Почечный канальцевый ацидоз у подростков. Гиперпролинурия и ихтиоз с глухотой
Добавил пользователь Алексей Ф. Обновлено: 10.01.2026
ЛОР-болезни:
Популярные разделы сайта:
Наследственная тугоухость с поражением почек. Синдром Альпорта, Шарко — Мари — Тута
Сочетание наследственно обусловленной тугоухости с заболеваниями почек отмечено еще в прошлом веке. Изучению этого вопроса посвящено большое количество работ, в основном зарубежных авторов. В нашей стране данное сочетание наследственной патологии слуха и почек стало привлекать внимание исследователей в последние 30 лет. Интерес к проблеме возрастает.
Приводим данные о наиболее изученных синдромах, когда наследственно обусловленная тугоухость выявляется в сочетании с патологией почек, а также других органов и систем.
1. Синдром Альпорта [Alport]. Гистологически выявляются уменьшение количества клеток (около 50%) спирального узла, дегенеративные изменения сосудистой полоски и волосковых клеток спирального органа. Составляет 1 % наследственной тугоухости. Одновременное поражение органа слуха и почки обусловлено сходством улитки и почки по балансу жидкости и электролитов, наличием общих ото- и нефротоксических веществ [Quick, Arnold, Weidaver] и некоторой идентичностью антигенных структур этих органов. У 10% мужчин развиваются хронический нефрит с уремией, патология хрусталика (лентиконус, сферофакия, катаракта). Наследуется по доминантному типу, сцепленному с Х-хромосомой.
2. Нейросенсорная тугоухость с гипогенитализмом, патологией стероидогенеза, почечной недостаточностью и артериальной гипертензией (одна из форм адреногенитально-го синдрома) [Harriet]. Нарушение гормональной регуляции обмена веществ вследствие недостаточности 17-ОН- и 11-ОН-гидроксилирования стероидов в коре надпочечников и гиперплазии клубочковой зоны. Нарушение обмена сопряжено с эндокринной патологией, проявляющейся у мальчиков крипторхизмом, у девочек—первичной аменореей. Первичные признаки половой зрелости могут отсутствовать. Прогрессирующий нефросклероз и вторичная артериальная гипертензия определяют неблагоприятный исход после 30 лет. Синдром Альпорта исключается на основании отсутствия гипогснитализма и артериальной гипертензии, ХХ-дисгенезия гонад — как заболевание с аутосомпо-рецессивной передачей.
3. Синдром Шарко — Мари — Тута, нефрит и нейросенсорная тугоухость. Документальных данных нет. Причиной являются нейросенсорные нарушения, по-видимому, вторичные. Прогрессирующая дистальная мышечная атрофия с детского возраста. Когтеобразная кисть. Гематурия в отличие от синдрома Альпорта сочетается с протеинурией, что обусловлено атрофией канальцев при интерстициальном нефрите, наличием «пенистых» клеток, гиалинизацией клубочков. Нейросенсорная глухота особенно на высоких частотах чаще всего обнаруживается в возрасте 7 лет.

4. Нейросенсорная тугоухость с нефритом, тромбоцитопенией [Epstein et al., Ekstcin ct al.]. Тромбоцитарный аппарат не участвует в развитии общебиологических реакций. Поражение почек наряду с гематурией характеризуется протеин- и цилиндрурией. Спленэктомия неэффективна. Хроническая почечная недостаточность резистентна к консервативному лечению. Имеются попытки изменить гомеостаз с помощью трансплантации почки. Нарушение слуха более выражено на высоких частотах. Передается по доминантному типу.
5. Нейросенсорная тугоухость и почечный канальцевый ацидоз. Определяется дефицит карбоангидразы. Клиническая картина почечного канальцевого ацидоза характеризуется рвотой, обезвоживанием, полидипсией, полиурией, гипостенурией, рахитом, гиперхлоремическим ацидозом вследствие кальциноза почек. С рождения тугоухость на высоких частотах. Наследование по аутосомно-рецессивному типу.
6. Нейросенсорная тугоухость, ихтиоз, заболевание почек и гиперпролинурия [Goyer et al., Perry et al.]. Нарушение тератогенеза поддерживается наличием кист в почках после рождения. Гиперпролинсмия. В семьях возможны различные варианты перечисленных синдромов и симптомов. Наблюдаются утомляемость, хроническое расстройство питания, коричневатый шелушащийся ихтиоз, протеинурия с гематурией, иногда макрогематурия, кистообразное перерождение почек, склонность к мочекаменной болезни, склероз клубочков, интерстициальный нефрит с отложениями в межклеточном пространстве плотных веществ, в дальнейшем развитие хронической почечной недостаточности. Гиперпролинурия является маркером возможных нарушений обмена веществ, ведущих к развитию тугоухости. Передается по доминантному типу.
7. Синдром Мукле — Веллса. Глухота у детей с крапивницей, амилоидозом и нефритом [Muckle, Wells]. Отсутствие спирального органа и чувствительного эпителия преддверия. Окостенение основной мембраны. Отложение амилоида в печени и почках. Амилоидоз. Гиперглицинурия. К юношескому возрасту появляются уртикарии, лихорадка, боль в конечностях. В 30 лет может возникнуть нефротический синдром с почечной недостаточностью. Глухота выявляется с детства. Синдром передается по доминантному типу.
8. Тугоухость кондуктивная с аномалиями почек, генеталий [Wiater et al.] и пальцев [Brayn, Gayer]. К этому приводят тератогенные воздействия. Предполагается аутосомно-рецессивный тип наследования. Тугоухость сочетается с гипоплазией и агенезией почек, гипониазией маточных труб, яичников и влагалища, укорочением булавовидных I пальцев на руках и ногах с раздвоением дистальных фаланг и язычка.
Почечный канальцевый ацидоз у подростков. Гиперпролинурия и ихтиоз с глухотой
Детский почечный канальцевый ацидоз с врожденной глухотой
Cohen с соавт. сообщили о 4 детях, родившихся от кровнородственных браков в двух разных семьях. Обе эти семьи в свою очередь происходили из большого инбредного рода, отягощенного нейросенсорной глухотой и детским канальцевым ацидозом. Другие случаи болезни были представлены Nance и Sweeney, а также Donckerwolcke с сотр..
Клинические данные. Данные осмотра. У всех больных отмечалась выраженная задержка роста. Их рост был ниже нормального на 30%.
Почки. С рождения или вскоре после рождения у детей появлялись рвота, обезвоживание, полидипсия, полиурия и гипостенурия.
Орган слуха. С младенчества выявлялась выраженная нейросенсорная глухота, более глубокая па высоких частотах.
Вестибулярная система. Результаты исследований не опубликованы.
Лабораторные данные. Произведенная в младенчестве внутривенная пиелография выявила почечный кальциноз. К 2-летнему возрасту у больных были обнаружены рентгенологические признаки рахита.
При исследовании рН крови и электролитов сыворотки был установлен гиперхлоремический ацидоз. В результате пробы нагрузкой хлоридом аммония содержание углекислоты в крови значительно снизилось, по показатель рH мочи оставался высоким. Удельный вес мочи был низким. В сыворотке крови были значительно повышен уровень хлоридов. Отмечалось поражение дистальпых канальцев.
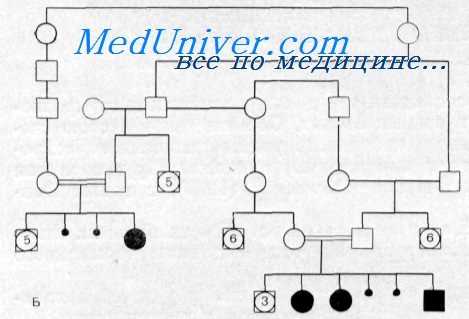
Наследственность. Из-за кровного родства родителей сибсы, описанные Cohen с соавт., были двоюродными кузенами, а также троюродными кузенами по отношению друг к другу. Мальчик, представленный Nance и Sweeney, также являлся потомком кровнородственного союза. Наследование синдрома отчетливо аутосомно-рецессивное.
У больных был обнаружен специфический дефицит карбоангидразы В (Cohen et al.).
Диагноз. Заболевание должно быть отдифференцировано от синдрома подросткового или юношеского почечного канальцевого ацидоза с медленно прогрессирующей нейросенсорной глухотой, наследующегося также по аутосомно-рецессивному типу.
Младенческий почечный канальцевый ацидоз существует и как изолированное заболевание с аутосомно-рецессивным типом наследования, а также может наблюдаться при большом числе синдромов, описанных в обзоре Nance и Sweeney.
Лечение. Дети получали щелочные растворы и большое количество жидкости. Применяли также слуховые аппараты.
Прогноз. Если заболевание рано диагностировать и правильно лечить, то продолжительность жизни больных не сокращается. Отставание в росте, однако, остается постоянным симптомом.
Выводы. Синдром характеризуется: 1) аутосомно-рецессивным наследованием; 2) младенческим почечным канальцевым ацидозом, манифестирующимся гиперхлоремией и неспособностью повысить кислотность мочи до нормы; 3) низким ростом; 4) врожденной глубокой нейросенсорной глухотой.
Konigsmark описал 17-летнюю девушку и ее 20-летнего брата с почечным канальцевым ацидозом и медленно прогрессирующей лсйросенсорной глухотой, выявившимися впервые в юности. Несколько лет спустя об этих же самых сибсах сообщил Walker.
Клинические данные. Данные осмотра. Рост и физическое развитие нормальные.
Почки. У женщины сибса, обследованной Konigsmark, были выявлены почечные камни с обеих сторон, появившиеся в 12-лстпем возрасте. Согласно данным Walker, у нее наблюдались повторные приступы почечной колики, после которых выходили многочисленные маленькие камушки. Установленный при лабораторном исследовании почечный канальцевый ацидоз протекал легко.
Орган слуха. Нейросенсорная глухота прогрессировала медленно, достигла только умеренной степени и была резче выражена па высоких частотах. Walker сообщил, что дефект слуха у мальчика был впервые замечен в 5-лстием возрасте. Различение речи было нормальным. Результаты SISI-теста составляли 100%, предполагая кохлеарную локализацию глухоты.
Вестибулярная система. Результаты исследований не приведены.
Лабораторные данные. Рентгенологическое исследование выявило почечный кальциноз. Был обнаружен гиперхлоремический ацидоз. В результате нагрузки хлоридом аммония показатель рН мочи не снизился.
Наследственность. Болезнь наследуется по аутосомно-рецессивному типу.
Диагноз. На основании времени выявления заболевание должно быть отдифференцировано от детского почечного канальцевого ацидоза с врожденной нейросенсорной глухотой.
Лечение. Больным были назначены щелочные растворы и обильное питье.
Прогноз. Прогноз относительно благоприятный. Почечный канальцевый ацидоз характеризуется легким течением. Несмотря на то что он сочетается с почечным кальцинозом, лечение может быть эффективным.
Выводы. Синдром характеризуется: 1) аутосомно-рецессивным наследованием; 2) легким почечным канальцевым ацидозом, выявляющимся в юности или молодости; 3) медленно прогрессирующей нейросенсорной глухотой.
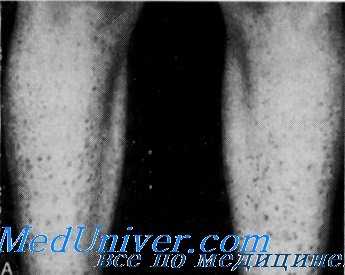
Гиперпролинурия и ихтиоз с глухотой
Goyer, Reynolds, Burke и Burkholder обнаружили у 23 членов одной семьи заболевание почек, глухоту, гиперпролниурию и ихтиоз в различных комбинациях.
Клинические данные. Данные осмотра. В возрасте 12 лет пробанд был помещен в клинику по поводу прогрессирующей утомляемости И недостаточной прибавки в весе.
Почки. У 8 больных была обнаружена нефропатия различной степени выраженности. В 13-летнем возрасте у пробанда развились альбуминурия и гематурия. Уровень мочевого азота в его крови постепенно повышался. У его 9-летнсго брата была выявлена легкая протеинурия, а у второго брата в 14-летнем возрасте появилась массивная гематурия. Внутривенная пислограмма показала большую кисту в левой почке, а в правой — маленький камушек и кортикальную кисту. Мать перенесла массивную гематурию в возрасте 22 лет. Позже у нее были обнаружены почечные камни.
Покровная система. У многих членов семьи была сухая кожа, а у 6 из 23 больных родственников пробанда отмечался коричневатый шелушащийся ихтиоз.
Лабораторные данные. Единственной рентгенологической патологией, выявленной в семье, были двусторонние почечные кисты, обнаруженные у 15-летнего брата пробанда, и, кроме того, отмечены камни почек.
У пробанда примерно в 3 раза против нормы был повышен уровень пролина в плазме крови. Уровень пролина и гидроксипролина в моче был также повышен. У его брата в крови уровень пролина был нормальным, но содержание пролина в моче было повышенным, что позволило предположить сниженную способность к реабсорбции пролина в канальцах. У 2 других членов семьи отмечалось повышение уровня пролипа в плазме.
Патология. Биопсия почки пробанда обнаружила склероз клубочков. Интерстициальпая ткань была интенсивно инфильтрирована лимфоцитами. В артериях нашли различную степень гипертрофии срединной оболочки и субэндотелиального склероза. Электрономикрофотографии выявили утолщение срединной соединительнотканной сосудистой оболочки. В межклеточном веществе — плотные отложения.
Наследственность. Родословная показывает, что в пяти поколениях семьи имеется 23 больных с различными симптомами данного синдрома. У 2 из 3 детей, родившихся от кровпо-родствеиного союза больных родителей, отмечались все компоненты синдрома, кроме глухоты. У одного из родителей была только глухота, а у другого — только заболевание почек. Имеются основания предполагать, что это заболевание, передающееся доминантным геном, характеризуется варьирующей экспрессивностью.
Диагноз. У больных с синдромом гиперпролинемии, аномалий почек и глухоты (гиперпролинемия тип I) не наблюдается ихтиоза, встречающегося при описанном ниже синдроме. Более того, это заболевание наследуется по аутосомно-рецессивному тину. При синдроме гиперпролинемии с микроскопической гематурией, описанном как аутосомно-доминантное заболевание в американской семье, не наблюдалась глухота (Perry et al.).
Ихтиоз, сходящееся косоглазие и нейросенсорная глухота были описаны у девочки, рожденной от кровно-родственного брака. В этой семье у 7 из 10 сибсов наблюдались комбинации двух или большего числа симптомов болезни. Но ни почечной патологии, ни гиперпролинурии отмечено не было (Fischman, Crystal).
Лечение. Можно использовать слуховые аппараты. Терапия почечной патологии не указана, кроме случаев уремии.
Прогноз. Данный синдром вопреки уремии не ведет к сокращению продолжительности жизни. Глухота, вероятно, медленно прогрессирует, приводя к довольно глубокой потере слуха в поздних десятилетиях жизни.
Выводы. Характеристика данного синдрома включает:
1) аутосомно-доминантное наследование с варьирующей экспрессивностью;
2) нефропатию различной степени выраженности;
3) гиперпролинурию;
4) ихтиоз и
5) медленно-прогрессирующую нейросенсорную глухоту.
Синдром Альпорта: нефрит с нейросенсорной глухотой
В 1927 г. Alport описал встречающийся преимущественно у мужчин синдром хронического нефрита с интермиттирующей или массивной гематурией, прогрессирующей. почечной недостаточностью и прогрессирующей нейросеисорной глухотой. С тех пор вышло несколько сот публикаций, посвященных синдрому Альпорта, составляющему, как считают, около 1% среди наследственной глухоты.
Клинические данные. Почки. Частым синдромом является гематурия. В течение нескольких лет она бывает выражена резко, а затем ослабевает. Гематурия может выявиться на первом году жизни («красная пеленка») в сочетании с асимптоматической альбуминурией и пиурией, но чаще она появляется в течение первого или второго десятилетия жизни. Гипертензия и почечная недостаточность встречаются главным образом у мальчиков в возрасте от 13 до 19 лет.
Больные мужчины обычно умирают до 30-летнего возраста вследствие медленно прогрессирующей уремии. У женщин заболевание почек выражено менее тяжело, но отмечается тенденция к возникновению тяжелой токсемии при беременности (Crawfurd, Toghill, Chiricosta at al.). He являются редкостью и спонтанные аборты (около 15%). Однако продолжительность жизни у больных женщин обычно нормальная.
Орган зрения. Глазная патология отмечается примерно у 10% больных с синдромом Альпорта. Наблюдаются коническая форма хрусталика, сферофакия и особенно часто кортикальные катаракты (Sohar, Perrian, Arnott, Crawfurd, Toghill, Arenberg et al., Purriel et al., Hauser).
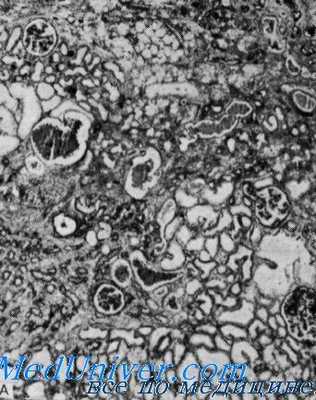
Орган слуха. Степень глухоты варьирует. Во втором десятилетии жизни обычно выявляется симметричная прогрессирующая нейросенсорная глухота в основном на средние и высокие частоты. Дефект слуха относительно легкий и помещать больных в специальные школы для глухих приходится редко (Klotz, Johnson, Hagan, Gekle et al.).
Дефект слуха чаще наблюдается у мальчиков. Он встречается также и у девочек, но в меньшей пропорции. Cassidy с соавт., изучив 7 семей с синдромом Альпорта, обнаружил глухоту примерно у 55% мужчин и у 40% женщин. Аналогичные данные сообщили Chiricosta с сотр.. Слабость восприятия низких тонов была отмечена Ferguson и Rance и Hauser.
Spear с сотр. произвели аудиометрическое исследование 16 больным из четырех семей. У детей младше 10-летнего возраста слух обычно был нормальным. Начиная со второго десятилетия жизни, выявлялась нейросенсорная глухота около 50 дБ, выраженная резче на высоких частотах. Различение речи обычно было нормальным. SISI-тест в 3 из 7 случаев был положительным, в то же время tone-decay-тест был положительным только в 1 случае из 9. Такие изменения, обнаруженные также Miller с соавт., позволяют предположить кохлеарное происхождение глухоты.
Вестибулярная система. Калорическая стимуляция вызнала сниженную реакцию (Miller et al., Celes-Blaubach et al.).
Лабораторные данные. У больных с тяжелым течением почечного заболевания при помощи внутривенной пиелографии была обнаружена атрофия некоторых участков почек.
Анализ мочи показал различную степень гематурии, протеинурии и пиурии. В моче могут быть обнаружены клеточные слепки, содержащие красные кровяные шарики. Параллельно тяжести заболевания повышается уровень мочевого азота в крови.
Почечный тубулярный ацидоз ( Ацидоз почечных канальцев , Почечный канальцевый ацидоз )
Почечный тубулярный ацидоз – это разновидность тубулопатии, которая сопровождается дисбалансом электролитов из-за сбоя экскреции ионов водорода (I), нарушением обратного всасывания бикарбонатов (II), аномальной продукцией альдостерона или взаимодействия с ним (IV). Тип III встречается казуистически редко. Течение может быть бессимптомным или с проявлениями электролитных расстройств: слабостью, тошнотой, костными деформациями. Диагностика почечной ацидемии базируется на измерениях рН мочи и электролитов, данных генетических анализов, УЗИ. Лечение предполагает восстановление кислотно-щелочного баланса: ощелачивающие средства, устранение гипо- гиперкалиемии и пр.
МКБ-10
N25.8 Другие нарушения, обусловленные дисфункцией почечных канальцев. Почечно-канальцевый ацидоз БДУ.
Общие сведения
Почечный канальцевый ацидоз (RTA, тубулярный, трубчатый, ацидоз почечных канальцев) определяется нефрологами как синдром, при котором нарушается выведение ионов водорода или реабсорбция фильтрованного бикарбоната, что приводит к хроническому метаболическому нарушению гомеостаза с гипохлоремией. Наиболее распространенным вариантом является генерализованный тип заболевания, второе место занимает поражение дистальных канальцев нефронов, форма с патологией проксимальных канальцев встречается реже. Точный уровень заболеваемости отследить сложно, поскольку латентные формы часто не распознаются. Наследственные почечные тубулопатии встречаются реже приобретенных.
Причины
Основная причина патологии – утрата способности почек подкислять мочу. Существует множество состояний, которые вызывают ацидоз почечных канальцев, при одних заболеваниях поражаются дистальные отделы, при других – проксимальные, иногда патология носит сочетанный характер или вызывается рядом нарушений, связанных с альдостероном. Характерными причинами для каждого типа являются:
- Первый (классический). При врожденных состояниях кислотно-щелочное равновесие изменяют мутации генов ATP6V1B1, ATP6V0A4 S1C4A, что инициирует дистальный почечный тубулярный ацидоз (синдром или болезнь Батлера-Олбрайта). Вторичная форма становится результатом приема лекарств, трансплантации почек и некоторых заболеваний (серповидноклеточной анемии, системной красной волчанки, синдрома Шегрена). Медуллярная губчатая почка, цирроз и хроническая обструктивная уропатия также рассматриваются как потенциальные триггеры.
- Второй (проксимальный). Проксимальный почечный канальцевый ацидоз сопровождает синдром Фанкони, нефропатию на фоне множественной миеломы, как осложнение развивается при длительном приеме ацетазоламида, сульфонамидов, ифосфамида, тетрациклина. Реже имеет другую этиологию: дефицит витамина Д, хроническую гипокальциемию с вторичным гиперпаратиреозом, ряд наследственных патологий: непереносимость фруктозы, окулоцереброренальный синдром, цистиноз.
- Четвертый (обобщенный, генерализованный). Почечный тубулярный ацидоз обусловлен дефицитом альдостерона или нарушением его взаимодействия с рецепторами. Как первопричину нарушения рассматривают прием калийсохраняющих диуретиков, циклоспорина, гепарина, других препаратов. Из нефрологических заболеваний тубулопатию провоцируют ХБП, хронический интерстициальный нефрит, ВИЧ-инфекция с поражением почек, в урологии – все ситуации, связанные с обструкцией мочевыводящих путей. Тубулярный почечный ацидоз (IV) вызывает аутоиммунная дисфункция (СКВ), серповидноклеточная анемия, диабет.
К утяжеляющим факторам относят диарею, отравление солями тяжелых металлов, оперативные вмешательства на органах ЖКТ с установлением дренажей, которые выводят основания вместе с жидким содержимым или секретом (например, поджелудочной железы). Длительная диарея и увеличение объема стула приводят к дополнительной потере бикарбонатов. Прием некоторых лекарственных препаратов – кальция хлорида, магния сульфата, холестирамина нарушают гомеостаз, в сочетании с почечной патологией усугубляют состояние.
Патогенез
При первом типе имеет место сбой секреции или всасывания ионов водорода в дистальных канальцах, что вызывает высокую кислотность крови с гиперхлоремией. При этом нарушается способность почек поддерживать нормальный градиент концентрации водородных ионов между кровью и тубулярной жидкостью. Гипотетически это может быть связано с уменьшением активности клеток, секретирующих ионы водорода, дефектом энергетического механизма, дефицитом систем транспортировки, патологическими изменениями А и Р-клеток нефрона, ответственных за выработку Н+. Гиперкальциурия, сниженная экскреция цитратов приводит к нефролитиазу.
При II типе присутствует дисфункция проксимальных трубочек с нарушением процессов реабсорбции бикарбонатов, НСО3-, но ацидогенетическая функция дистальных структур нефрона остается сохранной. Патогенез проксимального тубулярного ацидоза объясняют дефицитом или полным отсутствием карбоангидразы И-(С), снижением активности карбоангидразы 1-(В) крови и/или митохондриальной НСО3-АТФ-азы в мембранах эпителия проксимальных канальцев. Механизмы развития остеомаляции и остеопении (в том числе, рахита у детей) включают гиперкальциурию, гиперфосфатурию, дисметаболизм витамина Д.
Тип IV инициирует дефицит альдостерона или невосприимчивость к нему дистальных канальцев. Характерен дисбаланс электролитов: натрия, калия, хлора и гидрокарбоната. Гипокалиемия способствует уменьшению секреции аммиака, приводя к метаболической ацидемии. Это расстройство – наиболее распространенный вариант почечной дистальной тубулопатии – имеет вторичный или спорадический характер по отношению к нарушению оси ренин-альдостерон-ренальной системы.
Классификация
- Первый (тубулярный классический, дистальный). Заболевание вызвано уменьшением выработки молекул водорода дистальными канальцами, повышением экскреции HCO3- . Часто сопровождается нефрокальцинозом, камнеобразованием. Выделяют врожденный (аутосомно-доминантный, аутосомно-рецессивный с тугоухостью, аутосомно-рецессивный без нарушения слуха) и приобретенный варианты.
- Второй (тубулярный проксимальный). Возникает из-за патологии проксимального отдела почечных трубок. Ацидемия менее выражена из-за сохранности дистальных интеркалированных клеток, вырабатывающих кислоту. Основной особенностью синдрома является деминерализация костной ткани (остеомаляция, рахит) из-за истощения фосфатного обмена. Различают аутосомно-рецессивный, аутосомно-доминантный, спорадический (транзиторный детский и персистирующий взрослый) почечный ацидоз.
- Третий (тубулярный ювенильный). Представляет собой сочетание классического и второго типа почечной ацидемии. Аутосомно-рецессивная патология, связанная с генетической мутацией. Регистрируется крайне редко. Тубулопатию поддерживает унаследованный дефицит углекислой ангидразы 2.
- Четвертый (тубулярный гиперкалиемический). Вызван дефицитом или резистентностью к альдостерону. Сопутствующая гиперкалиемия приводит к патологическим процессам в миокарде. Почечная функция не нарушена или страдает незначительно. Состояние встречается преимущественно у взрослых, в большинстве наблюдений имеет вторичный характер.
Симптомы
Симптомы вариативны, связаны с причинными факторами. При первичном генетически обусловленном дистальном почечном тубулярном ацидозе до появления развернутых клинических проявлений у детей обнаруживается бледность кожных покровов, мышечная гипотония, полиурия и, как следствие, полидипсия. В ряде случаев наследственную патологию сопровождает тугоухость, глазные аномалии. Температура тела незначительно повышена, есть склонность к запорам. Иногда единственным симптомом становится отставание в физическом развитии от сверстников.
В тяжелых случаях без лечения примерно по достижении 2-летнего возраста происходит деформация костей скелета. К 3-4 годам, если патология не диагностирована, и ребенок не получает лечения, прогрессирует задержка роста и интеллектуальных способностей. Кости нижних конечностей Х-образно искривляются, грудная клетка приобретает бочкообразную форму, голова выглядит непропорционально большой. Патологические переломы, костные боли, снижение мышечного тонуса свидетельствуют о прогрессировании патологического процесса.
Реакциями со стороны нервной системы при длительно существующих обменных нарушениях являются раздражительность, агрессивность. Типично раннее наступление пубертата. На фоне развившегося нефрокальциноза присоединяются рецидивирующие инфекции мочевыводящих путей, которые проявляются резями при учащенном мочеиспускании, поясничными болями, повышением температуры. Нефролитиаз и самостоятельное отхождение конкрементов сопровождаются почечной коликой, тошнотой, рвотой.
Проксимальный тип тубулярного почечного ацидоза имеет схожую симптоматику, но рахитоподобные изменения развиваются раньше, а нефрокальциноз выявляется значительно реже. Для второго варианта тубулопатии характерно более тяжелое течение, без лечения патология может прогрессировать до ацидемической комы. Типичен эксикоз (обезвоживание) с обмороками, резким цианозом, одышкой. Изменению подвергается волосяной покров: волосы колючие, жесткие, при соответствующей терапии становятся мягче. Иногда патологическое состояние проходит самостоятельно без какого-либо лечения к 3-10 годам.
У взрослых электролитные расстройства протекают бессимптомно или порождают более мягкую симптоматику. Гипокалиемия вызывает мышечную слабость, снижение рефлексов, параличи. Проявления со стороны опорно-двигательного аппарата включают остеомаляцию, боли. Подобные признаки более типичны для проксимального почечного ацидоза, но могут определяться и при дисфункции дистальных канальцев. Четвертый тип чаще бессимптомный, диагностируется при выявлении незначительной ацидемии. Существует риск нарушений сердечного ритма, особенно если состояние сопровождается выраженной гиперкалиемией.
Осложнения
Без надлежащего лечения хроническая кислотность крови осложняется задержкой роста, нефролитиазом, переломами. У пациентов наблюдают рецидивирующий пиелонефрит, нефрокальциноз, постепенную утрату почечной функции вплоть до терминальной стадии ХПН. Основным последствием дистальной тубулопатии является гипокалиемия, которая сопровождается аритмией, нередко становящейся причиной летального исхода. У детей отсутствие терапии ведет к тяжелым деформациям костей, задержке психического развития. Выраженная гиперкалиемия (IV) осложняется перепадами АД, бронхоспазмом, кишечными коликами.
Диагностика
Ведущая роль в диагностике принадлежит лабораторным анализам. Инструментальные исследования назначаются для определения основной патологии при вторичном почечном тубулярном ацидозе. При наличии в роду наследственных заболеваний необходима консультация генетика, при признаках обструктивной нефропатии требуется осмотр уролога. У детей с наследственными формами, сопровождающимися врожденной глухотой, в обследовании принимает участие сурдолог. Алгоритм диагностики индивидуален, результаты анализов и инструментальных методов вариативны.
Инструментальная диагностика включает УЗИ почек и мочевого пузыря, КТ и МРТ для исключения обструктивных уропатий, нефролитиаза. При тубулярном ацидозе (I) на сонограммах всегда определяется патология почек, для проксимального типа она нехарактерна. На рентгенограммах у детей визуализируются рахитические изменения скелета, поражение трубчатых костей голеней, кистей. Минеральную плотность костной ткани определяют с помощью денситометрии. Деминерализация костей без явного рахита или остеомаляции сопутствует 1 и 4 типам, витамин Д-ассоциированный рахит, остеомаляция характерны для 2 варианта.
Дифференциальную диагностику проводят между возможными типами нарушения кислотно-щелочного равновесия, почечными патологиями иного генеза. Синдром Фанкони имеет схожие проявления с проксимальным и дистальным тубулярным ацидозом. Генетически обусловленные формы дистального почечного ацидоза различают с псевдогипоальдостеронизмом, первичной гипероксалатурией, нефрокальцинозом иного генеза. Иногда требуется дифференциация с почечной недостаточностью, сахарным диабетом, салицилатной нефропатией.
Лечение почечного тубулярного ацидоза
Лечение направлено на коррекцию дисбаланса электролитов (калия, кальция, фосфатов), предотвращение осложнений, коррелирует с типом патологии. Операции выполняют только для исправления тяжелых деформаций костей. Основным мероприятием остается назначение ощелачивающей терапии, без лечения при тубулярном почечном ацидозе у детей замедляется рост, страдает умственное развитие. Щелочные агенты помогают достичь относительно нормальной концентрации бикарбонатов плазмы, скорректировать нарушения.
Медикаментозная терапия
Лечение классического почечного тубулярного ацидоза необходимо для поддержки роста у детей, ликвидации костных изменений, профилактики накопления интерстициальной тканью кальция. Назначают бикарбонат натрия, калия-натрия гидроген цитрат. При остеопатии используют активные метаболиты витамина Д3, что требует осторожности из-за риска развития гиперкальциурии. Терапия проксимального варианта включает пероральное введение цитрата калия. При резистентности в схему добавляют гидрохлортиазид. При ювенильном варианте применяют цитрат натрия или цитрат калия. Дозы подбирают индивидуально.
Основное лечение четвертого варианта направлено на восполнение или замещение альдостерона. Пациентам рекомендуют исключить из питания продукты, богатые калием. Дополнительно вводят NaCl на фоне мониторинга К крови, альдостерона, ренина плазмы. Необходимо прекратить прием лекарств, поддерживающих гиперкалиемию, осуществлять лечение основной патологии (ЗГТ минералокортикоидами). Назначают диуретики (фуросемид, гидрохлортиазид). При отсутствии ответа на лечение используют флудрокортизон, бикарбонат натрия для компенсации ацидемии и усиления экскреции К.
Прогноз и профилактика
Своевременно начатое лечение улучшает исход для всех случаев. При дистальной тубулопатии прием препаратов до присоединения нефрокальциноза благоприятно влияет на прогноз. Проксимальная транзиторная форма с возрастом может купироваться самостоятельно, но подщелачивающая терапия обеспечивает нормальный рост и препятствует деминерализации, рахиту, у взрослых лечение предотвращает развитие остеомаляции. Прогноз почечного ацидоза (IV) определяется тяжестью основного заболевания. За пациентами наблюдают, контролируют электролиты крови каждые 12 недель, проводят УЗИ почек 1 раз в 6 месяцев.
Меры профилактики для врожденной патологии отсутствуют, при отягощенном семейном анамнезе показана консультация генетика. Важно своевременное лечение урологических и нефрологических заболеваний, которые могут приводить к тяжелым формам тубулопатий. Особого наблюдения требуют пациенты с почечным канальцевым ацидозом на фоне уремии (почечная недостаточность с нарушением скорости клубочковой фильтрации, высоким уровнем креатинина, мочевины). Больные, получающие адекватное лечение, обычно не имеют выраженных симптомов, многие из них могут вести нормальную жизнь.
1. Федеральные клинические рекомендации по оказанию медицинской помощи детям с тубулопатиями/ Баранов А.А. – 2015.
2. Ренальный тубулярный ацидоз (обзор литературы)/ Вашурина Т.В., Сергеева Т.В.// Нефрология и диализ – 2003 - №2.
Читайте также:
- Метастатическое поражение височной кости - лучевая диагностика
- Резистентность возбудителя. Роль изменчивости микроорганизмов в эпидемиологии.
- Лютеиновая фаза менструального цикла. Регуляция
- Послеоперационные осложнения у больных ожирением в гинекологии
- Осложнения в послеоперационном периоде сочетанной травмы.