Молекулы адгезии в подострой фазе воспаления.
Добавил пользователь Дмитрий К. Обновлено: 29.01.2026
Изобретение относится к области медицины и предназначено для прогнозирования подострой стадии хронической истинной экземы у индивидуумов русской национальности, уроженцев Центрального Черноземья РФ. Осуществляют забор венозной крови, выделение геномной ДНК из периферической крови методом полимеразной цепной реакции, анализ полиморфизмов генов фактора некроза опухоли α (TNFα), лимфотоксина α (Ltα) и рецептора фактора некроза опухоли 2 типа (TNFR2). При наличии аллеля -308А ТNFα, или генотипа -308GА ТNFα, или аллеля +1663G ТNFR2, или сочетания аллеля -308А ТNFα с аллелем +1663G ТNFR2, или сочетания аллеля -308А ТNFα с аллелем +250G Ltα, или сочетания аллеля +250G Ltα с аллелем +1663G ТNFR2 прогнозируют повышенный риск развития подострой стадии хронической истинной экземы, а при наличии генотипа -308GG ТNFα прогнозируют пониженный риск развития подострой стадии хронической истинной экземы. Изобретение обеспечивает получение новых критериев оценки риска развития подострой стадии хронической истинной экземы. 7 пр.
Способ прогнозирования подострой стадии хронической истинной экземы у индивидуумов русской национальности, уроженцев Центрального Черноземья РФ, включающий забор венозной крови, выделение геномной ДНК из периферической крови методом полимеразной цепной реакции синтеза ДНК, анализ полиморфизмов генов фактора некроза опухоли α (-308G/A TNFα), лимфотоксина α (+250A/G Ltα) и рецептора фактора некроза опухоли 2 типа (+1663A/G TNFR2) и прогноз повышенного риска развития подострой стадии хронической истинной экземы при наличии аллеля -308А ТNFα или генотипа -308GА ТNFα или аллеля +1663G ТNFR2 или сочетания аллеля -308А ТNFα с аллелем +1663G ТNFR2 или сочетания аллеля -308А ТNFα с аллелем +250G Ltα или сочетания аллеля +250G Ltα с аллелем +1663G ТNFR2; прогнозируют пониженный риск развития подострой стадии хронической истинной экземы при наличии генотипа -308GG ТNFα.
Изобретение относится к области медицинской диагностики, может быть использовано для прогнозирования подострой стадии хронической истинной экземы.
Истинная экзема - повсеместно встречающееся, чаще острое, реже хроническое рецидивирующее заболевание кожи, возникающее в любом возрасте, характеризующееся полиморфизмом морфологических элементов сыпи [Клиническая дерматовенерология: руководство для врачей: в 2-х т. /под ред. Ю. К. Скрипкина, Ю. С. Бутова. - Москва: Гэотар- Медиа, 2009. - Т. 2. - 921 с.]. Название данного дерматоза объясняется свойством экзематозных пузырьков быстро вскрываться, подобно пузырькам кипящей воды (по-гречески eczeo - вскипать). Важный признак острой экземы - наличие многочисленных сгруппированных и быстро вскрывающихся с образованием серозных «колодцев» мелких пузырьков, имеющих некоторое сходство с пузырьками на поверхности кипящей воды [Оспанова С. А. Совершенствование терапии экземы с учетом эндогенной интоксикации: автореф. дис. … канд. мед. наук: 14.00.11 /С. А. Оспанова; Казах. науч.-исслед. кож.-венерол. ин-т. - Алматы, 2008. - 24 с.]. Заболевание характеризуется хроническим рецидивирующим течением и значительно ограничивает трудоспособность больных.
В развитии экземы существуют три стадии воспалительного процесса: острая стадия, подострая стадия и хроническая стадия. Подострая стадия экземы характеризуется умеренной эритемой, выраженной лихенизацией, становятся видны более четкими границы поражения, уменьшается выделение экссудата, однако, на месте эпителизирующихся эрозий начинает появляться обильное шелушение, что способствует социальной дезадаптации пациентов, снижая их качество жизни, а также неблагоприятно сказывается на физическом и психическом состоянии больного и членов его семьи.
Этиологический и патогенетический аспекты развития экземы, освещенные в современных научных изданиях, носят весьма противоречивый характер. Патогенез экземы привлекал внимание многих исследователей. Они выдвигали различные теории ее происхождения и связывали возникновение экземы с преобладающими нарушениями в той или иной системе организма. Так, до сих пор окончательно не определены механизмы развития иммунных отклонений в организме в целом и непосредственно в коже больных [Барабанов, А. Л. Некоторые вопросы патогенеза экземы /А. Л. Барабанов, В. Г. Панкратов //Медицинская панорама. - 2004. - №6. - С. 5-8]. В то же время недостаточно изучены метаболические нарушения и их взаимосвязь с иммунной реактивностью, способствующие снижению активности факторов неспецифической защиты при развитии различных форм экземы. На данном этапе исследователи пришли к выводу, что необходим комплексный подход к изучению патогенеза экземы.
На разных этапах развития учения об экземе особое значение в этиологии и патогенезе заболевания придавали нервной системе (неврогенная теория), роли эндокринных желез, аллергическому состоянию организма (аллергическая теория), наследственным факторам. Следует признать, что этиология и даже патогенез экземы чрезвычайно сложны, не всегда одинаковы и во многих своих аспектах остаются неизученными [Modern aspects of cutaneous neurogenic inflammation /M. Steinhoff, S. Ständer, S. Seeliger [et al.] //Arch. Dermatol. - 2003. - Vol.139, №11. - P. 1479-1488].
По современным представлениям, в развитии экземы главную роль играют Т-лимфоциты, несущие на своей поверхности специфические рецепторы к антигену и выделяющие ряд противовоспалительных цитокинов: ИЛ-1, ИЛ-2, ФНО-α. Выброс биологически активных веществ вызывает развитие тканевых реакций воспаления, что клинически проявляется аллергическим ответом в виде гиперемии, отека, зуда. Антигенная стимуляция Th1 приводит к синтезу ИЛ-2, причем ИЛ-2 продуцирующая способность CD4-клеток у больных выше, чем у здоровых людей.
Провоспалительные цитокины вызывают индукцию экспрессии молекул адгезии на лейкоцитах и эндотелиальных клетках, вследствие чего стимулируется приток лейкоцитов из сосудистого русла в очаг воспаления путем их трансэндотелиальной миграции. Дальнейшее продвижение и накопление иммунокомпетентных клеток в очаге воспаления контролируется хемокинами, которые продуцируются макрофагами и эндотелиальными клетками. Клеточный инфильтрат в очаге воспаления, состоящий из нейтрофилов, эозинофилов и макрофагов, способствует дальнейшему развитию аллергического воспаления. Полиморфный инфильтрат в коже при экземе - результат действия образовавшихся противовоспалительных цитокинов, в том числе ФНО-α. Таким образом, в аллергических реакциях замедленного типа участвуют Th1- лимфоциты, макрофаги, эндотелиальные клетки и секретируемые ими цитокины.
Таким образом, в аллергических реакциях замедленного типа, лежащих в основе формирования истинной экземы, участвуют Th1- лимфоциты, макрофаги, эндотелиальные клетки и секретируемые ими цитокины.
Среди множества биологических эффектов и реакций, опосредуемых цитокинами, главными являются иммунные реакции, воспалительный ответ, регуляция гемопоэза, ангиогенез, репаративные процессы в тканях. Их действие является антиген-неспецифическим, они способны воздействовать на любые клетки, содержащие соответствующие рецепторы и находящиеся в адекватной физиологической активности. К цитокинам относят и факторы некроза опухолей (TNF).
С практической точки зрения представляется крайне необходимым выделение критериев индивидуального прогнозирования подострой стадии хронической истинной экземы на основании исследованных полиморфных вариантов генов факторов некроза опухолей и их рецепторов и других возможных факторов риска с целью выявления больных ХИЭ, предрасположенных к развитию подострой стадии.
В изученной научно-медицинской и доступной патентной литературе авторами не было обнаружено способа прогнозирования подострой стадии хронической истинной экземы на основе данных о генетических полиморфизмах -308G/A TNFα,+250A/G Ltα и+1663A/G TNFR2 и их сочетаний. Для оценки сложившейся патентной ситуации был выполнен поиск по охранным документам за период с 1990 по 2012 гг. Анализ документов производился по направлению: способ прогнозирования повышенного риска развития осложнений хронической истинной экземы. Аналогичных патентов нет.
Задачей настоящего исследования является создание способа прогнозирования подострой стадии хронической истинной экземы у индивидуумов русской национальности, уроженцев Центрального Черноземья Российской Федерации, на основе данных о генетических полиморфизмах -308G/A TNFα,+250A/G Ltα и+1663A/G TNFR2 и их сочетаний.
Технический результат использования изобретения - получение критериев оценки риска развития подострой стадии хронической истинной экземы у индивидуумов русской национальности, уроженцев Центрального Черноземья Российской Федерации, по данным о генетических полиморфизмах -308G/A TNFα,+250A/G Ltα и+1663A/G TNFR2 и их сочетаний.
В соответствии с поставленной задачей был разработан способ прогнозирования подострой стадии хронической истинной экземы, включающий:
- забор венозной крови;
- выделение геномной ДНК из периферической крови методом полимеразной цепной реакции синтеза ДНК;
- анализ полиморфизмов генов фактора некроза опухоли α (-308G/A TNFα), лимфотоксина α (+250A/G Ltα) и рецептора фактора некроза опухоли 2 типа (+1663A/G TNFR2);
- прогнозируют повышенный риск развития подострой стадии ХИЭ при наличии аллеля -308А ТNFα, или генотипа -308GА ТNFα, или аллеля+1663G ТNFR2, или сочетания аллеля -308А ТNFα с аллелем+1663G ТNFR2, или сочетания аллеля -308А ТNFα с+аллелем 250G Ltα, или сочетания аллеля+250G Ltα с аллелем+1663G ТNFR2;
- прогнозируют пониженный риск развития подострой стадии ХИЭ при наличии генотипа -308GG ТNFα.
Новизна и изобретательский уровень заключается в том, что впервые установлена вовлеченность генетических полиморфизмов фактора некроза опухоли α (-308 G/A TNF α), лимфотоксина α (+250 A/G Ltα), рецептoра фактoра некроза опухоли 2-го типа (+1663А/G ТNFR2) и их сочетаний в формировании подострой стадии хронической истинной экземы у индивидуумов русской национальности, уроженцев Центрального Черноземья Российской Федерации.
Способ осуществляют следующим образом:
Выделение геномной ДНК осуществляют стандартным методом фенольно-хлороформной экстракции. 1-й этап: к 4 мл крови добавляют 25 мл лизирующего буфера, который содержит 320 мМ сахарозы, 1% тритон Х -100, 5 мМ МgСl2 и 10 мМ трис-НCl (pH=7,6) [Мathew С.С., 1984]. Полученную смесь при 4ºС, 4000 об/мин в течение 20 минут перемешивают и центрифугируют. После центрифугирования сливают надосадочную жидкость, к полученному осадку добавляют 4 мл раствора, с 25 мM ЭДTA (рН=7,8) и 75 мМ NаСl, затем ресуспензируют. После этого прибавляют 0,4 мл 10% SDS, 35 мкл протеиназы K (10 мг/мл) и в течение 16 часов при 37ºС инкубируют образец.
На 2-м этапе в течение 10 минут при 4000 об/мин из полученного лизата последовательно экстрактируют ДНК равными частями фенола, фенол-хлороформа (1:1) и хлороформа с центрифугированием. Производят отбор водной фазы после каждого центрифугирования. Из раствора 2-мя объемами охлажденного 96% этанола осаждают ДНК. Полученную ДНК растворяют в деионизованной, бидистиллированной воде и хранят при t=-20 ° С. Выделенную ДНК используют для полимеразной цепной реакции (ПЦР) синтеза ДНК.
Анализ -308G/А TNFα,+250G/A Ltα и+1663A/G TNFR2 осуществляют методами ПЦР синтеза ДНК на CFX 96 с системой ПЦР в режиме real time. Используют ДНК-полимеразу Thermus aquaticus (производитель фирма «Силекс-М») и олигонуклеотидные праймеры и зонды (синтезированные фирмой «Синтол»).
Реакционная смесь (объем 25 мкл) включает: 67 мМ трис-НCl (pН=8,8), 2,5 мМ МgCl2, 0,1 мкг геномной ДHK, по 10 пM каждого праймера, по 5 пkмoль каждого зонда, по 200 мkM dАТР, dGТР, dСТР, dТТР и 1 единицу активной Тaq-полимеразы. Для каждой пары праймеров рассчитывают оптимальный температурный и временной режим отжига и подбирают соответствующую концентрацию MgCl2.
Генотипирование производят по методу Tag Man зондов на основании данных значений УОФ (далее, уровень относительной флуоресценции) для каждого зонда при проведении ПЦP в амплификаторе IQ5.
Возможность использования предложенного способа для оценки риска развития подострой стадии хронической истинной экземы подтверждает анализ результатов обследования 376 индивидуумов: 54 больных с подострой стадией ХИЭ (14,36%) и 322 человек контрольной группы (85,64%).
В исследуемые группы включались лица русской национальности, которые являлись уроженцами Центрального Черноземья Российской Федерации и не имели родства между собой. Клиническое и клинико-лабораторное обследование больных проводили на базе поликлинического отделения ОГБУЗ «Кожно-венерологический диспансер».
Средний возраст больных хронической истинной экземой составил 45,91±2,68 (варьировал от 18 лет до 86 лет), контрольной выборки - 43,62±3,46 (варьировал от 20 до 76 лет) (р>0,05). Следовательно, группа больных не отличалась от группы контроля по возрасту, месту рождения и национальности.
Концентрация генотипа -308GG ТNFα равна 61,70% и является наименьшей (в 1,3 раза) по сравнению с контрольной группой (80,00%, χ 2 =6,89, р=0,009, рcor=0,027, OR=0,40, 95% CI 0,20-0,81), следовательно можно прогнозировать пониженный риск развития подострой стадии ХИЭ.
Примеры, подтверждающие осуществимость предложенного изобретения среди лиц русской национальности, уроженцев Центрального Черноземья Российской Федерации.
1. У больного хронической истинной экземой М. выявлен аллель -308А ТNFα. При дальнейшем наблюдении у данного больного развилась подострая стадия данного заболевания.
2. У больного хронической истинной экземой Л. выявлен генотип -308GА ТNFα. При дальнейшем наблюдении у данного больного развилась подострая стадия данного заболевания.
3. У больного хронической истинной экземой О. выявлен аллель +1663G ТNFR2. При дальнейшем наблюдении у данного больного развилась подострая стадия данного заболевания.
4. У больного хронической истинной экземой Е. выявлено сочетание аллеля -308А ТNFα с аллелем+1663G ТNFR2. При дальнейшем наблюдении у данного больного развилась подострая стадия данного заболевания.
5. У больного хронической истинной экземой В. выявлено сочетание аллеля -308А ТNFα с аллелем+250G Ltα. При дальнейшем наблюдении у данного больного развилась подострая стадия данного заболевания.
6. У больного хронической истинной экземой Р. выявлено сочетание аллеля+250G Ltα с аллелем+1663G ТNFR2. При дальнейшем наблюдении у данного больного развилась подострая стадия данного заболевания.
7. У больного хронической истинной экземой Д. выявлен генотип -308GG ТNFα. При дальнейшем наблюдении у данного больного не было подострой стадии данного заболевания.
Таким образом, поставленная задача по созданию способа прогнозирования подострой стадии хронической истиннной экземы решена.
Молекулы адгезии в подострой фазе воспаления.
Молекулы адгезии в подострой фазе воспаления.
На эпителиальных клетках бронхов также экспрессированы адгезионные молекулы ICAM-1, экспрессия которых может повышаться под влиянием цитокинов IFNy, TNF-oc, IL-1(3. Экспрессия молекул ICAM-1 усиливается на эпителии при воспалении воздухоносных путей. На таких активированных эпителиальных клетках усилена адгезия эозинофилов, которая, в свою очередь, приводит к усиленной дегрануляции эозинофилов. Секретируемые при этом катионные белки могут участвовать в повреждении респираторного эпителия.

При остром воспалении легких нейтрофилы эмигрируют из альвеолярных капилляров и двигаются в ответ на воспалительные стимулы, преодолевая интерстициальный матрикс между эндотелием капилляров и альвеолярным эпителием. Этот процесс включает адгезию нейтрофилов и их локомоции. Альвеолярный интерстиций состоит из фибробластов. Адгезия нейтрофилов к альвеолярным фибробластам повышается под влиянием PAF за счет повышения экспрессии интегринов (CD11/CD18). Миграционная активность нейтрофилов возрастала под влиянием хемоаттрактанта IL-8, который секретировали альвеолярные фибробласты, активированные под влиянием TNF-oc. Фибробласты служат не только субстратом адгезии, но и источником стимулирующих молекул, обеспечивая миграцию нейтрофилов через воспаленный альвеолярный интерстиций. Для бактериальной пневмонии характерен ранний выход нейтрофилов в интерстиций и в просвет альвеол. При этом контакт нейтрофилов с фибробластами опосредован взаимодействием интегринов (CD11/ CD18) с молекулами ICAM-1(CD54), экспрессированными на фибробластах.
Конститутивная экспрессия адгезионных молекул ICAM-1 на альвеолярных эпителиальных клетках в 22 раза выше, чем на эндотелиальных кчетках капилляров. ICAM-1 экспрессированы, в основном, на пневмоцитах типа I, а не на их предшественниках пневмоцитах типа II. При пневмонии экспрессия ICAM-1 повышается в 178 раз на пневмоцитах типа II, т.е. происходит их ускоренная дифференцировка в ответ на повреждение эпителия альвеол. Кроме того, непосредственно в очаге пневмонии были обнаружены нейтрофилы с измененной экспрессией адгезионных молекул: L-селектина и CD18. Эмиграция нейтрофилов в просвет альвеол при пневмонии, вызванной Streptococcus pneumoniae, связана со снижением экспрессии L-селектина и повышением экспрессии CD18. Нейтрофилы в воздушном пространстве альвеол легкого имеют повышенный уровень экспрессии адгезионных молекул CD18, что может способствовать фагоцитозу патогенных бактерий. Адгезия моноцитов к эндотелиальным клеткам легочных капилляров также опосредована взаимодействием интегринов на моноцитах с молекулами ICAM на эндотелиальных клетках. Аккумуляции моноцитов в легких способствует и то, что диаметр легочных капилляров (6-8um) меньше диаметра многих моноцитов крови (7-10um), поэтому прохождение моноцитов через легочные капилляры требует деформации, пластичности моноцитов, которая в первую очередь нарушается при воспалении. Моноциты застревают в узких легочных капиллярах, прилипают к эндотелиальным клеткам и проделывают трансмиграцию в просвет альвеол. Бактериальные компоненты и продукты (в частности ЛПС) повышают адгезивность моноцитов, усиливая экспрессию адгезионных молекул.
Адгезионные молекулы могут стать клинически значимыми мишенями для противовоспалительной терапии. В настоящее время ведется поиск агентов, селективно ингибирующих адгезионные молекулы.
- Вернуться в оглавление раздела "Пульмонология."
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Молекулы участвующие в воспалении легких и бронхов: цитокины, интерферон-гамма, кахектины, молекулы адгезии
Миграционный ингибирующий фактор (MIF) был впервые описан в 60-е годы как продукт активированных Т-лимфоцитов.
Только через 25 лет удалось клонировать соответствующий ген, получить рекомбинантный белок и специфические моноклональные антитела.
Биологическая активность MIF может быть охарактеризована как негативный хемотаксический эффект: торможение миграции фагоцитирующих клеток (гранулоцитов, моноцитов, макрофагов).
Благодаря такому действию этот цитокин участвует в мобилизации фагоцитирующих клеток в очаг инфекции или воспаления на последнем этапе аккумуляции клеток в очаге. Кроме того, у MIF описаны и другие свойства провоспалительного цитокина. Наряду с TNF-a и IL-1 он участвует в каскаде реакций эндотоксического шока, возможно, контролируя уровень TNF-a. Этот цитокин участвует в качестве эффекторной молекулы в развитии клеточного иммунного ответа, реакций гиперчувствительности замедленного типа (ГЗТ).
Уровень продукции MIF, как правило, повышается при инфекциях и воспалительных процессах. Изучение способности мононуклеаров крови к усиленной продукции MIF давно используется в качестве одного из тестов для оценки функциональной активности Т-лимфоцитов и специфической сенсибилизации клеток (реакция торможения миграции лейкоцитов (РТМЛ)).
В последние годы показано, что продуцентами MIF кроме активированных Т-лимфоцитов могут быть моноциты и макрофаги, которые отвечают продукцией и секрецией MIF, наряду с другими провоспалительными цитокинами, на индукцию липополисахарида (ЛПС). Кроме того, пресинтезированный MIF был обнаружен в передней доле гипофиза и была показана способность клеток передней доли гипофиза отвечать продукцией MIF на индукцию ЛПС. Усиленную секрецию цитокина in vivo вызывал кортикотропин-релизинг фактор (CRF), что было расценено как компонент стрессорной реакции.
В связи с этим возникло предположение о том, что MIF может выполнять функции контр-регулятора иммунного ответа по отношению к глюкокортикоидам, которые известны как наиболее сильные ингибиторы воспаления и клеточного иммунного ответа. В физиологических концентрациях глюкокортикоиды индуцируют секрецию MIF макрофагами и Т-лимфоцитами, хотя секрецию других провоспалительных цитокинов те же глюкокортикоиды подавляют. Очевидно, MIF контролирует противовоспалительные эффекты глюкокортикоидов.
Так, например, MIF блокировал протективный эффект дексаметазона на модели эндотоксического шока. Показана способность MIF противостоять ингибирующему действию глюкокортикоидов на секрецию макрофагами провоспалительных цитокинов: TNF-а, IL-1в, IL-6, IL-8. Уровень MIF может повышаться как следствие глюкокортикоидной терапии. Повышенный уровень MIF контролирует иммуносупрессирующие эффекты глюкокортикоидов: эндогенных или введенных для лечения. Отсюда анти — MIF стратегия может быть полезна для повышения иммуносупрессивного и противовоспалительного действия глюкокортикоидов.
Интерферон-гамма
Важнейшим провоспалительным цитокином является IFN-y, который продуцируется активированными Т-лимфоцитами и активированными ЕК. Продукция IFN-y Т-лимфоцитами запускается при распознавании комплекса антигенного пептида с собственными молекулами гистосовместимости (главный комплекс гистосовместимости (МНС) 1 или 2 класса) соответствующим Т-клеточным рецептором (ТКР) и регулируется другими цитокинами: типичным стимулятором — IL-2 и типичным ингибитором — IL-10. Уровень продукции IFN-y при иммунном ответе в значительной степени определяется доминированием определенной субпопуляции: ТН1 или ТН2.
Продукция IFN-y естественными киллерами запускается при их взаимодействии с клетками-мишенями (опухолевыми, зараженными вирусами) и усиливается некоторыми цитокинами, в частности IL-12, который является продуктом активированных макрофагов или Т-лимфоцитов.
Среди функций IFN-y одной из важнейших является активация эффекторных функций макрофагов: их микробицидности и цитотоксичности, продукции ими цитокинов, супероксидных и нитроксидных радикалов, простагландинов.
IFN-y повышает экспрессию антигенов МНС 1 и 2 классов на разных клетках, он может даже индуцировать экспрессию этих молекул на тех клетках, которые не экспрессируют их конститутивно. Тем самым IFN-y повышает эффективность презентации антигенов и способствует их распознаванию Т-лимфоцитами.
В случаях достаточно ранней продукции IFN-y естественными киллерами он участвует в обеспечении прочной адгезии лимфоцитов к эндотелиальным клеткам в посткапиллярных венах перед их выходом из сосудов: он повышает на эндотелиальных клетках экспрессию адгезионных молекул ICAM-1, что ведет к повышенной адгезии лимфоцитов, экспрессирующих соответствующий лиганд — интегрин LFA-1. Кроме того, IFN-y повышает проницаемость эндотелия для макромолекул. В сочетании с TNF-a он индуцирует продукцию хемокинов семейства RANTES. Преинкубация с IFN-y сенсибилизирует клетки к индукции TNF-a. Kpоме того, он может в качестве синергиста TNF-a участвовать в развитии синдрома кахексии.
Описаны весьма противоречивые эффекты IFN-y на лимфоциты. Для большинства клеток он является мягким ингибитором пролиферации, а митоген-индуцированную пролиферацию Т-лимфоцитов он стимулирует, вместе с тем он слегка супрессирует активирующее действие IL-2 и IL-4 на пролиферацию ТН2, но не ТНТ. IFN-y повышает функциональную активность цитотоксических Т-лимфоцитов (CD8+), характер влияния на функции Т-хелперов зависит от уровня экспрессии соответствующих рецепторов. Описана даже индукция апоптоза Т- и В-лимфоцитов под влиянием IFN-y (табл.6.21.).
Таблица 6.21. Провоспалительные цитокины
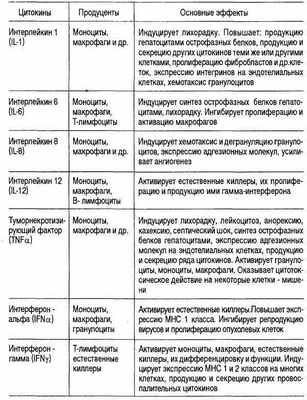
Туморнекротизирующий фактор альфа (кахектин)
Туморнекротизирующий фактор альфа (TNF-a), он же кахектин-полипептидный цитокин, выполняющий регуляторные и эффекторные функции в иммунном ответе и воспалении.
Основные продуценты TNF-a — моноциты и макрофаги, но есть и другие продуценты: лимфоциты крови, естественные киллеры (ЕК), гранулоциты крови, Т-лимфоцитарные клеточные линии. Главными индукторами синтеза TNF-a считаются ЛПС и другие компоненты микроорганизмов. Кроме того, роль индукторов могут взять на себя другие цитокины: IL1, IL2, IFNa/в, GM-CSF. Значительно меньшие количества TNF-a могут продуцировать некоторые опухолевые клетки в ответ на различные стимулы.
Разные проявления активности TNF-a опосредуются через специфические рецепторы. Показано существование двух разных рецепторов для TNF-a: миелоидного происхождения клетки несут рецептор 55 kDa, а эпителиального происхождения — рецептор 75 kDa. Показана причастность рецептора 55 kDa к цитотоксическому и ростстимулирующему действию TNF-a. Внеклеточная часть рецептора 55 kDa может существовать вне клеток как растворимый рецептор, способный связывать TNF-a. Он может образоваться в результате шеддинга рецепторов с мембраны клеток (гранулоцитов) под влиянием цитокинов (GM-CSF), компонентов комплемента (С5а) или лекарственных препаратов.
Многие биологические эффекты TNF-a связаны с активацией или ингибицией экспрессии определенных генов. Он активирует транскрипцию генов других провоспалительных цитокинов, действуя через фактор транскрипции NFkB. В других клетках-мишенях он действует через другие факторы транскрипции на экспрессию других генов.
Основные проявления биологической активности TNF-a: избирательная цитотоксичность в отношении некоторых опухолевых клеток, угнетение синтеза ключевого фермента липогенеза — липопротеинкиназы, участие в регуляции иммунного ответа и воспаления. Он входит в группу провоспалительных цитокинов и выполняет важнейшие функции в период запуска воспаления: активирует эндотелий, способствует адгезии лейкоцитов к эндотелию за счет индукции экспрессии на эндотелиальных клетках адгезионных молекул и последующей трансэндотелиальной миграции лейкоцитов в очаг воспаления, активирует лейкоциты (гранулоциты, моноциты, лимфоциты), индуцирует продукцию других провоспалительных цитокинов: IL-1, IL-6, IFNp, GM-CSF, обладающих синергидным с TNF-a действием.
Местная продукция TNF-a в очаге инфекции или воспаления обеспечивает хемотаксис гранулоцитов и моноцитов в очаг, усиление фагоцитоза и микробицидности фагоцитов, усиленную их дегрануляцию, продукцию и секрецию реактивных кислородных радикалов (супероксидных и нитроксидных), повышенную цитотоксичность фагоцитов.
Т-лимфоциты в процессе активации приобретают усиленную экспрессию рецепторов для IL-2 и TNF-a. В синергизме с IL-2 TNF-a усиливает продукцию Т-клетками IFNy.
TNF-a участвует не только в защитных реакциях, но и в процессах деструкции и репарации, сопутствующих воспалению. TNF-a служит одним из медиаторов деструкции тканей, обычной при длительном, хроническом воспалении. Вместе с тем, способность TNF-a стимулировать рост фибробластов и индуцировать ангиогенез делает возможным его участие в процессах репарации тканей.
Роль TNF-a в патологии может быть связана с его способностью индуцировать пролиферацию фибробластов и депозицию коллагена. Это было показано на примере легочного фиброза при силикозах (табл.6.21.).
Молекулы адгезии
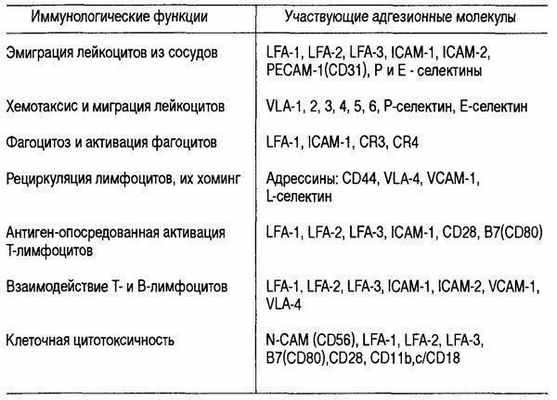
Движение лейкоцитов в очаг воспаления начинается с серии адгезионных событий, каждое из которых касается лейкоцитов определенного- типа: нейтрофилов, моноцитов или лимфоцитов. Циркулирующие лейкоциты обычно вступают лишь в мимолетные контакты с эндотелиальными клетками посткапиллярных венул: лейкоциты как бы "скользят" по поверхности эндотелия сосудистой стенки.
Эта фаза обеспечивается взаимодействием вначале Р-, а затем L- и Е-селектинов с углеводными компонентами мембран клеток. L-селектин экспрессирован на большинстве лейкоцитов. Р-селектин эндотелиальных клеток опосредует адгезию нейтрофилов и моноцитов к эндотелию. Е-селектин экспрессируется на активированных эндотелиальных клетках и поддерживает адгезию лимфоцитов.
Лигандами селектинов служат сиалил-фукозилированные олигосахариды в составе многих гликопротеинов и гликолипидов мембран клеток, например, муциноподобные молекулы. Муциноподобный домен содержит клеточная адгезионная молекула — мукозный адрессин (MAdCAM-1), которая за счет взаимодействия с L-селектином обеспечивает возврат лимфоцитов в мукозноассоциированную лимфоидную ткань. Некоторые из этих лиганд появляются на поверхности клеток только после их активации.
Фаза скольжения происходит без активации лейкоцитов, однако скользящие лейкоциты при контактах с поверхностью эндотелия получают сигналы активации, что ведет к их иммобилизации. Наступает вторая фаза прочной адгезии, опосредованная усилением способности лейкоцитарных интегринов связываться с лигандами из суперсемейства иммуноглобулинов на эндотелиальных клетках. В качестве сигналов активации могут служить воздействия цитокинов (хемокинов): MIP-ip, МСР-1, IL-8, MIF, PAF, С5а-фракции комплемента, которые способны связываться с глюкозамингликанами поверхности эндотелиальных клеток и действовать на "скользящие" лейкоциты.
Интегрины — это большое семейство молекул клеточной поверхности, представители которых обнаружены на большинстве типов клеток. Интегрины опосредуют взаимодействие клеток с их микроокружением, обеспечивая адгезию клетка — клетка и клетка — матрикс. Интегрины — это гетеродимеры гликопротеинов, состоящие из различных комбинаций а- и в-цепей.
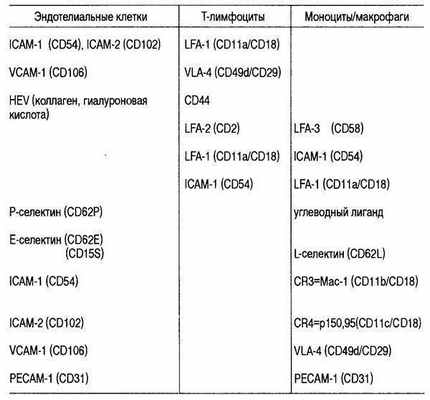
Описано более 20 разных представителей интегринов. На лейкоцитах экспрессированы: LFA-1, Macl, р150,95. Лигандами для LFA-1 являются: ICAM-1, ICAM-2, ICAM-3, для Macl — ICAM-1. Эти интегрины опосредуют адгезию к эндотелию нейтрофилов, базофилов, эозинофилов, моноцитов и лимфоцитов. В отличие от нейтрофилов остальные типы клеток могут адгезироваться к цитокин-активированным эндотелиальным клеткам через интегрины VLA-4 к лигандам VCAM-1.
На поверхности эндотелиальных клеток лигандами интегринов служат молекулы, имеющие структурную гомологию с иммуноглобулинами. К ним относятся интерклеточные адгезионные молекулы: ICAM-1, ICAM-2, ICAM-3, васкулярно-клеточная адгезионная молекула — VCAM-1. Последняя эксирессируется преимущественно на активированных эндотелиальных клетках.
Следующая после прочной адгезии стадия трансмиграции лейкоцитов через эндотелий контролируется частично теми же интегринами, взаимодействующими с молекулами ICAM-1, расположенными и на внутренней, и на латеральной, и на базальной поверхности эндотелиальных клеток. Описаны и другие молекулы, облегчающие трансмиграцию лейкоцитов: например CD31 (РЕСАМ-1), обнаруженные и на эндотелиальных клетках, и на тромбоцитах, нейтрофилах, моноцитах, лимфоцитах. За трансмиграцию моноцитов отвечает интегрин CD18, но после активации эндотелиальных клеток под влиянием IL-1 и TNF-a трансмиграция идет при участии интегринов а4в1, взаимодействующих с молекулой VCAM-1.
Все стадии адгезии и трансмиграции зависят от активации эндотелиальных клеток, которая проявляется усилением экспрессии на них адгезионных молекул. Экспрессия Е-селектина усиливается в самые ранние стадии воспаления тромбином, тистамином или активированной системой комплемента, и не требует синтеза белка de novo. Роль стимулято: ров на этой стадии могут играть различные оксиданты.
В подострой фазе воспаления активация эндотелиальных клеток опосредована, в основном, цитокинами: IL-1, TNF-a, которые индуцируют экспрессию Е-селектина, VCAM-1 и усиливают экспрессию ICAM-1. Такое же действие оказывает ЛПС. Эта активация связана с необходимостью синтеза белка de novo. Кинетика экспрессии трех вышеперечисленных молекул различна: TNF-a раньше всего усиливает экспрессию Е-селектина — за 2-4 часа до максимальной экспрессии VCAM-1 и ICAM-1.
Эффекты TGFp противоречивы: он стимулирует хемотаксис лейкоцитов, но препятствует их трансмиграции через эндотелий. С дефектом TGFp может быть связано развитие многофокусного воспаления с лейкоцитарной инфильтрацией многих тканей, с избыточной продукцией провоспалительных цитокинов.
На эпителиальных клетках бронхов также экспрессированы адгезионные молекулы ICAM-1, экспрессия которых может повышаться под влиянием цитокинов IFNy, TNF-a, IL-ip. Экспрессия молекул ICAM-1 усиливается на эпителии при воспалении воздухоносных путей. На таких активированных эпителиальных клетках усилена адгезия эозинофилов, которая, в свою очередь, приводит к усиленной дегрануляции эозинофилов. Секретируемые при этом катионные белки могут участвовать в повреждении респираторного эпителия.
При остром воспалении легких нейтрофилы эмигрируют из альвеолярных капилляров и двигаются в ответ на воспалительные стимулы, преодолевая интерстициальный матрикс между эндотелием капилляров и альвеолярным эпителием. Этот процесс включает адгезию нейтрофилов и их локомоции. Альвеолярный интерстиций состоит из фибробластов. Адгезия нейтрофилов к альвеолярным фибробластам повышается под влиянием PAF за счет повышения экспрессии в2-интегринов (CD11/CD18).
Миграционная активность нейтрофилов возрастала под влиянием хемоаттрактанта IL-8, который секретировали альвеолярные фибробласты, активированные под влиянием TNF-a. Фибробласты служат не только субстратом адгезии, но и источником стимулирующих молекул, обеспечивая миграцию нейтрофилов через воспаленный альвеолярный интерстиций. Для бактериальной пневмонии характерен ранний выход нейтрофилов в интерстиций и в просвет альвеол. При этом контакт нейтрофилов с фибробластами опосредован взаимодействием интегринов (CD11/ CD18) с молекулами ICAM-1(CD54), экспрессированными на фиб-робластах (табл.6.25.).
Таблица 6.25. Адгезионные молекулы и соответствующие лиганды при взаимодействии клеток
Конститутивная экспрессия адгезионных молекул ICAM-1 на альвеолярных эпителиальных клетках в 22 раза выше, чем на эндотелиальных клетках капилляров. ICAM-1 экспрессированы, в основном, на пневмоцитах типа I, а не на их предшественниках пневмоцитах типа II. При пневмонии экспрессия ICAM-1 повышается в 178 раз на пневмоцитах типа II, т.е. происходит их ускоренная дифференцировка в ответ на повреждение эпителия альвеол.
Кроме того, непосредственно в очаге пневмонии были обнаружены нейтрофилы с измененной экспрессией адгезионных молекул: L-селектина и CD18. Эмиграция нейтрофилов в просвет альвеол при пневмонии, вызванной Streptococcus pneumoniae, связана со снижением экспрессии L-селектина и повышением экспрессии CD18. Нейтрофилы в воздушном пространстве альвеол легкого имеют повышенный уровень экспрессии адгезионных молекул CD18, что может способствовать фагоцитозу патогенных бактерий.
Адгезия моноцитов к эндотелиальным клеткам легочных капилляров также опосредована взаимодействием Р2 интегринов на моноцитах с молекулами ICAM на эндотелиальных клетках. Аккумуляции моноцитов в легких способствует и то, что диаметр легочных капилляров (6-8цт) меньше диаметра многих моноцитов крови (7-10цт), поэтому прохождение моноцитов через легочные капилляры требует деформации, пластичности моноцитов, которая в первую очередь нарушается при воспалении.
Моноциты застревают в узких легочных капиллярах, прилипают к эндотелиальным клеткам и проделывают трансмиграцию в просвет альвеол. Бактериальные компоненты и продукты (в частности ЛПС) повышают адгезивность моноцитов, усиливая экспрессию адгезионных молекул (табл.6.26.).
Таблица 6.26. Иммунологические функции, в которых участвуют адгезионные молекулы
Адгезионные молекулы могут стать клинически значимыми мишенями для противовоспалительной терапии. В настоящее время ведется поиск агентов, селективно ингибирующих адгезионные молекулы.
ПОЛИТРАВМА / POLYTRAUMA
Ревматологи и рентгенологи на рубеже веков дифференцировали две основные формы хронического артрита: 1) атрофический артрит с синовиальным воспалением, с формированием эрозий и/или атрофией хряща и кости (например, ревматоидный артрит) и 2) гипертрофический артрит, характеризующийся очаговой потерей хряща, без формирования типичного воспалительного каскада, а также рост (гипертрофия) прилегающей кости и мягких тканей [3]. Последняя группа стала синонимом остеоартроза. Этот термин подчеркивал отсутствие явного воспаления и даже использовался в качестве суррогата нормальной ткани суставов. Остеоартроз считался по своей природе невоспалительным заболеванием подвижных суставов, характеризующимся ухудшением свойств суставного хряща и образованием новой кости на поверхностях суставов и краях, в основе которого, как полагали, лежит замедление восстановительных процессов в поврежденном хряще. В процессе экспериментов на животных это мнение было подтверждено обнаружившемся отсутствием кровоснабжения хрящевой ткани, низким метаболизмом хондроцитов и их неспособностью к восстановлению поврежденного хряща. Изменения в динамическом равновесии между синтезом и деградацией матрикса хондроцитами было расценено ведущим механизмом в развитии дегенерации суставного хряща, приводящим к остеоартрозу. Таким образом, остеоартроз определялся как первичное невоспалительное заболевание суставов, развивающееся у лиц старше 45-50 лет, основным клиническим признаком которого является боль механического типа, и имеющее определенные диагностические признаки поражения суставов при проведении визуализирующих диагностических методов исследования [3].
Однако в последнее время доказано, что такой взгляд является некорректным, и более обоснован термин «остеоартрит» (ОА) – патологическое ремоделирование суставных тканей, корригируемое различными провоспалительными факторами, которые продуцируются синовием и субхондральной костной тканью [3]. Хроническое воспаление –характерный признак ОА , при котором в патологический процесс вовлечены все компоненты суставной ткани: хрящ, синовиальная оболочка, суставная капсула, связки, сухожилия и субхондральная кость [15]. В этиопатогенезе ОА ведущими биомеханическими факторами являются патологические изменения в суставном хряще, обусловленные аномальной нагрузкой [16]. Таким образом, травма-индуцированные повреждения структуры суставного хряща приводят к длительному воспалительному процессу [12].
Посттравматический остеоартрит
Посттравматический ОА (ПТОА) – разновидность ОА, где установленным этиологическим фактором является травма [24]. Основными травматическими повреждениями, приводящими к ПТОА, являются разрывы и значительные повреждения менисков и/или связочного аппарата, хрящевой ткани, внутрисуставные переломы, особенно если они сопровождаются гемартрозом. Травматическое повреждение сустава, ассоциированное с нарушением биомеханики, значительно увеличивает риск возникновения ПТОА [8]. Возникновение ПТОА характерно преимущественно для молодых пациентов и характеризуется достаточно быстрым прогрессированием [10].
В отличие от возраст-зависимого и/или метаболического ОА, при ПТОА, учитывая знание времени травматического события, которое способствовало повреждению сустава, можно понять и оценить патогенетические механизмы после его повреждения.
Следует отметить, что нарушение функционирования, нестабильность сустава часто возникают не только после травмы, но и после хирургических вмешательств по рестабилизации сустава. Согласно данным литературы, хирургические операции, проведенные для стабилизации сустава, являются факторами, ассоциированными с прогрессирующей дегенерацией суставов [12], отмечено, что в среднем через 20 лет после проведения операции по удалению мениска у 3/4 пациентов развивался тибиофеморальный или пателлофеморальный ОА [38]. Обнаружено, что у таких пациентов в течение значительного времени в синовиальной жидкости (СЖ) наблюдается повышение уровня провоспалительных маркеров, таких как интерлейкин-6 (ИЛ-6) и фактор некроза опухоли a (ФНОa), что позволило предложить, что эти показатели способствуют развитию ПТОА и прогрессированию ОА [26].
Обнаружено, что менискэктомия усугубляет дальнейшее повреждение суставного хряща. Согласно данным литературы, после артроскопической менискэктомии из-за разрыва мениска формируется «менискэктомия-индуцированный остеоартрит», частичная менискэктомия увеличивает риск ОА в четыре раза, что оценивалось через 16 лет после операции. При проведении сравнения степени восстановления хряща определено, что оно происходило более эффективно и быстро в случае дегенеративного поражения мениска, чем в случае отсутствия его, что можно объяснить вторичным повреждением суставных тканей [29].
Наиболее частыми причинами, приводящими к ПТОА, являются внутрисуставные переломы, травмы мениска, связочного аппарата и хрящевой ткани [30]. Среди суставов чаще всего травмируются голеностопный и коленный суставы. Общей особенностью травм суставов, которые вызывают ПТОА, является внезапное приложение механической силы (удара) к суставной поверхности. Степень механического повреждения зависит от интенсивности удара. Исследования показывают, что более сильное энергетическое воздействие вызывает большее локальное повреждение тканей, что измеряется экспериментально по доле клеток, высвобождающих активные формы кислорода, гибели хондроцитов и разрушении матрикса [10, 11]. Различные уровни прикладываемой энергии удара вызывают разные типы повреждения суставов с различными ответными реакциями на восстановление и с различным потенциалом заживления: 1) повреждение клеток и/или матрикса, которое не вызывает макроскопическое разрушение структуры хряща или кости; 2) повреждение клеток и/или матрикса наряду с макроскопическим разрушением структуры сустава хряща без смещенного перелома кости (эти повреждения могут быть связаны с микроразрушениями кальцифицированного хряща и в некоторых случаях субхондральной или трабекулярной кости; 3) переломы со смещением суставной поверхности, распространяющиеся на хрящ и кость [12, 13]. Низкоэнергетические травмы, включая ушибы суставов, вывихи и повреждения связок, обычно вызывают первые два типа повреждения суставной поверхности, в то время как травмы с более высокой энергией воздействия вызывают внутрисуставные переломы со смещением [8, 9].
Существует достаточно доказательств того, что разрыв передней крестообразной связки (ПКС) и разрыв мениска являются двумя основными факторами риска для развития ПТОА коленного сустава [10]. Повреждения ПКС часто возникают у молодых пациентов, особенно у спортсменов, что приводит к боли, функциональным нарушениям и снижению физической активности так называемых молодых пациентов со старыми коленями.
Повреждения приводят к скоплению крови в полости сустава (формирование гемартроза), кроме того изменения формируются и на клеточном уровне в виде апоптоза хондроцитов и остеобластов, высвобождения большого количества провоспалительных медиаторов. Исследования острой посттравматической стадии показали повышенную экспрессию молекул, участвующих как в катаболических, так и в анаболических процессах [7, 37].
Патологические изменения провоспалительных цитокинов в синовиальной оболочке сустава
Согласно ряду исследований СЖ, у относительно молодых пациентов с травматическим повреждением ПКС обнаружены высокие уровни ИЛ-1β, ИЛ-6, ИЛ-8 и ФНО a , в большей степени за счетИЛ-8 и ФНОa [44]. В первую очередь в СЖ снижается концентрация ИЛ-1, а уровни ИЛ-6 и ФНОa остаются повышенными в течение более длительного времени (около 6 месяцев после травмы) [11].
После разрыва ПКС в течение нескольких первых недель обнаружены повышенные уровни ИЛ-10, ИЛ-1Ra в СЖ, которые снижались в течение 3-6 недель [36], а через 6 месяцев обнаружено сохранение повышенных концентраций ИЛ-1β в СЖ, причем уровень повышения прямо коррелирует со степенью хрящевых повреждений [36]. На моделях животных с ПТОА выявлено, что ИЛ-10 и ИЛ-4 защищают суставной хрящ от дальнейшего провоспалительного ответа и предотвращают последствия активации воспаления в ответ на гемартроз, из чего можно сделать вывод о возможном хондропротективном действии этих цитокинов [47]. ИЛ-1Ra может останавливать отрицательные эффекты ИЛ-1 в поврежденном суставе [23].
В течение первых двух недель после травмы наблюдаются три фазы: ранняя, характеризующаяся гибелью клеток и воспалительными явлениями; подострая, с сохранением воспаления, но более низкой интенсивности; и поздняя, характеризующаяся прежде всего увеличением деградации суставного матрикса [40]. Предполагается, что активация дополнительного протеолитического каскада и toll-подобных рецепторов (TLR), таких как TLR-2 и TLR-4, происходит совместно с цитокинами/хемокинами как первая линия защиты врожденного иммунитета [15.]
Наряду с активацией провоспалительного ответа после травмы отмечается снижение концентрации лубрицина в СЖ, что приводит к увеличению риска более быстрого развития деструктивных изменений в суставе вследствие нарушения вязкоэластичных свойств СЖ. Уровень лубрицина после травмы остается низким на протяжении достаточно длительного периода времени (около 12 мес.) [48]. Уменьшение концентрации лубрицина взаимосвязано с повышенным уровнем ФНО a , выявлено, что ингибирование последнего приводит к повышению концентрации протеогликана-4 [11]. Кроме того, повышенные уровни провоспалительных цитокинов, таких как ФНОa, ИЛ-1β и тромбоцитарного фактора роста β (TGF-β), тормозят и угнетают образование других суставных лубрикантов – гиалуроновой кислоты, общих протеогликанов, олигомерного матриксного протеина хряща [4].
Острое синовиальное воспаление, связанное с повреждением суставов, тесно связано с клеточной инфильтрацией и коррелирует с тяжестью/степенью повреждения. Исследования на животных моделях подтверждают роль как инфильтрирующих макрофагов, так и Т-лимфоцитов в прогрессировании посттравматического заболевания. Как было показано на эксплантатах крупного рогатого скота, синовиальное воспаление также приводит к окислительному повреждению хондроцитов суставного хряща и матрикса посредством повышенной секреции активных форм кислорода (АФК) и снижения антиоксидантной защиты [44, 49]. В дополнение к непосредственному повреждению жизнеспособных хондроцитов, АФК синергируются с провоспалительными цитокинами и оксидом азота для стимулирования экспрессии катаболических генов через внеклеточную сигнал-регулируемую киназу-1/2 ( ERK ) и N -терминальную – Junкиназу (JNK) [49].
Патологические изменения матриксных ферментов в синовиальной оболочке сустава
Формирование ПТОА
Соотношение противо- и провоспалительных цитокинов в сторону преобладания последних приводит к хронизации воспаления и в конечном итоге к ПТОА [29]. В подострой и хронической фазе (от 2 месяцев до 1 года) после травмы уровни провоспалительных цитокинов остаются значительно повышенными.
В хронической фазе ведущая роль в формировании ПТОА отводится прогрессирующей потере гликозаминогликанов, а повреждение хряща способствует высвобождению или расщеплению многих других белков, таких как ММП и коллаген 2-го типа [17]. Многие из этих внеклеточных белков происходят из перицеллюлярного матрикса и могут быть результатом его повреждения. В связи с этим в СЖ после повреждения выявляется множество матричных белков, повышены также уровни фрагментов олигомерных белков коллагена и хряща, генерируемых различными аггрекиназами. Поскольку эти фрагменты остаются в течение нескольких лет после травмы, они могут способствовать развитию ПТОА [43]. Более низкая концентрация лубрикантов (гиалуроновая кислота и лубрицин), наблюдаемая в СЖ вследствие протеолиза нейтрофильными ферментами и накопления воспалительных медиаторов, приводит к нарушению смазочной функции. В хронической фазе происходит прогрессирование метаболических и деструктивных изменений в суставных тканях, что приводит в конечном итоге к переходу клинически бессимптомного периода ПТОА в симптоматический период, с болью в суставах и нарушением их функции.
Повреждение суставного хряща инициирует экспрессию фактор a роста эндотелия сосудов (VEGF) [20]. Повышение концентрации VEGF приводит к снижению экспрессии хондромодулина-1, антиангиогенного фактора, последние активно участвуют в поддержании функции и трофики суставного хряща [19].
Патологические изменения суставного хряща и суставной кости при формировании ПТОА
Выраженность патологических изменений, которые формируются при ПТОА, зависит от степени травмирующего фактора.
В остром периоде травмы основными факторами, способствующими развитию ПТОА, являются экстравазация плазмы в СЖ со снижением концентрации любрицина и гиалуроновой кислоты, снижение синтеза протеогликанов, сверхэкспрессия матричных металлопротеиназ (ММП) и провоспалительных медиаторов функционирующими клетками [27].
В острой посттравматической стадии вследствие травмы появляется повреждение структуры суставных тканей, происходит запуск апоптозахондроцитов и остеобластов [45]. Нарушение биомеханических и физико-химических свойств ткани приводит к значительным изменениям в хондроцитах, изменяя их способность экспрессировать белки, участвующие в метаболических путях, и приводя к гибели клеток. Поскольку хондроциты отвечают за поддержку функций суставного хряща, их гибель через апоптотические механизмы занимает одно из ведущих мест в формировании ПТОА [41]. Это также подтверждается тем фактом, что более высокий процент апоптотических клеток был обнаружен в хряще пациентов с внутрисуставными переломами по сравнению с пациентами с ОА и ревматоидным артритом (РА) без травм [32]. Исследования in vitro и in vivo выявили связь между гибелью клеток и такими факторами, как энергия удара, близость к суставной поверхности и наличие перелома [1]. В таблице суммированы основные звенья патогенеза ПТОА.
Таблица. Патогенез посттравматической деградации хряща в формировании посттравматического остеоартрита во временном аспекте
Ремоделлирование суставной ткани
Инфильтрация лейкоцитами и медиаторами воспаления
Дегидратация экстрацеллюлярного матрикса
Модели in vivo и in vitro
В последнее десятилетие появилось достаточно много научных работ, посвященных экспериментальным моделям ПТОА на животных и человеческих культурах тканей, что демонстрирует актуальность данной проблемы. Вероятнее всего, это связано с тем, что более глубокое изучение молекулярных и клеточных процессов, которые приводят к деградации хряща, особенно в острую посттравматическую фазу, открывает новые перспективы для раннего фармакологического вмешательства и профилактики развития ПТОА.
В инициации ПТОА участвуют множество различных механических и биохимических процессов. Поэтому трудно в точности воспроизвести in vitro повреждение тканей и активировать специфические клеточные пути. В большинстве исследований исследуется роль травмы с использованием моделей человеческого хряща и с изучением выживаемости клеток, экспрессии генов и медиаторов воспаления. Экспланты хряща подвергаются определенной ударной нагрузке или повторяющимся травмам с помощью различных устройств, и таким образом оценивается аддитивный эффект цитокинов, ингибиторов и лекарственных препаратов на травма-индуцированный воспалительный процесс [34].
Модели животных имеют решающее значение для понимания развития ПТОА и оценки новых возможных методов лечения [13]. Экспериментальный ПТОА обычно индуцируется либо посредством хирургического вмешательства, либо путем непосредственной физической травмы сустава. В первом случае рассекается связка надколенника, а медиальные латеральные мениски удаляются микрохирургическим методом, причем суставной хрящ остается неповрежденным. Хирургическая дестабилизация медиального мениска (ДММ) в настоящее время является наиболее широко выполняемой процедурой для формирования модели ПТОА [14]. ДММ приводит к дегенеративным повреждениям суставного хряща большеберцовой кости в течение 10–12 недель после процедуры, проявляющимся склерозом субхондральной кости и умеренным синовитом. В модели на мышах с ДММ признаки воспаления возникают очень рано и через 7-10 дней после хирургической процедуры в суставных тканях и СЖ обнаруживаются огромные инфильтраты воспалительных моноцитов и активированных макрофагов [21 ].
У модели мышей с внутрисуставным переломом большеберцовой кости на 3-й день после травмы обнаруживали высокие уровни ИЛ-1β, ИЛ-6, ИЛ-8 и MCP-1, сохраняющиеся вплоть до 16-й недели [35], а через 7 дней – появление значительных эрозивных изменений хряща в месте перелома, потерю костной массы и острый синовит в течение 7 дней [9].
Недавние исследования на животных показали, что специфические генетические мутации, которые изменяют синтез различных молекул, могут выступать как предиктивные биомаркеры в развитии хронического посттравматического артрита и ПТОА. В частности, модификации в генах, участвующих в деградации хрящевого матрикса, воспалении, дифференцировке и апоптозе хондроцитов, способствуют возникновению ПТОА [30].
Исследования эпигенетичеких феноменов человека позволили выявить некоторые патогенетические механизмы в развитии ПТОА. Так, прогрессированию заболевания способствует снижение CpG метилирования PH домена богатой лейцином повторяющейся протеинфосфатазы 1 (PHLPP1), что ведет к усилению экспрессии PHLPP1. PHLPP1 представляет собой Ser / Thr фосфатазу, которая снижает активность нескольких киназ, стимулирующих анаболическую функцию хряща. Кроме того, было показано, что дефицит PHLPP1 у мышей, хирургически дестабилизированных путем рассечения медиальной менисковой связки, защищает от начала развития ПТОА за счет увеличения клеточного содержимого и толщины суставного хряща [6 ].
Лечение и профилактика
Таким образом, травма является этиологическим фактором ПТОА, который развивается после нее. Однако даже при проведении хирургического вмешательства риск формирования ПТОА есть у каждого второго пациента после травмы и составляет чуть более 50 % [24]. Наиболее опасен острый посттравматический период, когда происходят максимальные патологические изменения в синовии, суставном хряще и субхондральной кости, которые сохраняются до 1 года. Лечение ПТОА является сложной задачей. В настоящее время отсутствуют биохимические маркеры, которые предсказывают или коррелируют с прогрессированием заболевания, и лечение ограничивается восстановлением и стабилизацией сустава. Противовоспалительная терапия, в частности внутрисуставное ингибирование цитокинов, может обеспечить эффективный подход для снижения или предотвращения развития ПТОА. Идеальная терапия должна быть разнообразной и включать положительные воздействия на метаболизм хондроцитов и стимуляцию внутреннего восстановления, в то же время подавляя катаболические пути, которые приводят к гибели хондроцитов и потере матрикса. Был идентифицирован ряд молекулярных мишеней и возможных лекарственных агентов, эффективных на животных моделях с травмами суставов и ПТОА
Однако необходимы дальнейшие исследования, чтобы определить конкретные маркеры для раннего выявления прогрессирования заболевания и изучить инновационные возможности для предотвращения будущего хронического заболевания.
Информация о финансировании и конфликте интересов
Исследование не имело спонсорской поддержки.
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Читайте также: