Udd et al., 1997; Liquori et al., 2001). ДМ2 также характеризуется клиническими и электрофизиологическими проявлениями миотонии, катарактами, облысением лобной области у мужчин, нарушением сердечной проводимости и эндокринопатиями. В отличие от ДМ1, тяжелой врожденной формы ДМ2 не существует.
Иногда миотоническую дистрофию необходимо дифференцировать с другими формами наследственных дистальных миопатий, в том числе с миозитом с включенными тельцами, миофибриллярной миопа-тией и миопатией Мийоши. У младенцев дифференциальная диагностика сложнее, хотя постановке правильного диагноза помогают общий вид младенца и поражение лица. Пациенты с врожденными миопатиями и с врожденными мышечными дистрофиями могут выглядеть очень похожими на больных с миотонической дистрофией. Обследование матери, особенно на предмет миотонии, является главным диагностическим исследованием, хотя иногда у матери через месяцы или годы после рождения больного младенца может и не выявляться миотония, даже при ЭМГ.
Если длина последовательности ЦТГ DMPK в нормальных пределах, и если после молекулярного тестирования на ZNF9 исключена ДМ2, для выявления других причин заболевания мышц часто необходимо выполнить ЭМГ, определить уровень сывороточной КК и/или провести исследование биоптата мышечной ткани.
Большое значение имеет наблюдение пациентов с миотонической дистрофией, включая ежегодное выполнение ЭКГ и/или холтеровского мониторирования с целью выявления нарушений сердечной проводимости, а также измерение уровня глюкозы крови натощак и содежания гликозилированного гемоглобина для скрининга диабета. Миотония редко бывает настолько тяжелой, чтобы требовалось лечение (Ricker et al, 1999). Используются прокаинамид, хинин и мексилетин.
Читайте также: