Причины апластической анемии - конституционная апластическая анемия Фанкони
Добавил пользователь Skiper Обновлено: 31.01.2026
Химическая и лекарственная апластическая анемия - лекарства поражающие костный мозг
Апластическая анемия, обусловливаемая химическими и физическими агентами характеризуется прямым отношением количества воздействующего на костный мозг токсического вещества и интенсивности развиваемой им гипоплазии (Мунтяну, Rohr); наличием — в клинической картине — отдельных признаков поражения участков паренхимы органов большого значения, таких как печень, почки, центральная нервная система, что реже наблюдается при остальных видах апластической анемии.
К токсическим веществам, оказывающим большое влияние на костный мозг и очень хорошо изученным, относятся бензол, хлорамфеникол и ряд других медикаментов.
Ниже приведены вещества, определяющие гипоплазию костного мозга и оценка представляющего ими риска.
Физические и химические факторы, обусловливающие развитие апластической анемии:
А. Факторы, наличие которых в определенном количестве, в любом случае способствуют развитию аплазии костного мозга, в том числе, ионизирующие излучения, соединения иприта (азотный иприт и пр.), цитостатические медикаменты (антиметаболиты, антифолиевые препараты), бензол, его соединения.
Б. Факторы, вызывающие лишь случайно костномозговую аплазию, в том числе:
1. Противомикробные:
Мышьякобензолы
Хлорамфеникол
Сульфамид
Стрептомицин
2. Противосудорожные:
Мезантоин
Триметадион
3. Антитиреоидные: Метилтиоурацил
4. Антигистаминные: Фенерган
5. Инсектициды: ДДТ
6. Различные факторы:
Соли золота
Фенилбутазон
Нитрофенол
Мепробамат
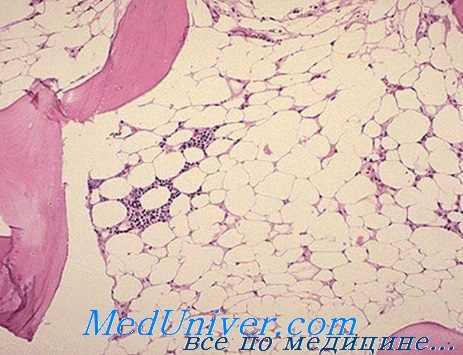
Бензол и его соединения как причина апластической анемии. Несчастные случаи, вызываемые бензолом, начали обнаруживаться в начале нашего века, в связи с промышленным применением этого вещества в качестве органического растворителя (Robr). В принципе бензол угнетает костный мозг, что означает приостановление синтеза РНК в уже дифференцированных элементах.
Видимо бензол не угнетает колонии клеток-штамм, в чем следует искать объяснение полной обратимости нарушений в случаях непродолжитекьности токсического воздействия (Nomiyama).
Влияние на костный мозг тесно связано с количеством воздействующего на больного вещества. Исходно отмечается раздражение костного мозга и общая, в частности эритроидная, гиперплазия, сопровождающаяся лейкоцитозом, нейтрофилией и отклонением влево в периферической крови; затем, постепенно, развивается костномозговая гипоплазия с обширными участками жирного преобразования, наличием редких миелоидных элементов, выраженной реакцией лимфатической или плазмоцитной ткани, а в отдельных случаях моноцитной реакцией, которую часто нелегко различить от лейкемического поцесса (Мунтяну).
После развития костномозговой гипоплазии имеются лишь немногие шансы на его восстановление. Вот почему внимание направлено на предупреждение заболевания путем ограничения концентрации бензола в атмосферном воздухе соответствующих заводов, максимально до 34 частей на 1 млн. и выявление первых признаков поражения костного мозга с помощью наблюдения за развитием эритроцитного макроцитоза у рабочих данных предприятий (Мунтяну).
Однако бензол оказывает и ряд других влияний на костный мозг, в том числе обусловливает: выраженную гиперплазию эритроидного ряда и приостановление деления (мегалобластоз), картину, называемую бензоловой эритропатией, имеющей большое сходство с острой эритемией; хронический миелосклероз с миелоидной метаплазией; гипоплазию лимфатической тнани, равно как и острую лейкемию (Rohr, Wintrobe).
Хлорамфеникол это антибиотик, вызвавший большой энтузиазм в 1948 г. когда был проверен его чрезвычайный эффект при заболеваниях брюшным тифом, а затем и отдельными риккетсиозами. Но в дальнейшем широкое применение препарата в борьбе с инфекцией грамм-отрицательными возбудителями начало выявлять его вредные последствия, поскольку он способствует развитию весьма тяжелой необратимой костномозговой аплазии.
В клинике риск развития аплазии после лечения хлорамфениколом составляет 1/10 тыс.—1/20 тыс. Однако по иным статистическим данным, например Объединения американских врачей, хлорамфеникол составил причину развития 44% всех заболеваний панцитопенией в США (Williams).
Формула хлорамфеникола включает бензоловое ядро с боковой цепью дихлорацетамида (Мунтяну). В связи с этим создалось мнение о том, что токсический эффект следует отнести за счет бензолового ядра. За справедливость такого предположения говорит и тот факт, что, подобно бензолу, медикамент поражает исходно эритроидный ряд, приостанавливая созревание, блокируя включение железа в гем с последующим ростом показателей сывороточного железа, костномозговых сидеробластов и развитием ретикулоцитопении.
Дальнейшие исследования выявили одновременное по существу поражение всех рядов крови, при этом первым морфологическим признаком поражения костного мозга оказалось появление вакуолей в костномозговых клетках, которые лучше выражены в эритробластах (Мунтяну, Ward).
Эти данные совпадают с результатами экспериментальных наблюдений, по которым медикамент угнетает синтез белков в клетках костного мозга в результате соревнования с ARNm (Ward), а также и синтез РНК (Williams).
В дальнейшем были обнаружены довольно тяжелые осложнения спустя некоторое время после прекращения лечения этим медикаментом. В связи с этим была выдвинута гипотеза о возможных, вызываемых им изменениях в геноме клеток-штамм, результатом чего являются необратимые сдвиги механизма деления последних, развитие необратимой костномозговой аплазии и острой лейкемии (Mukherji) или ночной пароксизмальной гемоглобинурии (Дачие и Левис).
В клинической практике целесообразно помнить, что этот медикамент вызывает тяжелые, смертельные аплазии и его применение следует ограничивать заболеваниями, вызываемыми строго чувствительными к нему возбудителями.
Прочие химические агенты, приводящие к апластической анемии. К категории иных химических факторов, обусловливающих костномозговую аплазию относятся ДДТ, сульфонамиды, противосудорожные средства, равно как и соединения золота. Также доказано, что атебрин создает чрезмерную чувствительность и тем самым способствует развитию костномозговой аплазии.
Ночная пароксизмальная гемоглобинурия как последствие апластической анемии. По результатам ряда наблюдений ночная пароксизмальная гемоглобинурия развивается после костномогзовой аплазии (Dacie и Lewis). Описаны также 2 случая костномозговой аплазии, сопровождающейся ночной пароксизмальной гемоглобинурией, а в конечном итоге — острой миелобластической лейкемией («трехзтапная» эволюция) (Holden и Lichtman).
Утверждается, что, по началу, токсический агент поражает (вызывает мутацию) клетки-штамм (Dacie и Lewis). Выживающие при этом клетки-штамм порождают аномальные популяции: эритроцитов — чувствительных к комплементу, гранулоцитов — лишенных лейкоцитной щелочной фосфатазы и тромбоцитов с недостатком ферментов.
Апластическая анемия отмечена и при беременности (Evans), гипопитуитаризме, почечной недостаточности и пр.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Анемия Фанкони ( Врожденная панмиелопатия Фанкони , Наследственная панмиелопатия )
Анемия Фанкони – это генетическое заболевание, которое передается по аутосомно-рецессивному типу и характеризуется нарушением кроветворения, формированием злокачественных новообразований, пороками развития, ломкостью хромосом. Проявляется частыми кровотечениями, кровоподтеками на коже, вялостью, бледностью, склонностью к инфекциям. Диагностика проводится лабораторными методами, назначаются цитогенетическое, молекулярно-генетическое и клиническое исследования крови, миелограмма. Основные способы лечения – пересадка костного мозга, медикаментозное поддержание кроветворения, переливание крови.
МКБ-10
Общие сведения
Синонимичные названия анемии Фанкони – врожденная панмиелопатия Фанкони, наследственная панмиелопатия. Заболевание названо по фамилии швейцарского педиатра Гвидо Фанкони, который в 1927 году описал врожденную апластическую патологию на основе симптомов у трех братьев. Анемия Фанкони является редкой генетической болезнью, наследуется согласно аутосомно-рецессивному принципу. Эпидемиологические показатели низкие – 1 больной ребенок на 350 тысяч новорожденных. Распространенность одинакова среди представителей женского и мужского пола, выше в сообществах с разрешенными близкородственными браками, например, у некоторых южноафриканских народов.
Причины
Заболевание является наследственным, развивается при передаче дефектного гена от родителей к ребенку. Выявлено 15 генов, мутации которых проявляются анемией Фанкони. Из них 14 расположены в аутосомах и являются рецессивными, 1 тип гена находится в X-хромосоме (сцепленной с полом). Все эти гены отвечают за производство определенного фермента, участвующего в репарации ДНК.
Аутосомно-рецессивное наследование подразумевает, что и отец, и мать должны быть носителями патологической генетической информации. При этом сами они, как правило, здоровы. Вероятность рождения больного ребенка в такой паре составляет 25%. Генетическая панмиелопатия диагностируется у детей и взрослых, получивших от каждого из родителей один и тот же измененный ген. В крайне редких случаях анемия провоцируется передачей дефектной Х-сцепленной хромосомы. Женщины могут быть носительницами мутации, заболевание проявляется только у мальчиков. Риск развития патологии у сына при наличии у матери мутированного гена – 50%.
Патогенез
В норме в клетках организма существуют специальные ферментные системы, которые исправляют разрывы молекул ДНК, поврежденных в процессе биосинтеза или воздействия химических, физических реагентов. При анемии Фанкони обнаруживается генетический дефект в кластере белков, ответственных за репарацию ДНК, что приводит к повышенной ломкости хромосом. В итоге у пациентов развиваются нарушения функций костного мозга – неоплазии и апластическая анемия. Онкологические заболевания чаще всего представлены острым миелоидным лейкозом – злокачественной опухолью миелоидного ростка крови, провоцирующей накопление измененных белых клеток, подавляющих рост эритроцитов, тромбоцитов и нормальных лейкоцитов. При апластической анемии в результате дисплазии костного мозга резко угнетается рост и созревание всех трех видов клеток крови.
Симптомы анемии Фанкони
Более чем у половины пациентов наблюдаются врожденные аномалии развития внутренних органов и скелета. Костные деформации проявляются специфическим внешним видом: больные низкорослые, с уменьшенным размером головы, отсутствием или заметным укорочением большого пальца на руках, недоразвитием лучевой кости, врожденным вывихом бедра и/или наличием шейного ребра, косолапостью, недоразвитым подбородком («птичьим лицом»). Характерна гиперпигментация кожи в виде светлых и коричневатых пятен.
Неврологические расстройства представлены косоглазием, недоразвитием одного или двух глаз, опущением верхнего века, глазным дрожанием, глухотой, умственной отсталостью. Больные зачастую имеют незрелые половые органы, у них отсутствует одно или оба яичка. К распространенным аномалиям строения органов относятся пороки мочевыделительной системы: удвоение мочеточников или лоханки, подковообразные почки, почечные кисты, смещенное наружное отверстие уретры (гипоспадия). Врожденные пороки сердца включают атрезию трехстворчатого клапана, дефект межпредсердной перегородки, митральный стеноз, дефект межжелудочковой перегородки. Пациенты страдают от почечной и сердечной недостаточности.
Ключевые симптомы связаны с постепенным нарастанием нарушений в работе костного мозга. Чаще они дебютируют в детском возрасте (в 5-10 лет). Из-за снижения количества тромбоцитов развивается повышенная кровоточивость: при ранениях кровь долго не сворачивается, легко возникают носовые кровотечения, выделения при менструациях обильны, на теле обнаруживается много «беспричинных» кровоподтеков. Уменьшение числа эритроцитов проявляется анемией с характерной слабостью, быстрой утомляемостью, головокружениями, обмороками, бледностью кожи, учащенным сердцебиением и одышкой. Недостаток лейкоцитов способствует ухудшению сопротивляемости инфекциям. Впоследствии формируется лейкоз, миелодиспластический синдром, онкологические болезни.
Осложнения
Наиболее распространенным осложнением считаются частые инфекционные заболевания. У пациентов развивается ОРВИ, ангина, ринит, бронхит, грипп, тиф, герпес. Рецидивирующий характер болезней и их тяжелое течение приводят к деструкции органов, сопровождаются риском сепсиса. Другим осложнением наследственной анемии являются злокачественные новообразования – лейкемия, эпителиальные опухоли органов шеи и головы, половых органов. Рак у таких больных тяжело поддается лечению из-за повышенной ломкости и сниженной репарации ДНК. Это явление ограничивает применение лучевой терапии, цитотоксических препаратов. Нарушение свертываемости становится причиной больших кровопотерь.
Диагностика
Обследование больных проводят онкологи, гематологи, педиатры, врачи-генетики. Диагностика начинается с анализа анамнестических данных и жалоб. Врач выясняет, имеется ли данное наследственное заболевание у близких родственников, уточняет время появления первых признаков болезни, ранние обращения к врачам. При осмотре оценивает общее состояние пациента, выявляет наличие аномалий развития, гиперпигментированных пятен, кровотечений, кровоподтеков. В большинстве случаев не составляет труда обнаружить типичные деформации костей, недоразвитие глаз. Для подтверждения диагноза и различения анемии Фанкони с приобретенной анапластической анемией проводится ряд лабораторных исследований:
- Клинический анализ крови. Характерны изменения клеточного состава крови. На ранних этапах нарушения кроветворения диагностируется тромбоцитопения и лейкопения, на более поздних – панцитопения (резкое снижение объема эритроцитов, лейкоцитов и тромбоцитов). Возможен умеренный гемолиз без гипербилирубинемии, но с ретикулоцитозом. Значение СОЭ увеличено до 60-80 мм/ч.
- Цитогенетическое исследование клеток. Выполняется проба с диэпоксибутаном, митомицином C, указывающая на частоту и спектр хромосомных аберраций. В пользу генетической анемии рассматриваются показатели ДЭБ-теста более 45%, пограничный уровень – 11-45% (процент клеток с хромосомными разрывами).
- Молекулярно-генетический анализ. Исследуются гены, мутации в которых могут привести к развитию заболевания. В 60-70% случаев мутации обнаруживаются в паре генов FANCA, в 14% – в аллели FANCC, в 10% – в генах FANCG. Частота мутаций в других парах – 0,2-3%.
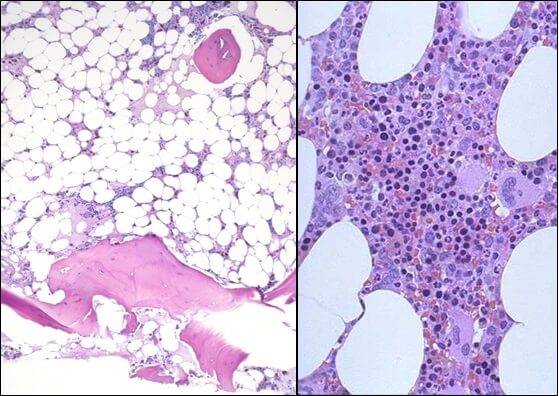
- Миелограмма. По данным исследования определяется увеличение количества плазматических клеток и макрофагов, фагоцитирующих жиры. Содержание недифференцированных клеток – в пределах нормы. Снижена концентрация клеток миелоцитарного ростка, увеличен показатель лимфоцитов.
Лечение анемии Фанкони
Основная терапия направлена на восстановление процесса кроветворения. Методы лечения подбираются индивидуально, зависят от тяжести заболевания, возраста пациента, наличия и выраженности врожденных аномалий. Дополнительно проводится лечение инфекций и онкопатологий, осуществляются реабилитационные мероприятия. Для устранения анемии используются следующие методы:
- ТКМ.Трансплантация костного мозга является наиболее эффективной в долгосрочной перспективе, но имеет противопоказания, нередко сопровождается развитием осложнений. Оптимальный возраст для проведения операции – до десяти лет. Донорами могут выступать здоровые сестра и братья, подходящие по критериям совместимости. Предварительная интенсивная терапия (кондиционирование) связана с риском токсического воздействия на органы. После трансплантации сохраняется высокая вероятность острого или хронического иммунного конфликта между клетками донора и реципиента.
- Медикаментозная стимуляция кроветворения. При невозможности проведения трансплантации пациентам показано консервативное лечение, временно улучшающее их состояние. Выработка кровяных клеток стимулируется андрогенами (мужскими половыми гормонами) и гематопоэтическими факторами роста – эритропоэтином, фактором стволовых клеток, интерлейкинами-1-12. Параллельно применяются иммунодепрессанты. Медикаментозная терапия способна на протяжении многих лет поддерживать высокое качество жизни больных, но ее эффективность постепенно снижается.
- Переливание компонентов крови. При выраженных побочных эффектах или противопоказаниях к этиотропной терапии (трансплантации, стимуляции кроветворения) назначаются процедуры гемотрансфузии. Переливаются отмытые эритроциты – донорские красные кровяные тельца, освобожденные от поверхностных белков. При кровотечениях и снижении уровня тромбоцитов пациентам вводится тромбоцитарная масса.
Прогноз и профилактика
Продолжительность жизни больных определяется степенью нарушения функции костного мозга. Иногда пациенты доживают до 40 лет без лечения, но нередко умирают в детстве от тяжелой анемии или онкологических заболеваний. Прогноз наиболее благоприятен при своевременном проведении аллогенной трансплантации костного мозга, после которой есть шанс полного восстановления нормального кроветворения и увеличения срока жизни. Поскольку заболевание генетическое, предотвратить его развитие невозможно. Профилактика сводится к медико-генетической консультации супружеских пар из групп риска, планирующих беременность, а также к проведению пренатальной диагностики патологии, в ходе которой из пуповинной вены плода производится забор крови и выполняется ДЭБ-тест. При его положительном результате рассматривается вопрос о прерывании беременности.
1. Генетическая диагностика анемии Фанкони. Обзор литературы/ Панферова А.В. Тимофеева Н.М. Ольшанская Ю.В.// Онкогематология. - 2016.
3. Свободнорадикальный статус клеток крови и костного мозга у детей с острыми лейкозами и анемией Фанкони: Автореферат диссертации/ Чивилева И.Ю. - 1996.v
Апластическая анемия ( Гипопластическая анемия )
Апластическая анемия – угнетение функции кроветворения красного костного мозга (эритроцитопоэза, лейкопоэза и тромбоцитопоэза), приводящее к пангемоцитопении. К основным клиническим проявлениям гематологического синдрома принадлежат головокружение, слабость, обмороки, одышка, покалывание в груди, кожные геморрагии, кровотечения, склонность к развитию инфекционно-воспалительных и гнойных процессов. Заболевание диагностируется на основании характерных изменений гемограммы, миелограммы и гистологического исследования трепанобиоптата. Лечение патологии включает проведение гемотрансфузий, иммуносупрессивной терапии, миелотрансплантации.
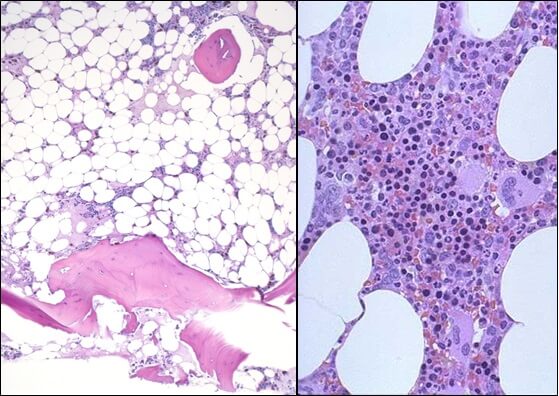
Апластическая (гипопластическая) анемия – тяжелое расстройство гемопоэза (чаще всех его звеньев), сопровождающееся развитием анемического, геморрагического синдромов и инфекционных осложнений. Развивается в среднем у 2 человек на 1 млн. населения в год. Приблизительно с одинаковой частотой патология поражает мужчин и женщин. Возрастные пики заболеваемости приходятся на возраст 10–25 и старше 50 лет. При данной патологии в костном мозге чаще нарушается образование всех трех типов клеточных элементов крови (эритроцитов, лейкоцитов и тромбоцитов), иногда - только одних эритроцитов; в зависимости от этого различают истинную и парциальную апластическую анемию. В гематологии данный вид анемии относится к числу потенциально фатальных заболеваний, приводящих к гибели 2/3 заболевших.
По происхождению апластическая анемия может быть врожденной (связанной с хромосомными аберрациями) и приобретенной (развившейся в течение жизни). Принято считать, что угнетение миелопоэза связано с появлением в красном костном мозге и крови цитотоксических T-лимфоцитов, производящих фактор некроза опухолей и γ-интерферон, которые в свою очередь подавляют ростки кроветворения. Запускать этот механизм могут различные внешнесредовые (химические соединения, физические явления, лекарственные вещества), а также эндогенные факторы (вирусы, аутоиммунные реакции). К числу наиболее значимых причин относят:
- Прием миелотоксических препаратов. Достоверно установлена связь анемии с приемом некоторых противоопухолевых, противосудорожных, антибактериальных, антитиреоидных, противомалярийных препаратов, транквилизаторов, препаратов золота и др., обладающих потенциальным миелотоксическим эффектом. Лекарственные вещества могут вызывать как прямое повреждение стволовых кроветворных клеток, так и опосредованное - через аутоиммунные реакции. Анемии, связанные с таким механизмом развития, называются лекарственными.
- Контакт с химическими и физическими агентами. Супрессию костного мозга может вызывать взаимодействие с органическими растворителями, соединениями мышьяка, бензольными соединениями, пестицидами, облучение всего тела. В некоторых случаях недостаточность гемопоэза является временной и обратимой - главными факторами здесь являются концентрация/доза вещества и время контакта. супрессию костного мозга.
- Вирусные инфекции. Из вирусных агентов наибольшее значение уделяется возбудителям гепатитов В, С и D. В этом случае гипопластическая анемия обычно развивается в течение полугода после перенесенного вирусного гепатита. При изучении патогенеза было замечено, что репликация вируса происходит в мононуклеарах крови и костного мозга, а также в иммунных клетках. Предполагается, что подавление миелопоэза в этом случае является своеобразным иммунным ответом, возникающим против клеток, несущих на своей поверхности вирусные антигены. Такой вид анемии выделяется в отдельную форму – постгепатитную. Среди других вирусных инфекций называются ЦМВ, инфекционный мононуклеоз, грипп.
Также описаны случаи панцитопении, вызванные инфицированием туберкулезом, интоксикацией, лучевой болезнью, лимфопролиферативными заболеваниями (тимомой, лимфомой, хроническим лимфобластным лейкозом), беременностью. Почти в половине наблюдений причину анемии выявить не удается - такие случаи относят к идиопатической форме.
В основе апластической анемии может лежать либо первичное повреждение гемопоэтических стволовых клеток, либо нарушение их эффективной дифференцировки. При наследственных анемиях недостаточность гемопоэза опосредована кариотипическими аберрациями, приводящими к нарушению репарации ДНК и невозможности репликации стволовых клеток костного мозга. В случае приобретенной анемии под влиянием этиофакторов наблюдается активация Т-клеток, которые начинают продуцировать цитокины (интерферон-гамма, ФНО), поражающие клетки-предшественники гемопоэза. В стволовых клетках костного мозга повышается экспрессия генов, отвечающих за апоптоз и активизацию клеточной гибели. Основные клинические проявления обусловлены пангемоцитопенией – снижением в составе крови всех ее форменных элементов (эритроцитов, лейкоцитов, тромбоцитов).
Классификация
Кроме различных этиологических вариантов (лекарственного, постгепатитного, идиопатического), различают острую (до 1 мес. течения), подострую (от 1 до 6 мес.) и хроническую (более 6 мес.) форму заболевания. Анемию, протекающую с избирательным угнетением эритропоэза, называют парциальной красноклеточной аплазией. На основании выраженности тромбо- и гранулоцитопении данная форма анемии подразделяется на 3 степени тяжести:
- очень тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,2х109/л)
- тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,5х109/л), по данным трепанобиопсии – низкая клеточность костного мозга (менее 30% от нормы)
- умеренную (тромбоцитов более 20,0х109/л; гранулоцитов более 0,5х109/л)
Симптомы апластической анемии
Поражение трех гемопоэтических ростков (эритро-, тромбоцито- и лейкопоэза) обусловливает развитие анемического и геморрагического синдромов, инфекционных осложнений. Дебют апластической анемии обычно происходит остро. Анемический синдром сопровождается общей слабостью и утомляемостью, бледностью кожи и видимых слизистых, шумом в ушах, головокружением, покалыванием в груди, одышкой при нагрузке.
Основным проявлением тромбоцитопении выступает геморрагический синдром. Больные отмечают появление петехий и экхимозов на коже, повышенную кровоточивость десен, спонтанные носовые кровотечения, меноррагии. Возможно возникновение гематурии, маточных и желудочно-кишечных кровотечений. Следствием лейкопении и агранулоцитоза служит частое развитие инфекционных процессов – стоматитов, пневмоний, инфекций кожи и мочевыводящих путей. Для апластической анемий нехарактерны похудание, лимфаденопатия, гепато- и спленомегалия – при этих признаках следует искать другую причину пангемоцитопении.
Врожденная апластическая анемия (синдром Фанкони) обычно развивается у детей в возрасте до 10 лет и кроме аплазии костного мозга характеризуется другими нарушениями: микроцефалией, гипоплазией почек, низкорослостью, аномалиями развития верхних конечностей (гипоплазией первой пястной и лучевой кости), гипоспадией, гиперпигментацией кожи, крайней степенью тугоухости и др. При наследственной анемии Эстрена-Дамешека отмечается тотальное поражение кроветворения и панцитопения при отсутствии врожденных аномалий развития. Для анемии Даймонда-Блекфена или парциальной красноклеточной аплазии характерно только снижение количества эритроцитов.
Летальный исход может быть обусловлен кровоизлияниями во внутренние органы, массивными кровотечениями, инфекционными осложнениями, анемической комой. Наиболее грозное из геморрагических осложнений – кровоизлияние в головной мозг (геморрагический инсульт). Больные склонны к частым и тяжело протекающим вирусным и бактериальным инфекциям респираторного тракта. Значительное или стремительное снижение уровня красных кровяных телец может привести к анемической коме. При молниеносной форме крайне быстро развиваются тяжелейшая анемия, иммунодефицит, коагулопатии, имеющие фатальные последствия.
Оценка гематологического статуса включает внимательный клинический осмотр и проведение тщательной лабораторной диагностики. При физикальном обследовании выявляется выраженная бледность или желтушность кожи, артериальная гипотония, тахикардия. Основу диагностического алгоритма составляет проведение общего и биохимического анализа крови, стернальной пункции, трепанобиопсии:
- Исследования крови. Для гемограммы при гипопластической анемии типичны эритро-, лейкоцито- и тромбоцитопения, нейтропения и относительный лимфоцитоз. Оценка биохимических показателей (печеночных проб, нефрологического комплекса, сывороточного железа, билирубина) информативна для исключения других анемий.
- Исследованиепунктата костного мозга. В миелограмме обнаруживается уменьшение количества миелокариоцитов и мегакариоцитов, снижение клеточности. В трепанобиоптате определяется замещение красного костного мозга жировым (желтым).
В рамках диагностического поиска апластическую анемию необходимо дифференцировать с мегабластными (В12-дефицитными, фолиеводефицитными) анемиями, идиопатической тромбоцитопенической пурпурой, пароксизмальной ночной гемоглобинурией, острым лейкозом.
Лечение апластической анемии
Больные с апластической анемией госпитализируются в специализированные отделения. Им обеспечиваются полная изоляция и асептические условия для предупреждения возможных инфекционных осложнений. Проведение эффективного лечения является сложной проблемой практической гематологии. В зависимости от уровня цитопении используются следующие лечебные подходы:
- Иммуносупрессиная терапия. При умеренной цитопении назначается фармакотерапия, включающая комбинацию антитимоцитарного иммуноглобулина и циклоспорина А. Поддерживающая терапия проводится анаболическими стероидами или их сочетанием с циклоспоринами.
- Гемотрансфузии. В комплексе с курсом иммуносупрессивной терапии при низких показателях красной крови показано проведение заместительной гемотрансфузионной терапии (переливание тромбоцитов и эритроцитарной массы), плазмафереза. Данная мера не оказывает воздействия на патогенетическое звено заболевания, но позволяет восполнить дефицит кровяных телец, не вырабатываемых костным мозгом.
- Трансплантация КМ и СК. Наиболее благоприятные прогнозы на долгосрочную выживаемость оказывает выполнение аллогенной трансплантации костного мозга. Однако ввиду сложности подбора иммунологически совместимого донора процедура используется ограниченно. В качестве экспериментальных подходов рассматриваются аутологичные трансплантации, пересадка стволовых клеток периферической крови. Больным с нетяжелой формой анемии может быть показано проведение спленэктомии, эндоваскулярной окклюзии селезеночной артерии.
Прогноз определяется этиологической формой, тяжестью и остротой течения анемии. Критериями неблагоприятного исхода служат быстрое прогрессирование заболевания, тяжелый геморрагический синдром и инфекционные осложнения. После трансплантации костного мозга ремиссии удается достичь у 75–90% пациентов. Первичная профилактика данной разновидности анемии предполагает исключение влияния неблагоприятных внешнесредовых факторов, необоснованного применения лекарственных препаратов, предупреждение инфекционной заболеваемости и др. Пациентам с уже развившимся заболеванием требуется диспансерное наблюдение гематолога, систематическое обследование и длительная поддерживающая терапия.
2. Комплексная программа диагностики апластической анемии с определением прогностически значимых патогенетических особенностей заболевания. Методические рекомендации. - 2015.
4. Апластическая анемия: современные представления о патогенезе и терапии/ Айсариева Б. К., Раймжанов А. Р., Айтбаев К.// Молодой ученый. - 2011 - №9.
Причины апластической анемии - конституционная апластическая анемия Фанкони
Этиология апластической анемии весьма разнообразна. Основные причины заболевания и их классификация приведены в ниже.
Этиологическая классификация апластических анемий:
I. Генуинные
II. Конституционные
III. Вызываемые физическими или химическими факторами
IV. Вирусные заболевания (гепатит), бактериальные инфекции (туберкулез).
V. Иммунологические заболевания (диссеминированная красная волчанка, аллергия и пр.).
VI. Ночная пароксизмальная гемоглобинурйя.
VII. Прочие: беременность, эндокринные болезни (Симмондса), хронический панкреатит и пр.
Не забывать, что тот же возбудитель обусловливает различные аспекты костного мозга вплоть до очень тяжелой формы аплазии.

Генуинная форма апластической анемии
К этой группе заболеваний относятся все те случаи, при которых не обнаруживается какой-либо причинный фактор (50%).
Однако, за последние десятилетия отмечается рост показателя заболеваемости этой формой болезни, что, видимо, следует отнести за счет загрязненности среды (ионизирующие излучения), питания, чрезмерного потребления медикаментов, вирусных заболеваний.
Привлекает внимание очень тяжелое течение этой формы у детей, картина носит острый характер и нередко смертельный исход наступает быстро. Гистологическое исследование выявило различную степень поражения костного мозга — от нормопластического аспекта вплоть до весьма тяжелой аплазии.
Необходимо отметить также формы с минимальной бластической реакцией в строении костного мозга или с лимфоидной реакцией, которые нередко трудно различить от острой лейкемии с небольшим процентом » или от апластической формы хронической лимфатической лейкемии (Bryon, Dreyfus и Bessis).
Впрочем в литературе описаны случаи так называемой генуинной апластической анемии, которые, по существу, оказывались «предлейкемическим состоянием» (Dreyfus и Bessis).

Конституционная или семейная генуинная анемия - апластическая анемия Фанкони
Первый случай был описан Фанкони в 1927 г. К этой патегории относятся случаи семейного характера, развивающиеся у детей с костномозговой аплазией и дефектами, как, например, синдактилия, правое сердце, стрельчатое небо, микроцефалия, умственная отсталость.
Гематологическая картина отражает наличие периферической панцитопении и нормоцитной или умеренно макроцитной анемий; показатель плодного гемоглобина бывает завышенным. При этом костный мозг представляется гипопластическим, жирным, иной раз нормо- или гипоцеллюлярным (Fanconi, Rohr Williams). Синдром видимо определяет рецессивный ген, в то же время цитогенетические исследования выявили большое разнообразие структурнохромосомных сдвигов (Bloom и сотр.). Описаны случаи, преобразовавшиеся в дальнейшем в острую лейкемию (Wintrobe).
Отмечается также другой вид семейной панцитопении, когда этому заболеванию сопутствует недостаточность поджелудочной железы по причине ее кистовидного фиброзирования (Williams).
Анемия Даймонда-Блекфена ( Наследственная парциальная красноклеточная аплазия )
Анемия Даймонда-Блекфена – это наследственная форма красноклеточной аплазии с достоверно неизученным типом наследования (предполагается аутосомно-доминантный тип наследования, встречающийся у четверти больных). Симптомами заболевания являются анемические проявления, возникающие, как правило, на протяжении первого года жизни – бледность, слабость, повышенная утомляемость, уменьшение в крови количества эритроцитов. Диагностика производится на основании данных общего анализа крови, исследования уровня эритропоэтинов, биопсии и микроскопии костного мозга, в четверти случаев информативно генетическое исследование. Лечение осуществляется с помощью гемотрансфузий, глюкокортикостероидов.
Анемия Даймонда-Блекфена (наследственная парциальная красноклеточная аплазия) представляет собой генетическое поражение системы крови, при котором нарушается образование эритроцитов. Название патологии было дано по фамилиям врачей, которые в 1938 году совместно обследовали четверых детей с симптомами сильной анемии наследственного характера.
Это состояние относится к очень редким, на сегодняшний день достоверно описано около 500 случаев. Подсчитано, что встречаемость анемии Даймонда-Блекфена составляет около 4-6:1000000, ей в одинаковой степени подвержены как мальчики, так и девочки. Несоответствие количества доказанных случаев с рассчитанной встречаемостью объясняется тем, что у определенной части больных ошибочно диагностируется либо эритромиелобластный лейкоз, либо приобретенные формы парциальной красноклеточной аплазии. Наиболее распространенная форма заболевания наследуется по аутосомно-доминантному принципу, однако это объясняет только 25% всех случаев, тогда как по поводу остальных вариантов пока данных нет.
Непосредственной причиной четверти случаев анемии Даймонда-Блекфена служит мутация в гене RPS19 расположенном в 19-й хромосоме, который кодирует важный рибосомальный белок S19. Последний входит в состав малой (40S) субъединицы рибосомы человека. Такая мутация наследуется по аутосомно-доминантному механизму со встречаемостью 6 случаев на миллион человек. В других случаях были обнаружены мутации иных генов, однако они так или иначе связаны с рибосомальными белками – это гены RPS7, RPS24, RPL5, RPL32A и ряд других.
Распространенность таких мутаций, характер их наследования, доля в общем количестве заболевших анемией Даймонда-Блекфена, их влияние на прогноз и исход патологии в настоящий момент остается объектом изучения врачей-генетиков. Также интерес представляет собой вопрос, почему мутации генов рибосомальных белков оказывают влияние именно на эритропоэз и почти не влияют на другие ростки кроветворения.
Существует несколько теорий, пытающихся объяснить торможение образования эритроцитов в красном костном мозге. Наиболее распространенные указывают в качестве возможных причин анемии Даймонда-Блекфена дефекты микроокружения клеток-предшественников эритроцитов, их внутренние аномалии, супрессию со стороны иммунной системы или гуморальные факторы, останавливающие созревание эритробластов. Ни одна из теорий на сегодняшний день не получила достоверного и однозначного подтверждения.
При этом заболевании в красном костном мозге наблюдается неуклонное снижение эритроидных единиц, причем примерно в трети случаев данный процесс начинается во время внутриутробного развития, что позволяет диагностировать анемию Даймонда-Блекфена сразу после рождения. Соответственно, начинает снижаться количество выделяемых в кровь эритроцитов, в костном мозге накапливаются эритробласты, что может вводить в заблуждение (подобные изменения характерны для лейкоза). При этом у младенцев уровень фетального гемоглобина может не снижаться, поэтому данный показатель не считается диагностическим в случае анемии Даймонда-Блекфена. Возникает компенсаторный рост уровня эритропоэтинов в крови, однако они в данном случае не способны увеличить скорость образования эритроцитов. В конечном итоге развивается выраженная анемия.
Симптомы анемии
На первый план при синдроме Даймонда-Блекфена выступают анемические симптомы – бледность, слабость ребенка, у грудных младенцев часто развивается гипотрофия, наблюдается недобор массы. Примерно у половины больных помимо нарушений со стороны крови также возникает ряд физических отклонений – микроцефалия, гипертелоризм, птоз век, микрогнатия. Возможны аномалии скелета – увеличение размера лопаток и кистей, отсутствие некоторых пальцев, задержка роста костной ткани. В некоторых случаях возможны такие нарушения как «заячья губа».
Поражаются и органы зрения – развивается косоглазие, глаукома, катаракта. Многие указанные симптомы возникают в раннем возрасте ребенка и усугубляются выраженной анемией, поэтому своевременно начатое лечение может значительно ослабить или даже устранить многие из них.
В отличие от транзиторных и приобретенных анемий, синдром Даймонда-Блекфера незначительно влияет на работу печени и селезенки – их заметное увеличение может возникать лишь на конечных стадиях заболевания или в результате осложнений гемотрансфузионной терапии.
При осмотре ребенка, больного анемией Даймонда-Блекфена, определяется бледность кожных покровов, синюшность слизистых, на голове заметны венозные сосуды. Также могут наблюдаться сопутствующие заболеванию физические отклонения (микроцефалия, гипертелоризм и другие), при взвешивании часто выявляется недобор массы тела. Методы лабораторной диагностики включают:
- Анализы крови. ОАК показывает картину нормохромной анемии, часто макроцитарного характера, резкое снижение количества ретикулоцитов. В некоторых случаях наблюдается гранулоцитопения и тромбоцитопения, однако это не может служить достоверным диагностическим критерием анемии Даймонда-Блекфена. При биохимическом исследовании крови выявляется резкое увеличение уровня эритропоэтина.
- Биопсия костного мозга. При микроскопическом исследовании биоптата костного мозга выявляется в целом нормоклеточный тип с выраженным снижением клеток эритроидного ряда. При этом при некоторых формах анемии Даймонда-Блекфена в костном мозге могут накапливаться эритробласты, что часто ведет к ошибочной диагностике острого миелолейкоза.
- Генодиагностика. Современная генетика методом прямого секвенирования последовательности определяет мутации только одного гена, ассоциированного с анемией Даймонда-Блекфена - RPS19. С помощью данного метода диагностики мутации гена обнаруживаются только в 25-30% случаев клинически выявленного заболевания.
Лечение анемии Даймонда-Блекфена
При выраженной анемии показаны гемотрансфузии или переливание эритроцитарной массы для восполнения угрожающего жизни дефицита эритроцитов. В некоторых случаях переливание крови может потребоваться неоднократно – например, при отсутствии эффекта от глюкокортикостероидной терапии. В таких ситуациях необходимо принимать меры в отношении профилактики поражения печени и селезенки избытком железа и других посттрансфузионных осложнений – например, назначать хелационную терапию дефероксамином.
Основными препаратами для лечения анемии Даймонда-Блекфена являются глюкокортикостероиды (преднизолон, метилпреднизолон и другие). Терапию начинают с повышенных (ударных) дозировок препаратов внутрь, затем постепенно снижая их до уровня поддерживающей дозы – при этом одновременно должен расти уровень гемоглобина и улучшаться картина крови (в кровотоке появляются ретикулоциты, снижается количество макроцитов).
В зависимости от динамики заболевания терапию глюкокортикостероидами производят при помощи двух основных схем – в режиме пульс-терапии (до 7-ми дней приема повышенных доз с последующим двух-трех недельным перерывом) и поддерживающей терапии с ежедневным приемом небольших количеств глюкокортикостероидов. Выбор той или иной схемы зависит от реакции организма больного на препараты, наличия и выраженности побочных явлений, эффективности лечения. Описаны несколько случаев лечения анемии Даймонда-Блекфена с положительным исходом посредством пересадки костного мозга от близкого родственника.
Прогноз
Прогноз анемии Даймонда-Блекфена во многом неопределенный по причине слабого понимания процессов, которые приводят к ее развитию. Обширное исследование, в ходе которого изучили историю жизни более чем 200 больных, выявило, что почти у четверти детей, которых лечили посредством гемотрансфузий и назначения глюкокортикостероидов, еще до подросткового возраста возникла спонтанная ремиссия с восстановлением адекватного уровня гемоглобина. При этом больше половины детей остались зависимыми как от переливаний крови, так и от использования метилпреднизолона и после подросткового возраста. Оставшиеся 25% больных умерли еще в детстве, несмотря на все предпринятые терапевтические меры.
Во многом на прогноз влияет наличие или отсутствие сопутствующих физических отклонений и степень их выраженности. Лечение ростовыми факторами, препаратами железа и другими традиционными при анемии средствами неэффективно и может еще больше усилить нагрузку на печень и селезенку.
2. едеральные клинические рекомендации по диагностике и лечению анемии Даймонда-Блекфена у детей. - 2015.
3. Анемия Даймонда-Блекфана: модель трансляционного подхода к пониманию заболеваний у людей/ Влахос А.Бланк Л.Липтон Дж.М.// Российский журнал детской гематологии и онкологии. - 2014.
Читайте также: