Синдром Альпорта
Добавил пользователь Дмитрий К. Обновлено: 27.01.2026
PDF
PDF
DOI: 10.2215/CJN.00580116
РЕЗЮМЕ
Синдром Альпорта – наследственное заболевание, характеризующееся прогрессирующей почечной недостаточностью, потерей слуха и аномалиями глаз. Наследование Х-сцепленное (85%) или аутосомно-рецессивное (15%). Многие нефрологи считают, что при синдроме Альпорта заболевают преимущественно мужчины. Однако женщины при Х-сцепленных заболеваниях страдают в 2 раза чаще. У женщин заболевание, как правило, остается недиагностированным, но у 15-30% из них к 60 годам развивается почечная недостаточность, а к среднему возрасту часто отмечается потеря слуха. Половина их сыновей и дочерей также страдают данным заболеванием. Аутосомно-рецессивный вариант синдрома Альпорта встречается реже, но часто ошибочно принимается за Х-сцепленное заболевание. Рецессивный тип наследования подозревают, когда у женщины рано развивается почечная недостаточность или лентиконус. В их семьях могут быть близкородственные браки. Прогноз для других членов семьи очень отличается от Х-сцепленного заболевания. У представителей других поколений, включая родителей и потомков, заболевание отсутствует, и в среднем только один из четырех их сиблингов унаследует данную патологию. У всех женщин с синдромом Альпорта диагноз должен быть подтвержден с помощью генетического исследования (даже если функция почек у них в норме) из-за риска развития почечной недостаточности у них и рисков у их потомков. Вид мутации указывает на тип наследования и вероятность передачи заболевания их детям, кроме того, вид мутации характеризует почечный прогноз как при Х-сцепленном, так и при рецессивном типах наследования. У женщин с Х-сцепленным синдромом Альпорта необходимо как минимум ежегодно определять альбуминурию и измерять уровень артериального давления. Экспертное руководство по диагностике и лечению синдрома Альпорта ( Expert guidelines for the diagnosis and management of Alport syndrome ) при наличии альбуминурии рекомендует проводить терапию блокаторами ренин-ангиотензин-альдостероновой системы (РААС) (с адекватной контрацепцией из-за риска тератогенного действия ингибиторов ангиотензинпревращающего фермента) с целью торможения прогрессирования почечной недостаточности. Согласно современным рекомендациям, женщины с аутосомно-рецессивным синдромом Альпорта должны получать терапию блокаторами РААС с момента установления диагноза. Кроме того, эти женщины должны быть направлены на медико-генетическое консультирование и информированы о своих репродуктивных возможностях, также им необходим тщательный мониторинг во время беременности с целью выявления развития АГ.
КОММЕНТАРИИ
Синдром Альпорта – наследственное заболевание, характеризующееся прогрессирующей почечной недостаточностью, потерей слуха и аномалиями глаз, включая образование рубцов на роговице, лентиконус, истончение сетчатки и пигментный ретинит. Тип наследования – Х-сцепленный (85%) или аутосомно-рецессивный (15%). Подозрение о Х-сцепленном варианте заболевания возникает, если у мужчин в семье наблюдаются более тяжелые проявления, а болезнь проявляется не в каждом поколении. Об аутосомно-рецессивном типе наследования говорят, когда заболевание возникает только в одном поколении, в семье имеются близкородственные браки или у молодой женщины развивается ТПН и потеря слуха или лентиконус.
Х-сцепленный вариант синдрома Альпорта обусловлен мутациями в гене COL 4 A 5, кодирующем a 5 цепь коллагена IV типа. Аутосомно-рецессивное заболевание является результатом двух мутаций in trans (в разных хромосомах) в генах COL 4 A 3 или COL 4 A 4, которые кодируют a 3 и a 4 цепи коллагена IV типа соответственно. a 3, a 4 и a 5 цепи образуют гетеротример, который является основой базальных мембран гломерулярного фильтра, роговицы, капсулы хрусталика и сетчатки (подобным тканевым распределением объясняются клинические проявления).
При Х-сцепленном синдроме Альпорта в гене COL 4 A 5 описано более 2000 мутаций, и более 1000 в генах COL 4 A 3 и COL 4 A 4 при рецессивной форме.
Клинические проявления синдрома Альпорта у мужчин с Х-сцепленной формой и у мужчин и женщин с рецессивным вариантом заболевания идентичны и включают гематурию, протеинурию, ТПН, лентиконус, истончение сетчатки и ретинопатию. К редким проявлениям относят лейомиоматоз (опухоли мягких тканей пищевода, бронхов), аневризму аорты и гигантские разрывы сетчатки.
У женщин с Х-сцепленным вариантом синдрома Альпорта заболевание часто остается недиагностированным. Однако в среднем женщины заболевают в два раза чаще мужчин.
У больных женщин следует применять контрацепцию, чтобы избежать наступления беременности во время приема ингибиторов АПФ из-за риска тератогенного действия последних. Некоторые женщины с данным заболеванием принимали решение не заводить детей из-за риска рождения больного ребенка. Если семейная мутация известна, то теперь им может быть предложено проведение пренатальной диагностики или предимплантационной генетической диагностики, но эти процедуры требуют планирования беременности и являются дорогостоящими.
Жак ШАНАР (Pr. Jacques CHANARD)
Клинические рекомендации по диагностике и лечению синдрома Альпорта у детей Текст научной статьи по специальности «Клиническая медицина»
Похожие темы научных работ по клинической медицине , автор научной работы — Длин В.В., Игнатова М.С., Конькова Н.Е.
Редкие заболевания в практике «Взрослого» нефролога: наследственный нефрит (синдром Альпорта), болезнь тонкой базальной мембраны, олигомеганефрония
Нефротический синдром: гистопатологическая дифференциальная диагностика. Часть 1: определение, классификация, патофизиология, генетические формы
Текст научной работы на тему «Клинические рекомендации по диагностике и лечению синдрома Альпорта у детей»
ПРОГРАММА НЕПРЕРЫВНОГО ПОСЛЕДИПЛОМНОГО ISSN 1561-6274. Нефрология. 2015. Том 19. №3. ОБРАЗОВАНИЯ ПО НЕФРОЛОГИИ
© В.В. Длин, М.С. Игнатова, Н.Е. Конькова, 2015 УДК 616.61-053.32-056.7-07-08
КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ ПО ДИАГНОСТИКЕ И ЛЕЧЕНИЮ СИНДРОМА АЛЬПОРТА У ДЕТЕЙ
Творческое объединение детских нефрологов; Российский национальный исследовательский медицинский университет имени Н.И. Пирогова
Длин В.В. - д.м.н., профессор, нефролог Игнатова М.С. - д.м.н., профессор, нефролог Конькова Н.Е. - к.м.н., нефролог
CLINICAL PRACTICE GUIDELINES FOR THE DIAGNOSTICS AND TREATMENT OF THE ALPORT SYNDROME IN CHILDREN
Creative Association of Pediatric Nephrologists; Clinical Research Institute of Pediatrics Pirogov Russian National Research Medical University
Dlin V.V. - MD, PhD, DSc Ignatova M.S. - MD, PhD, DSc Kon'kova N.E. - MD, PhD
Методика оценки силы рекомендаций и уровня их предсказательности, использованная при составлении данных клинических рекомендаций*.
По силе рекомендации подразделяются на три категории в убывающем порядке: уровень 1 (эксперты рекомендуют); уровень 2 (эксперты предлагают); «недифференцированный уровень» (табл. 1). Сила предсказательности рекомендаций подразделена на 4 уровня (табл. 2).
ВВЕДЕНИЕ, ГЕНЕТИКА, ЭПИДЕМИОЛОГИЯ, ПАТОГЕНЕЗ
Рекомендация 1.1. Синдром Альпорта (СА, синоним: наследственный нефрит) - неиммунная генетически детерминированная гломерулопатия, обусловленная мутацией гено
в, кодирующих коллаген 4 типа базальных мембран, проявляющаяся гематурией и/или протеи-нурией, прогрессирующим снижением почечных функций, нередко сочетающаяся с патологией слуха и зрения (НГ).
При СА так же редко наблюдается лейомиоматоз и измененные тромбоциты.
Коллаген IV типа состоит из гетеротримеров, составленных из комбинаций 6-ти альфа цепей:
• альфа 3, 4 и 5 цепи в гломерулярных базальных мембранах (ГБМ);
• альфа 1 и 5 цепи в базальной мембране (БМ) капсулы Боумена и дистальной тубулярной БМ.
Гены COL4A1 и COL4A2 расположены на 13 хромосоме, COL4A3 и COL4A4 - на 2 хромосоме, COL4A5 и COL4A6 - на X хромосоме
Рекомендация 1.2. Синдром Альпорта - этиологически гетерогенное наследственное заболевание моногенной природы. Причина заболевания лежит в мутации одного из генов: СОЬ4А5, СОЬ4А4, СОЬ4А3. При классическом варианте СА мутация происходит в гене СОЬ4А5, расположенном на длинном плече Х-хромосомы (Хд22.2), который кодирует а5-цепь коллагена IV типа (НГ). Аутосомно-рецессивная форма синдрома Альпорта обусловлена мутацией генов С0Ь4А3 и СОЬ4А4, расположенных на хромосоме 2 и кодирующих соответственно а3- и а4-цепи коллагена этого типа. Аутосомно- доминантная форма СА сцеплена с генным локусом СОЬ4А3-СОЬ4А4.
Частота СА в популяции составляет 1:5000. Он служит причиной 1% всех случаев терминальной почечной недостаточности (тПН) в Европе. В 2,3% случаев почечная трансплантация проводится больным с СА. СА описан у представителей всех рас на всех континентах. Встречается СА очевидно чаще, чем о нем сообщается, в связи с различной пенетрантностью и экспрессивностью гена, му-
Оценка силы рекомендаций
Уровень Со стороны пациентов Со стороны врача Дальнейшее направление использования
Уровень 1 «Эксперты рекомендуют» Подавляющее большинство пациентов, оказавшихся в подобной ситуации, предпочли бы следовать рекомендуемым путем и лишь небольшая часть из них отвергли бы этот путь Подавляющему большинству своих пациентов врач будет рекомендовать следовать именно этим путем Рекомендация может быть принята в качестве стандарта действия медицинского персонала в большинстве клинических ситуаций
Уровень 2 «Эксперты полагают» Большая часть пациентов, оказавшихся в подобной ситуации, высказались бы за то, чтобы следовать рекомендуемым путем, однако значительная часть отвергла бы этот путь Для разных пациентов следует подбирать различные варианты рекомендаций, подходящие именно им. Каждому пациенту необходима помощь в выборе и принятии решения, которое будет соответствовать ценностям и предпочтениям данного пациента Рекомендации, вероятно, потребуют обсуждения с участием всех заинтересованных сторон до принятия их в качестве клинического стандарта
«Нет градации» (НГ) Данный уровень применяется в тех случаях, когда в основу рекомендации укладывается здравый смысл исследователя-эксперта или тогда, когда обсуждаемая тема не допускает адекватного применения системы доказательств, используемых в клинической практике.
Уровень Характеристика уровня предсказательности Значение/описание
А Высокий Эксперты абсолютно уверены в том, что при выполнении данной рекомендации, наблюдаемый эффект почти полностью совпадет с ожидаемым.
В Средний Эксперты ожидают, что при выполнении данной рекомендации, наблюдаемый эффект скорее всего будет близок к ожидаемому, однако не исключается возможность того, что он будет существенно отличаться от него.
С Низкий Предсказываемый эффект может существенно отличаться от реального.
Р Очень низкий Предсказание эффекта крайне ненадежно и очень часто будет отличаться от реального.
тация которого его обуславливает. Частота различных вариантов СА (аутосомно-доминантного и аутосомно-рецессивного) неодинакова в различных популяциях. По эпидемиологическим данным в России частота СА среди детской популяции составляла 17:100000 населения. При длительном наблюдении за членами семьи с СА было отмечено, что у женщин пожилого возраста, как и у представителей мужского пола, отмечается снижение функции почек.
Рекомендация 1.3. Генетическая диагностика важна, так как от характера и типа мутации зависит клинические проявления заболевания, скорость прогрессирования заболевания и возраст формирования терминальной хронической почечной недостаточности (тПН) (1А).
Классификация СА в настоящее время проводится по типу наследования:
• Х-сцепленный доминантный (классический);
Около 80% в популяции составляют пациенты с Х-сцепленным доминантным вариантом СА, около 15% - с аутосомно-рецессивным СА и около 5% - с аутосомно-доминантным СА.
При мутации COL4A5 выделяют более тяжелые варианты: делеции, нонсенс или сплайсинг мутации и более легкий вариант - миссенс мутация.
У лиц мужского пола с Х-сцепленным доминантным вариантом СА заболевание имеет прогрессирующее течение: тПН развивается у 50% в возрасте до 25 лет, у 90% - в возрасте до 40 лет и почти у 100% в возрасте до 60 лет. При делеции или нонсенс-мутации COL4A5 риск развития тПН в возрасте до 30 лет составляет 90%, при сплайсинг мутации - 70%, а при миссенс мутации - 50%. Важность определения типа мутации обосновывается выраженной фенотип-генотипической корреляцией, что важно для прогнозирования сроков оказания помощи (заместительная терапия) этим пациентам. В большинстве случаев сроки развития тПН у мальчиков с Х-сцепленным доминантным вариантом СА схожи с другими членами семьи мужского пола, что позволяет прогнозировать время наступления терминальной ХПН даже без определения генотипа. У женщин с Х-сцепленным доминантным вариантом СА такой взаимосвязи наступления сроков тПН не наблюдается, вероятно, из-за Х-инактивации. Решение вопроса об активной
терапии у женщин с Х-сцепленным доминантным вариантом СА зависит от факторов риска про-грессирования: протеинурия, макрогематурия и нейросенсорная тугоухость.
Аутосомно-рецессивный СА связан с мутацией в обоих аллелях COL4A3 или COL4A4. Генофе-нотипические корреляции не так выражены, но характерно развитие терминальной ХПН к 30-летнему возрасту.
Аутосомно-доминантный СА связан с мутацией в COL4A3 или COL4A4 и прогрессирует медленно и мало показаний для начала терапии в детском возрасте.
Рекомендация 2.1. Для диагностики СА необходимо присутствие трех из пяти признаков: гематурия или летальный исход от тПН в семье; гематурия и/или протеинурия в семье; специфические изменения БМ клубочков у больного при электронной микроскопии (ЭМ) биоптата; снижение слуха по данным аудиометрического исследования; врожденная патология зрения. Эти критерии обосновывают необходимость тщательного сбора данных о наличии однотипной клинической картины и летальных исходов от тПН у кровных родственников, проведение электронно микроскопического исследования нефробиоптатов, тщательное обследование пробанда и родственников для исключения нейросенсорной тугоухости и патологии зрения (наиболее характерно наличие лентиконуса) (НГ).
У всех больных имеется бессимптомная микрогематурия; у девочек и маленьких мальчиков она может возникать периодически. Примерно в 50% случаев через 1-2 дня после респираторной инфекции появляется макрогематурия. У мальчиков часто наблюдается протеинурия, но у девочек она может быть слабой, преходящей или вообще отсутствовать. Ко второму десятилетию жизни протеинурия обычно прогрессирует, преимущественно у лиц мужского пола, нередко превышает 1 г/24ч и может сопровождаться нефротическим синдромом.
К внепочечным проявлениям СА относятся тугоухость и нарушения зрения. Двусторонняя нейро-сенсорная глухота, которая никогда не проявляется в ранние сроки после рождения, отмечается у 90% гемизиготных мальчиков с Х-сцепленным синдромом Альпорта, у 10% гетерозиготных девочек с Х-сцепленным СА и у 67% больных с аутосомно-рецессивным синдромом. Вначале нарушается восприятие звуков высокой частоты, но постепенно
больные перестают слышать обычную разговорную речь, и им приходится пользоваться слуховым аппаратом. Глазные нарушения выявляются у 3040% больных с Х-сцепленным СА и включают передний лентиконус (смещение центральной части хрусталика в переднюю камеру глаза), крапинки на желтом пятне и эрозии роговицы. В редких случаях наблюдается лейомиоматоз пищевода, трахеи, бронхов и женских наружных половых органов, а также изменение тромбоцитов. Лейомиоматозом называется патологический процесс, характеристиками которого являются пролиферация гладких волокон мышц, в тканях и органах, которые содержат в норме гладкомышечную ткань.
Рекомендация 2.2. При аутосомно-рецессивных формах СА, а так же в части случаев аутосомно-доминантных, в том числе и при Х-сцепленных формах заболевания, этих признаков недостаточно и необходимо проведение молекулярно-генетического исследования (НГ).
Рекомендация 2.3. Электронно-микроскопическими критериями диагностики являются: истончение БМ, особенно ее средней пластинки lamina densa, одновременно наблюдается расщепление БМ и появление ее слоистости. При ЭМ биоптата почки одновременно с тонкими ГБМ выявляются утолщенные ГБМ с участками просветления, напоминающие пчелиные соты. БМ теряют свою структуру, внутри них появляются скопления тонкогранулярного вещества. По мере дальнейшего прогрессирования болезни происходит тяжелая деструкция ГБМ с ее дальнейшим утолщением и дистрофией (НГ).
В норме моноклональные антитела к альфаЗ-цепи коллагена IV типа (MAB3) и моноклональные антитела к альфа5-цепи коллагена IV типа (MAB5) окрашивают всю толщину клубочковой базальной мембраны, а также базальную мембрану дистальных канальцев. В семьях с X-сцепленным СА окрашивание на MAB3 и MAB5 отсутствует у мужчин и носит прерывистый характер у женщин. Такую картину можно наблюдать у 60-70% семей. У больных при аутосомно-рецессивном типе СА окрашивание на MAB5 видно на базальной мембране Боуменовой капсулы и собирательных протоков, тогда как окрашивание на MAB3 отрицательное.
Окрашивание MAB3 в норме в коже не выявляется, тогда, как MAB5 окрашивают эпидермальную базальную мембрану. Это окрашивание обычно отсутствует при X-сцепленном СА у мужчин, тогда как у женщин выявляется сегментарная локализация антигена. У больных при аутосомно-рецессивном типе наследования окрашивание
МАВ5 эпидермальной базальной мембраны является нормальным.
Рекомендация 2.4. Гематурия является наиболее ранним признаком СА. Микроальбуминурия, а в последующем протеинурия, выявляется в более поздний период и ее выраженность определяет скорость прогрессирования заболевания (НГ).
Рекомендация 2.5. Интерстициальный фиброз, как и при других гломерулопатиях, играет важную роль в прогрессировании СА. Поэтому меры направленные на предупреждение формирования и прогрессирования интерстициального фиброза при СА необходимо начинать в детском возрасте (НГ).
РАЗДЕЛ 3. ЛЕЧЕНИЕ
Рекомендация 3.1. Мальчикам с СА, связанным с мутацией COL4A5, которые имеют делецию, нонсенс или сплайсинг мутацию или имеют в семье случаи развития тПН до 30 летнего возраста должна проводиться более ранняя и активная терапия снижающая протеинурию, что позволяет предотвратить повреждение эпителиальных клеток почечных канальцев и их атрофию и предупреждает развитие интерстициального фиброза (НГ).
Рекомендация 3.2. Цель антипротеинурической терапии снижение протеинурии менее 0,5 мг/мг креатинина или на 50%, если начальное значение протеинурии менее 1,0 мг/мг креатинина (НГ).
Рекомендация 3.3. Первой линией антипротеинурической терапии СА являются ингибиторы ангиотензин-превращающего фермента (иАПФ). Используют рамиприл в дозе 1-2 мг/м2/24 ч, энала-прил в дозе вдвое выше (2-4 мг/м2/24 ч), лизиноприл, фозиноприл, квинаприл в дозе вчетверо выше (4-8 мг/м2/24 ч) (НГ).
Рекомендация 3.4. Второй линией антипротеинурической терапии СА являются блокаторы рецепторов ангиотензина (БРА). Используют ло-зартан в дозе 12,5 мг/м2/24ч, удваивая дозу каждые 3 месяца до достижения максимальной дозы (при отсутствии побочных эффектов) 50 мг/м2/24ч, ирбисартан - тройная доза лозартана (37,5 мг/ м2/24 ч), вальсартан - 1,5 доза лозартана (18,75 мг/м2/24 ч) (НГ).
ОСЛОЖНЕНИЯ ТЕРАПИИ. Лечение проводят под регулярным контролем уровня калия. Повышение его уровня может быть связано с действием иАПФ или БРА и требует снижения дозы препарата
на 50%, а при персистенции гиперкалиемии - полной отмены терапии.
При тПН начинают лечение гемодиализом или выполняют трансплантацию почки. Примерно у 5% больных, перенесших трансплантацию почки, развивается нефрит, обусловленный антителами к базальной мембране клубочков, главным образом у лиц мужского пола с Х-сцепленным синдромом, у которых тПН развивается до 30-летнего возраста.
РАЗДЕЛ 4. ВТОРИЧНАЯ ПРОФИЛАКТИКА
В семьях с Х-сцепленной формой СА при известных мутациях возможна пренатальная диагностика, что крайне важно, если плод мужского пола.
РАЗДЕЛ 5. ПЕРСПЕКТИВЫ В ТЕРАПИИ СА
Реальных перспектив разработки этиотропной терапии СА в настоящее время нет
1. Фокеева В.В. Наследственный нефрит. В кн.: Детская нефрология (руководство для врачей) /Под ред. М.С. Игнатовой, Ю.Е. Вельтищева Л.:Медицина, 1989.-с.244-256. [Fokeeva V.V. Nasledstvennyj nefrit. V kn.: Detskaja nefrologija (rukovodstvo dlja vrachej) /Pod red. M.S. Ignatovoj, Ju.E. Vel'tishheva L.:Medicina, 1989.-s.244-256].
2. Atkin C.L., Gregory V.S., Border W.A. Alport syndrome. In.: Diseases of the kidney /Ed. R.W. Schrier, C.M. Cottschalk.- 4th ed.-Boston:Little,1989.
3. Bekheimia M.R., Reed B., Gregory M.C. et al. Genotype-phenotype correlation in X-linked Alport syndrome. J.Am.Soc. Nephrol. 2010; 21: 876-883.
5. Grunfield J-P., Noel L.H., Hafez S, Droz D. Renal prognosis in women with hereditary nephritis. Clin Nephrol. 1985;23:267-271.
6. Jais J.P., Knebelmann B., Giatras I. et al. X-linked Alport syndrome: natural history in 195 families and genotype-phenotype correlations in males. J.Am.Soc.Nephrol. 2000; 11: 649657.
7. Jais J.P., Knebelmann B., Giatras I. et al. X-linked Alport syndrome: natural history and genotype-phenotype correlations in girls and womans belonging to 195 families: a "European Community Alport Syndrome Concerted Action" study. J.Am.Soc. Nephrol. 2003; 14:2603-2610.
8. Pochet J.M., Bobrie G., Landais P. Et al. Renal prognosis in Alport's and related syndromes: influence of the mode of inheritance. Nephrol.Dial. Transp. 1989;4:1016-1021.
9. Rheault M.N., Kren S.M., Hartich L.A. X-inactivation modifies disease severity in female carriers of murine X-linked Alport syndrome. Nephrol.Dial. Transp. 2010;25:764-769.
10. Rheault M.N. Women and Alport syndrome. Pediatr Nephrol. 2012; 27(1): 41-46.
Лечение наследственного нефрита (синдрома Альпорта) в Израиле — современные методы и высокие результаты
Развитие в Израиле научной и практической медицины, успешная разработка и осуществление лечения различных редко встречающихся заболеваний делают страну все более популярной. В израильские клиники обращаются тысячи пациентов из зарубежных государств, которых привлекает высокая эффективность терапии, мировой уровень подготовки специалистов, приемлемая стоимость предоставляемых услуг. Лечение наследственного нефрита (синдрома Альпорта) в Израиле предусматривает использование лекарственных препаратов последнего поколения, гемодиализ, современные методы физиотерапии. Проведение адекватного терапевтического курса на начальных этапах болезни более, чем у 80% пациентов предупреждает дальнейшее развитие патологического процесса и улучшает прогноз.
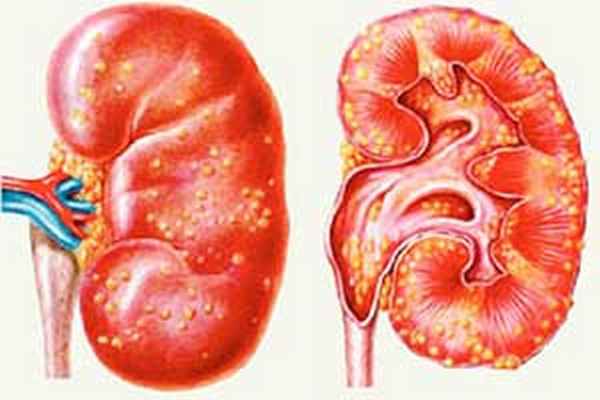
Во время консультаций и диагностического обследования собирается подробный семейный анамнез, проводится ряд лабораторных исследований, включая молекулярно-генетическое тестирование. В клиниках Израиля практикуется мультидисциплинарный подход, подразумевающий принятие решений группой специалистов различного профиля, каждый из которых является высококвалифицированным опытным врачом. При разработке схемы терапии принимаются во внимание специфика и выраженность симптомов, особенности больного. Прошедшие лечение пациенты оставляют отзывы, в которых говорится о быстром улучшении самочувствия, внимательности медицинского персонала, комфортных условиях.
Как проходит лечение заболевания
Наследственный нефрит, или синдром Альпорта, является наследственным почечным заболеванием, которое развивается в результате недостаточной выработки коллагена, входящего в базальные мембраны структур почек, хрусталика глаза и внутреннего уха. Таким образом, классической триадой симптомов выступают урологические расстройства (микрогематурия, хроническая почечная недостаточность), тугоухость и проблемы со зрением.
Этот тип наследственного нефрита был назван в честь американского доктора Артура Альпорта, впервые предположившего наследственный характер болезни. На такие выводы врача натолкнуло то, что у мужчин симптомы заболевания проявлялись практически всегда, в то время как у пациенток-женщин наблюдалась слабая выраженность или полное отсутствие симптоматики.
Исследования показали, что причины развития заболевания заключаются в мутациях генов, кодирующих структуру цепочек коллагена IV типа. Примерно в 85% случаев наследственный нефрит обусловлен мутационными изменениями гена, расположенного в Х-хромосоме. При этом внешние симптомы ярко проявляются у пациентов-мужчин, тогда как у женщин выработка коллагена компенсируется за счет локализованного во второй Х-хромосоме нормального гена.
Обязательным нарушением, выявляющимся практически у всех пациентов, независимо от пола, является микрогематурия. При синдроме Альпорта также отмечаются повышенное содержание в моче белка, почечная недостаточность, артериальная гипертензия. У пациентов диагностируется тугоухость и расстройство зрительной функции, в некоторых случаях прогрессирующее до слепоты. При позднем начале специфического лечения могут развиваться судороги, мышечная слабость, тремор, боли.
Сегодня не существует способов излечения от данного заболевания. Терапия направлена на снятие болезненных симптомов, нормализацию состояния пациента и предупреждение развития осложнений.
Лекарственная терапия
В схему лечения входят следующие виды медикаментозных средств:
- препараты из группы ингибиторов ангиотензин-превращающего фермента — их действие способствует активизации почечного кровообращения, расширению кровеносных сосудов, также средства применяются для профилактики фиброза и некоторых других осложнений;
- блокаторы рецепторов ангиотензина II — оказывают такой же эффект, что и предыдущие, но действуют более мягко;
- анаболические средства;
- иммуномодуляторы;
- комплексы витаминов.
Диализная терапия
Пациентам старше 60 лет, а также при запущенной стадии почечной недостаточности показано регулярное прохождение диализа — очистки крови от продуктов метаболизма и токсический соединений.
В программу лечения входят:
- гемодиализ — кровь забирается из вены больного, очищается в аппарате «искусственная почка» и вводится обратно в кровоток;
- перитонеальный диализ — введение в брюшную полость пациента раствора для диализа, который поглощает накопившиеся токсины и затем выводится из организма, при этом брюшина действует как мембрана.
Оксигенобаротерапия
Эта физиотерапевтическая процедура, также известная как гипербарическая оксигенация, направлена на насыщение клеток организма кислородом. Пациента помещают в барокамеру, куда под давлением подается обогащенная кислородом смесь газов. Поглощение клетками в несколько раз большего объема кислорода, чем в нормальных условиях, способствует ускорению метаболизма и восстановительных процессов, укреплению и активизации сердечно-сосудистой и дыхательной систем.
Образ жизни
Поскольку способы лечения данного заболевания отсутствуют, большое внимание уделяется профилактике обострений и развития серьезных осложнений. При синдроме Альпорта на протяжении всей жизни необходимо придерживаться ряда правил:
- соблюдать низкобелковую диету, исключая из рациона богатые белком продукты;
- быть физически активным, но при этом избегать излишних нагрузок;
- вовремя санировать очаги патологии, не допуская развития хронических инфекционных воспалений;
- прививки делать исключительно по эпидемиологическим показаниям;
- всегда сообщать о своем заболевании врачам, поскольку, прописывая медикаментозное лечение, доктор должен учитывать, что при синдроме Альпорта противопоказаны препараты с нефротоксическим эффектом.
Как проводится диагностика заболевания
Устанавливает окончательный диагноз и разрабатывает лечебную программу группа врачей, состоящая из специалистов-урологов, терапевтов и генетиков. На комплексное обследование пациента и принятие решения касательно способов терапии, обычно, требуется примерно три дня.
Первый день
После прибытия в клинику пациент проходит консультацию лечащего нефролога, на которой врач знакомится с больным, уточняет характер и время появления симптомов, собирает семейный анамнез. Во время поверхностного осмотра выявляются характерные для заболевания бледность кожи и слизистых, пониженный тонус мускулатуры. Прием завершается составлением списка необходимых исследований.
Второй день
- общий анализ мочи;
- биопсия почек;
- молекулярно-генетическое тестирование;
- консультации ЛОР-врача и окулиста.
Третий день
Полученные результаты рассматриваются врачебной комиссией, состоящей из нефролога и узкопрофильных специалистов, которые, изучив их, коллегиально приходят к решению по поводу диагноза и способов терапии.
Сколько стоит лечение
Цена терапевтического курса имеет большое значение при выборе клиники. В Израиле лечение обходится приблизительно на 30% дешевле, чем в западноевропейских странах, и на 50% — чем в США.
Преимущества лечения в Израиле
- Мировой уровень подготовки специалистов.
- Оснащение клиник новейшей аппаратурой.
- Применение инновационных методик лечения.
- Комфортные условия.
- Лояльные цены.
Сегодня в Израиле с успехом удается восстановить здоровье пациентам с наследственным нефритом, еще недавно считавшимся неизлечимым заболеванием, угрожающим жизни пациента. Самое главное — не медлить, обращаться за помощью и не позволять болезни победить.
Синдром Альпорта
Синдром Альпорта - генетически гетерогенное заболевание, характеризующееся нефритическим синдромом (т.е. гематурией, протеинурией, гипертонией, в конечном итоге почечной недостаточностью), часто с нейросенсорной глухотой и, реже, офтальмологическими симптомами. В основе заболевания лежит генная мутация, повреждающая коллаген IV типа. Диагностика производится на основании данных анамнеза, включая семейный анамнез, результатов анализа мочи и биопсии (почки или кожи). Лечение такое же, как и при хронической болезни почек, иногда включает пересадку почки Трансплантация почки Трансплантация почек является наиболее распространенным видом трансплантации цельных органов. (См. также Обзор трансплантации (Overview of Transplantation)). Основное показание для трансплантации. Прочитайте дополнительные сведения .
Синдром Альпорта – это нефритический синдром, вызванный мутацией гена COL4A3, COL4A4, COL4A5, (кодирующего альфа-5 цепь коллагена IV типа), что приводит к формированию измененных коллагеновых волокон IV типа. Механизм развития гломерулярной болезни в результате изменений структуры коллагена неизвестен, но предполагают, что в основе лежит нарушение структуры и функции; у членов большинства семей развивается утолщение и истончение гломерулярных и канальцевых базальных мембран с образованием многослойной плотной пластинки с очаговым или местным распределением (пример переплетения "плетенка"). В результате происходит рубцевание гломерулярной ткани и интерстициальный фиброз.
Болезнь обычно наследуется по Х-сцепленному механизму, хотя существуют аутосомно-рецессивные и, редко, аутосомно-доминантные варианты. Случаи наследования по Х-хромосоме могут быть клинически классифицированы как:
Ювенильная форма: почечная недостаточность развивается между 20 и 30 годами
Зрелая (взрослая) форма: почечная недостаточность развивается у людей > 30 лет
Клинические проявления
Классическое Х-хромосомное заболевание у мужчин и аутосомно-рецессивное заболевание клинически сходны. У пациентов развиваются почечные симптомы, характерные для острого нефритического синдрома (например, микрогематурия, гипертензия, а впоследствии макрогематурия с протеинурией), а прогрессирование заболевания до почечной недостаточности происходит в возрасте 20–30 лет (ювенильные формы).
Нейросенсорная тугоухость встречается часто, при этом характерно нарушение восприятия более высоких частот; она может оставаться недиагностированной в раннем детстве.
Реже, чем тугоухость, наблюдаются офтальмологические патологии: катаракта (чаще всего), передний лентиконус (простая протрузия конуса к переднему полюсу хрусталика из-за утолщения капсулы хрусталика), сферофакия (сферическая деформация хрусталика, предрасполагающая к подвывиху хрусталика), нистагм, пигментный ретинит, слепота.
Х-хромосомное заболевание встречается у гетерозиготных женщин, которые, потому что они имеют одну нормальную Х-хромосому, как правило, имеет менее тяжелые, более медленно прогрессирующие симптомы, чем у мужчин.
У некоторых мужчин с Х-хромосомным заболеванием почечная недостаточность развиваются после 30 лет с потерей слуха, которая происходит с опозданием или в легкой форме, а аутосомно-доминантное заболевание, как правило, не приводит к почечной недостаточности до возраста ≥ 45 лет (взрослые формы).
У больных с Х-хромосомным синдромом Альпорта, сенсоневральная потеря слуха, обычно, проявляется в детсве, в то время, как болезнь почек часто не проявляется до взрослого возраста.
Синдром Альпорта ( Гематурический нефрит , Наследственный нефрит 1 типа , Семейный гломерулонефрит )
Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза. Мужчины страдают развернутой формой болезни с тяжелой симптоматикой. Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо. Основные симптомы – микрогематурия, протеинурия, почечная недостаточность, сенсорная тугоухость, деформация и вывих хрусталика, катаракта. Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.
МКБ-10
Общие сведения
Семейные случаи гематурической нефропатии впервые привлекли внимание исследователей в 1902 году. Спустя почти 30 лет, в 1927 году американский врач А. Альпорт обнаружил частую сочетаемость гематурии с тугоухостью и уремией у мужчин, в то время как у женщин симптомы отсутствовали или были слабовыраженными. Он предположил наследственный характер болезни, которая впоследствии была названа синдромом Альпорта. Синонимы – наследственный нефрит 1 типа, гематурический нефрит, семейный гломерулонефрит. Распространенность невысока – 1 случай на 5 тысяч человек. На долю патологии приходится 1% больных с почечной недостаточностью, 2,3% пациентов, перенесших трансплантацию почек. Заболевание диагностируется у людей всех рас, но соотношение различных форм неодинаково.
Причины
По своей природе синдром является гетерогенным наследственным заболеванием – его развитие провоцируется дефектом генов, которые кодируют структуру различных цепей IV типа коллагена. Генетические изменения представлены делециями, сплайсинг, миссенс и нонсенс-мутациями. Их локализация определяет тип наследования болезни:
- X-сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.
- Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
- Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
Патогенез
Гломерулярная базальная мембрана имеет сложное строение, ее образует строгая геометрическая последовательность молекул коллагена 4-го типа и полисахаридные компоненты. При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул. На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество. Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз. Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями: мутацией гена, дефектом строения коллагена, деструкцией базальных мембран, патологией почек (иногда – нарушением слуха и зрения).
Симптомы
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни. Другой распространенный признак заболевания – протеинурия. Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки. У всех больных отмечается неуклонное прогрессирование симптома.
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоих полов с аутосомно-рецессивным вариантом наследования. Тяжесть гипертонии увеличивается вместе с нарастанием ХПН. У юношей, мужчин снижение функции почек достигает терминальной стадии к 16-35 годам, при медленном течении болезни – к 45-65 годам. Иногда выявляются диффузные гладкомышечные опухоли пищевода и бронхов, проявляющиеся в позднем детстве дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой, частыми бронхитами.
Часто у больных формируется нейросенсорная тугоухость. Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии. По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи. При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%. Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах. У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
Осложнения
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт. У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся мышечной слабостью, болями, судорогами, тремором, парестезиями, снижением чувствительности. Другим осложнением является тромбоцитопения с высоким риском кровотечений. Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой. К 60 годам 100% больных этой группы нуждаются в процедурах гемодиализа, перитонеального диализа, трансплантации донорской почки.
Диагностика
В диагностическом процессе принимают участие врачи-нефрологи, урологи, терапевты и генетики. При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН. Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов, вторичных нефропатий. Для подтверждения диагноза проводятся следующие процедуры:
- Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония, на поздних – артериальная гипертония.
- Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома, ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
- Исследование биоптата почек. При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
- Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.
- Аудиометрия, офтальмологическое исследование. Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога. При аудиометрии обнаруживается снижение слуха: в детском и подростковом возрасте – билатеральная высокочастотная тугоухость, во взрослом возрасте – низкочастотная и среднечастотная тугоухость. Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.
Лечение синдрома Альпорта
Специфическая терапия отсутствует. С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза. С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией ХПН назначается гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почек.
Прогноз и профилактика
Синдром прогностически благоприятен в случаях, когда гематурия протекает без протеинурии, нет расстройств зрения и тугоухости. Кроме этого, прогноз хороший у большинства женщин – даже при наличии гематурии болезнь прогрессирует медленно, не ухудшает общего состояния. Ввиду наследственного характера патологии предупредить ее развитие невозможно. В семьях, где установлено наличие X-сцепленной формы синдрома, возможно проведение пренатальной диагностики. Генетический скрининг особенно рекомендован женщинам, вынашивающим мальчиков.
1. Наследственный нефрит (синдром Альпорта) / Сунгатуллина И.Л. // Казанский медицинский журнал – 2002 – Т.83, №1.
2. Проект клинических рекомендаций по диагностике и лечению синдрома Альпорта у детей / Длин В.В., Игнатова М.С., Конькова Н.Е. – 2014.
3. Наследственные заболевания почек, протекающие с гематурией / Длин В.В., Игнатова М.С. // Российский вестник перинатологии и педиатрии – 2014 - №3.
Читайте также: