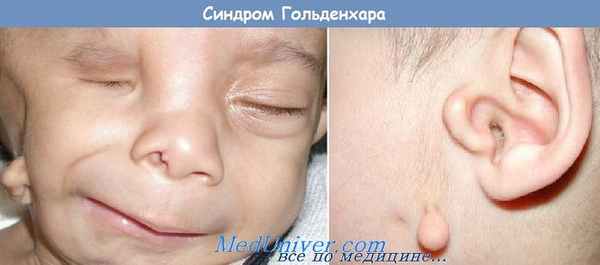
Синдром Голденхара. HELLP-синдром
Добавил пользователь Алексей Ф. Обновлено: 27.01.2026
Автор: Джекспаев Серекжан Абдуллович, отец Саяна Джекспаева 09.03.2013 г.р., просит оплатить лечение сына в Ярославской КБ имени Н.В. Соловьева. Диагноз: врожденная косолапость. Башкортостан.
Принято в работу.
Автор: Сикорская Наталья Александровна, мама Максима Мухи 30.06.2006 г.р., просит оплатить лечение сына в Ярославской КБ имени Н.В. Соловьева. Диагноз: врожденная левосторонняя косолапость. Могилевская область.
Принято в работу.
Автор: Халиков Абдурахим Магометович, отец Давуда Халикова 13.02.2013 г.р., просит оплатить лечение сына в Ярославской КБ имени Н.В. Соловьева. Диагноз: врожденная двусторонняя косолапость. Астраханская область.
Принято в работу.
Автор: Хрущ Светлана Миргабидзяновна, мама Александра Хруща 30.01.2013 г.р., просит оплатить лечение сына в Ярославской КБ имени Н.В. Соловьева. Диагноз: врожденная двусторонняя косолапость. Челябинская область.
Принято в работу.
Автор: Колегова Татьяна Сергеевна, мама Ивана Ежова 08.07.2011 г.р., просит оплатить лечение сына в Ярославской КБ имени Н.В. Соловьева. Диагноз: врожденная двусторонняя косолапость. Красноярский край.
Принято в работу.
Автор: Мурзабаева Язгуль Минниахатовна, мама Дамира Мурзабаева 17.03.2013 г.р., просит оплатить лечение сына в Ярославской КБ имени Н.В. Соловьева. Диагноз: врожденная косолапость. Башкортостан.
Принято в работу.
Автор: Казаченко Марина Григорьевна, мама Вячеслава Таркрашева 17.03.1996 г.р., просит оплатить операцию сыну в «АНО Клиника НИИТО» (Новосибирск). Диагноз: реберная дисплазия. Алтайский край.
Принято в работу.
Автор: Клямм Марина Алексеевна, мама Алисы Клямм 19.04.2009 г.р., просит оплатить операцию дочери в НИИ кардиологии ТНЦ. Диагноз: полная атриовентрикулярная блокада. Алтайский край.
Принято в работу.
Автор: Скворцов Евгений Сергеевич, государственный опекун Тимура Юркина 16.10.2007 г.р., просит оплатить операцию Тимуру в Ярославской КБ имени Н.В. Соловьева. Диагноз: синдром Голденхара. Ярославская область.
Принято в работу.
Автор: Фролова Юлия Павловна, мама Софьи Фроловой 17.09.2011 г.р., просит оплатить операцию дочери в Ярославской КБ имени Н.В. Соловьева. Диагноз: акушерский паралич слева. Ярославская область.
Принято в работу.
Автор: Быковских Юлия Валерьевна, мама Анны Быковских 31.01.1996 г.р., просит оплатить операцию дочери в «АНО Клиника НИИТО» (Новосибирск). Диагноз: паралитический неосложненный декомпенсированный прогрессирующий правосторонний лордосколиоз. Алтайский край.
Принято в работу.
Автор: Ночевская Марина Николаевна, мама Дарьи Кочетковой 17.04.2013 г.р., просит оплатить дочери расходные материалы для операции на сердце в НИИ кардиологии ТНЦ. Диагноз: врожденный порок сердца. Томская область.
Принято в работу.
Автор: Соколова Ольга Владимировна, мама Даниила Соколова 04.04.2012 г.р., просит оплатить операцию сыну в Ярославской КБ имени Н.В. Соловьева. Диагноз: врожденные аномалии развития правой кисти. Тульская область.
Принято в работу.
Автор: Жулдыбина Наталья Анатольевна, мама Анастасии Жулдыбиной 17.08.1998 г.р., просит оплатить дочери инвалидную коляску «Лиза». Диагноз: детский церебральный паралич. Кемеровская область.
Принято в работу.
Автор: Давыденко Татьяна Николаевна, мама Данилы Соколова 07.09.2004 г.р., просит оплатить операцию сыну в Ярославской КБ имени Н.В. Соловьева. Диагноз: послеожоговый рубец на лице. Ярославская область.
Принято в работу.
Автор: Шамшурина Ольга Александровна, мама Анны Шамшуриной 12.07.2010 г.р., просит оплатить операцию дочери в Немецком кардиоцентре (Берлин, Германия). Диагноз: врожденный порок сердца. Удмуртия.
Принято в работу.
Автор: Бабкина Екатерина Юрьевна, мама Елизаветы Бабкиной 10.06.2006 г.р., просит оплатить дочери обследование и подбор лечения в Финляндии. Диагноз: интоксикация неясной этиологии. Санкт-Петербург.
Принято в работу.
Автор: Коробкова Екатерина Николаевна, мама Егора Коробкова 05.02.2007 г.р., просит оплатить сыну курс восстановительного лечения в ООО «Реацентр Самара». Диагноз: задержка психоречевого развития. Тверская область.
Принято в работу.
Автор: Канзычакова Карина Леонидовна, мама Светланы Райковой 28.05.2012 г.р., просит оплатить операцию дочери в НИИ кардиологии ТНЦ. Диагноз: врожденный порок сердца. Хакасия.
Принято в работу.
Автор: Рахимова Эльвира Миргазияновна, мама Арины Рахимовой 13.12.2009 г.р., просит оплатить операцию дочери в НИИ кардиологии ТНЦ. Диагноз: врожденный порок сердца. Удмуртия.
Принято в работу.
Автор: Мясоедова Татьяна Олеговна, мама Аристарха Мясоедова 26.03.2007 г.р., просит оплатить сыну инсулиновую помпу и расходные материалы к ней. Диагноз: сахарный диабет 1 типа. Челябинская область.
Принято в работу.
Автор: Пуларгина Олеся Аркадьевна, мама Савелия Пуларгина 31.07.2010 г.р., просит оплатить сыну инвалидную коляску с электроприводом Stingray. Диагноз: детский церебральный паралич. Самарская область.
Принято в работу.
Автор: Долгова Анна Викторова, мама Александра Долгова 10.07.2001 г.р., просит оплатить дочери перевязочные средства. Диагноз: рецессивный дистрофический буллезный эпидермолиз. Пензенская область.
Принято в работу.
Ответы Минздрава на письма из Русфонда
Автор: Щербакова Светлана Владимировна, мама Александры Щербаковой 22.09.2011 г.р., просит оплатить реабилитацию дочери в больнице по лечению детского церебрального паралича (г. Юйцы, Китай). Диагноз: детский церебральный паралич. Челябинская область.
Ответ Минздрава
Sent:Sent: Wednesday, May 08, 2013 10:38 AM
Уважаемая Светлана Владимировна!
Предоставление государственной услуги по направлению граждан Российской Федерации на лечение за пределами территории Российской Федерации за счет средств федерального бюджета (далее – государственная услуга) осуществляется Минздравом России в соответствии с приказом Минздравсоцразвития России от 19.12.2011 № 1571н «Об утверждении Административного регламента Министерства здравоохранения и социального развития Российской Федерации по предоставлению государственной услуги по направлению граждан Российской Федерации на лечение за пределы территории Российской Федерации за счет бюджетных ассигнований федерального бюджета» (далее – Административный регламент).
В соответствии с пунктом 22 Административного регламента основанием для рассмотрения вопроса о предоставлении государственной услуги является поступление в Минздрав России письменного заявления о предоставлении государственной услуги от пациента (законного представителя пациента или доверенного лица; пациента), составленного по образцу, предусмотренному приложением № 1 к Административному регламенту (далее – заявление о предоставлении государственной услуги), с приложением следующих документов:
1) копия паспорта пациента или копия свидетельства о рождении пациента (для детей в возрасте до 14 лет);
2) копия выписного эпикриза из медицинской карты стационарного больного, выданного федеральным медицинским учреждением, в которое пациент был направлен в установленном законодательством Российской Федерации порядке, с указанием сведений о состоянии здоровья (основной и сопутствующий диагнозы, анамнез заболевания, результаты проведенных обследований и проведенное лечение) и рекомендаций о необходимости диагностики и/или лечения за пределами территории Российской Федерации, выданных на основании заключения врачебной комиссии федерального медицинского учреждения (далее – выписной эпикриз из медицинской карты стационарного больного).
Срок выдачи выписного эпикриза из медицинской карты стационарного больного не должен превышать 3 месяца до направления пациентом (законным представителем или доверенным лицом пациента) заявления в Минздрав России.
В соответствии с пунктом 23 Административного регламента в случае, если заявление о предоставлении государственной услуги представлено законным представителем пациента или доверенным лицом пациента, дополнительно к документам, указанным в пункте 22 Административного регламента, прилагаются:
1) копия паспорта законного представителя пациента или доверенного лица пациента;
2) копия документа, удостоверяющего полномочия законного представителя пациента или доверенного лица пациента.
В случае поступления от Вас в Минздрав России заявления и документов, указанных в Административном регламенте, данный вопрос в установленном порядке будет подготовлен для рассмотрения на комиссии по направлению граждан Российской Федерации на лечение за пределы территории Российской Федерации.
Заместитель директора Департамента
специализированной медицинской помощи и медицинской реабилитации
Е.Г. Камкин
Синдром Голденхара. HELLP-синдром
Синдром Голденхара. HELLP-синдром
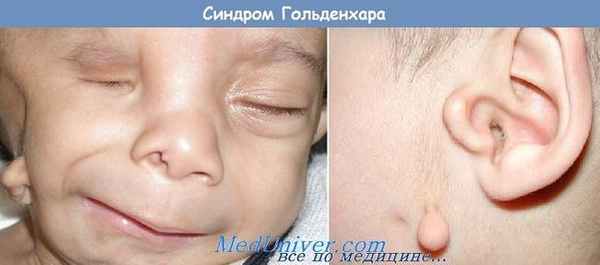
При данном синдроме отмечается наличие гемифациальной микросомии, эпибульбарных тератом, преаурикулярных отростков (околоушных придатков), поперечных расщелин лица, асимметрии черепа и аномалий строения позвоночника (дефектов сегментации позвонков).
Синонимы. Окулоаурикуло-вертебральная дисплазия, синдром Гольденхара-Горлина (GoldenharGorlin), фациоаурикуло-вертебральная дисплазия.
Распространенность. Частота встречаемости составляет 0,2 на 10 000 родов.
Этиология. Вероятно, заболевание возникает спорадически, с редкими случаями, предполагающими аутосомно-рецессивный или аутосомно-доминантный тип наследования.
Риск рецидива. Возможно, отсутствует, если не относится к аутосомно-доминантному или аутосомно-рецессивному наследованию.
Диагностика. Диагноз обычно устанавливается при обнаружении асимметрии лица (возможно, это будет еще более распространено с внедрением трехмерной эхографии) или расщелины верхней челюсти в сочетании с односторонней микрофтальмией. Другие признаки включают пороки сердца или мочевыводя-щей системы, а также возникновение липомы мозолистого тела.
Патогенез. Возможно, причиной является кровоизлияние у эмбриона в области первой и второй жаберных дуг в период, когда кровоснабжение в этой области из стременной артерии заменяется на кровоснабжение из наружной сонной артерии.
Сочетанные аномалии. Трахеопищеводный свищ, эпибульбарные тератомы, сирингогидромелия, задержка нервно-психического развития, тетрада Фалло (Fallot), дефект межжелудочковой перегородки, синдром асплении, инверсия желудочков сердца с отхождением аорты и легочного ствола от правого желудочка, атрезия ствола легочной артерии с дефектом межжелудочковой перегородки, полный поддиафрагмальный аномальный дренаж легочных вен, эктопические или слившиеся почки, агенезия почек, пузырно-мочеточниковый рефлюкс, обструкция лоханочно-мочеточникового соединения, удвоение мочеточников и мультикистоз почек.
Дифференциальный диагноз. Синдром Кауфмана (Kaufman) (окуло-церебро-фациальный синдром), акрофациальный дизостоз и ассоциация Чарджа (Charge).
Прогноз. Помимо умственной отсталости у таких пациентов может возникать много осложнений, связанных с верхними дыхательными путями и заболеваниями позвоночника.
Акушерская тактика. Если диагноз установлен на ранних сроках, может быть предложено прерывание беременности. После рождения возможно проведение косметической коррекции лицевых пороков.
HELLP-синдром
Название синдрома «HELLP» является аббревиатурой, образованной из начальных букв состояний, выявляемых при тяжелых вариантах течения гестоза, который характеризуется возникновением гемолиза, высоким уровнем печеночных ферментов и низким уровнем тромбоцитов (hemolys, elevated liver enzymes, and low platelets). Данное состояние представляет угрозу как для жизни матери, так и плода. Эхографические признаки заболевания плода включают задержку его внутриутробного развития, мышечную гипотонию, снижение двигательной активности, патологические значения допп-лерометрических показателей артерий пуповины и мозговых артерий.
HELLP-синдром поражает 2-12% беременных, у которых развивается гестоз. При этом большую предрасположенность к его развитию имеют, по-видимому, американки европейского типа, чем афроамериканки.
Этиология. HELLP-синдром является состоянием, специфичным для беременности, осложненной тяжелым течением гестоза.
Риск рецидива. Хотя риск рецидива точно неизвестен, каждую женщину с проявлениями HELLP-синдрома при последующей беременности следует вести как пациентку с повышенным риском.
Диагностика. Диагноз у матери устанавливается с помощью лабораторных исследований (явления гемолиза, повышенный уровень печеночных ферментов и тромбоцитопения с уменьшением количества тромбоцитов менее 100 000, которая является наиболее постоянным признаком), а также путем выявления клинических симптомов (отеков, гипертензии, тошноты, болей в животе вследствие возникновения субкапсулярного кровоизлияния или разрыва печени). Признаки у плода включают задержку внутриутробного развития и уменьшение объема околоплодных вод, что является реакцией плода на хроническую плацентарную недостаточность на фоне гипертензии у матери. В более тяжелом случае, при минимизации кислородного резерва, у плода могут быть обнаружены такие признаки, как мышечная гипотония, снижение его двигательной активности, дыхательных движений и появление патологических допплерометрических показателей кровотока. В тяжелых случаях нередки антенатальная или неонатальная гибель плода.
Сочетанные аномалии. Очень важным и опасным состоянием, которое может сочетаться с HELLP-синдромом, является синдром диссеменированного внутрисосудистого свертывания (ДВС).
Дифференциальный диагноз. Заболевания, которые могут развиваться аналогично, представляют собой нарушение функции печени или анемию, а также тромбоцитопеническую пурпуру, тромботическую тромбоцитопеническую пурпуру, гемолитический уремический синдром, заболевание желчного пузыря, вирусный гепатит и острый жировой гепатоз беременных.
Прогноз. Прогноз варьирует в зависимости от тяжести состояния как матери, так и плода. Материнская заболеваемость и смертность пропорциональна тяжести системной болезни, тогда как те же показатели у плода в большинстве случаев будут зависеть от его гестационного возраста.
Акушерская тактика. Пациенток с HELLP-синдромом следует рассматривать как страдающих тяжелым гестозом. Рекомендуются госпитализация их в специализированный центр и роды в доношенном сроке. До наступления времени родоразрешения беременность следует вести консервативно, с интенсивным мониторингом состояния как матери, так и плода. Индукцию родов выполняют при достижении зрелости легких плода.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Синдром Гольденхара - симптомы и лечение
Что такое синдром Гольденхара? Причины возникновения, диагностику и методы лечения разберем в статье доктора Гавран Надежды Александровны, генетика со стажем в 11 лет.
Над статьей доктора Гавран Надежды Александровны работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Гольденхара — это редкая врождённая аномалия, при которой изменяются размеры и форма лицевых структур. Обычно изменения локализуются на одной стороне лица, вызывая его асимметрию, но иногда встречается двустороннее поражение.

Данный синдром относится к спектру врождённых аномалий черепа и лицевых структур, имеющих общий термин "краниофациальная микросомия". Под ним понимается уменьшение какой-либо структуры тела в пределах черепно-лицевой области.
Синонимы синдрома: окулоаурикулярная дисплазия, фацио-аурикуло-вертебральная ассоциация, синдром 1-й и 2-й жаберных дуг, отомандибулярный дизостоз, гемифациальная микросомия и др.
Приблизительная частота встречаемости синдрома Гольденхара — 1 случай на 3500-25000 новорождённых [9] . У мальчиков он встречается в 2 раза чаще, чем у девочек.
Точные причины заболевания на сегодняшний день до конца не известны [1] [2] [3] [4] . Большинство случаев возникают случайно в семьях без отягощённой истории болезни. Однако у 1-2 % пациентов с синдромом Гольденхара есть близкие родственники с подобным нарушением. Это свидетельствует о роли генетических факторов в возникновении данной патологии [4] [5] . В частности предполагается участие гена MYT1, расположенного в локусе q13.33 хромосомы 20.
Другим возможным фактором развития синдрома Гольденхара являются хромосомные аномалии — потеря или удвоение участка хромосомы. Как правило, у людей с этими нарушениями могут наблюдаются такие сочетанные пороки развития, как аномалии сердца, лёгких, почек, конечностей и центральной нервной системы [1] [2] [5] [6] .
Некоторые исследователи полагают, что формированию синдрома способствует нарушение кровотока или внешние повреждающие факторы:
- приём некоторых лекарственных препаратов, противопоказанных при беременности;
- вредные привычки;
- химические и физические агенты, воздействующие на плод на 3-8 неделе внутриутробного развития [5][6] .
Также нельзя исключить роль таких акушерско-гинекологических факторов, как предшествующие аборты, сахарный диабет и ожирение [18] .
Первые описания врождённых аномалий лицевых структур обнаружены в древних письменах, датированных 2000 лет до н. э. В Колумбии и Мексике были найдены древние керамические изделия с изображениями различных вариантов гемифациальной микросомии, в том числе наследственной: на одном из изделий был изображён родитель с ребёнком на руках, которые имели схожие аномалии лица [10] .
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Гольденхара
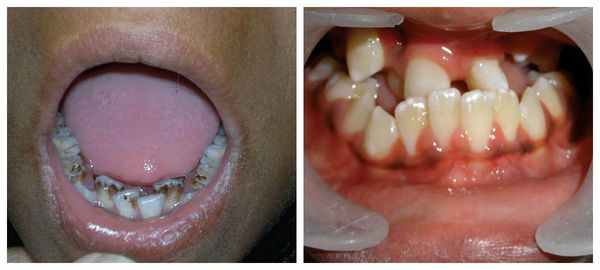
Для синдрома Гольденхара характерна асимметрия лица (одностороннее недоразвитие челюсти) в сочетании с аномалиями ушных раковин, доброкачественными опухолями глаз и поражением спинного мозга (как правило в шейном отделе позвоночника). В большинстве случаев эти нарушения локализуются с правой стороны [19] . Однако до 30 % людей с синдромом Гольденхара имеют двусторонние аномалии лицевых структур.
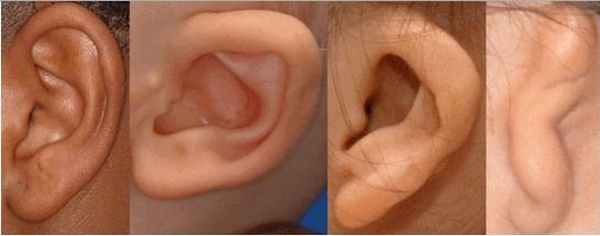
К лицевым аномалиям синдрома относятся:
- расщелины лица и нёба, аномалии лицевых мышц, верхней и нижней челюстей, скуловой и височной костей;
- аномалии ушных раковин: от недоразвития или полного отсутствия ушной раковины до образования околоушных кожных выростов при нормально сформированной ушной раковине;
- аномалии глаз (встречаются реже): одно- или двухстороннее уменьшение глазного яблока (микрофтальмия) вплоть до его отсутствия (анофтальмии), эпибульбарные дермоидные кисты глаз (доброкачественные опухоли) и ретинопатии [7] .
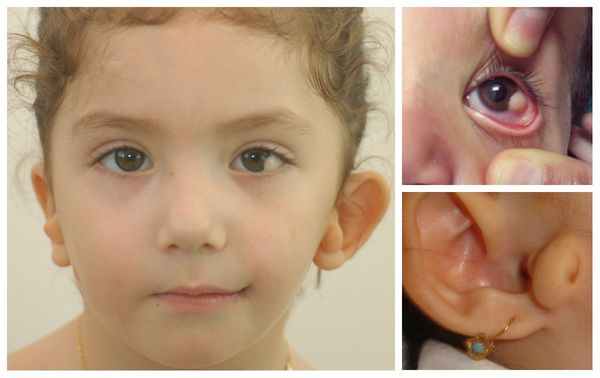
Перечисленные лицевые аномалии могут сопровождаться нарушением слуха, неправильной закладкой и прорезыванием зубов и другими нарушениями, которые могут повлиять на психофизическое развитие ребёнка.
Патогенез синдрома Гольденхара
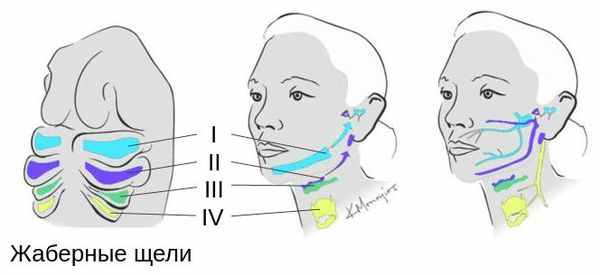
Лицевые структуры начинают формироваться на ранних сроках беременности. Со второй недели развития эмбриона на его головном конце образуется первичная ротовая ямка. К концу третьей недели она постепенно углубляется, достигает передней кишки (эндодермы) и, соединяясь с ней, образует начало пищеварительного тракта. В это же время по бокам головки эмбриона возникают два углубления — 1-я и 2-я жаберные щели, а ещё чуть позже — 3-я и 4-я щели. Между ними формируются жаберные или глоточные дуги, состоящие из нескольких частей: мешка, арки, бороздки и мембраны.
К концу первого месяца развития эмбриона первая жаберная дуга даёт начало пяти отросткам эктодермы: лобному, двум верхне- и нижнечелюстным. Непарный лобный отросток на третьей неделе разделяется на срединный и боковые носовые отростки, из которых к концу 10-11 недели внутриутробного развития формируются лоб, глазницы, нос, средние части верхней челюсти и верхней губы [11] [12] [14] . Нижнечелюстные отростки образуют единую структуру к концу четвёртой недели, а верхнечелюстные — на шестой неделе развития. Также на шестой неделе из парных латеральных закладок нижнечелюстной дуги формируется язык. На седьмой неделе верхнечелюстные отростки объединяются с лобными, в результате чего формируются губы.
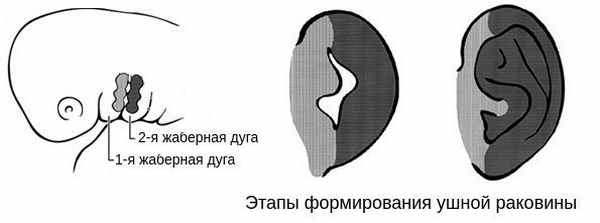
В образовании ушной раковины участвуют первая и вторая жаберные дуги. Из первой дуги образуется передняя треть наружного уха — козелок и ножки завитка. Срастание производных обеих дуг происходит очень рано: к восьмой неделе развития первичная ушная раковина оказывается уже сформированной, однако окончательный рельеф уха оформляется лишь к концу седьмого месяца развития эмбриона [13] .
Таким образом, верхняя и нижняя челюсти, жевательная и мимическая мускулатура, наружное ухо и костные структуры среднего уха формируются из первой и второй жаберных дуг с третьей по восьмую неделю развития эмбриона. Этот период является "критическим" в отношении возникновения пороков развития лица и челюстей. Нарушить нормальное развитие черепно-лицевых структур на данном этапе может сочетанное воздействие внешних факторов, хромосомных и генетических аномалий.
Классификация и стадии развития синдрома Гольденхара
Объём дефектов лицевых структур оценивается по классификации OMENS, в которой выделяют пять групп аномалий:
- O — поражение глазницы;
- M — недоразвитие нижней челюсти;
- E — аномалия уха;
- N — вовлечённость нерва;
- S — дефицит мягких тканей.
Степень тяжести данных дефектов определяется по классификации, созданной учёными Pruzansky S. и Kaban L. B.:
- 1 степень — уменьшение нижней челюсти и суставной ямки височной кости с сохранением анатомии других структур;
- 2а степень — деформация ветви нижней челюсти, суставного отростка и суставной ямки, сопровождается дефицитом жевательной мускулатуры, при этом функция височно-нижнечелюстного сустава сохраняется;
- 2б степень — недоразвитие и деформация мыщелка и суставной ямки, при этом височно-нижнечелюстной сустав не функционирует;
- 3 степень — отсутствие ветви нижней челюсти, мыщелка и суставной ямки с выраженным дефицитом мягких тканей на стороне поражения, височно-нижнечелюстной сустав не сформирован [16] .
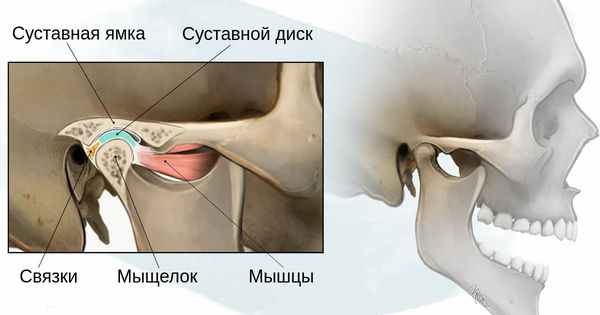
Основываясь на своих многолетних наблюдениях, стоматолог-хирург Г. В. Кручинский выделил три варианта синдрома Гольденхара, каждый из которых подразделил на несколько типов:
- Синдром первой и второй жаберных дуг:
- односторонний ушной тип — лицо симметрично, наблюдаются аномалии ушной раковины;
- односторонний челюстно-лицевой и ушной тип (редко бывает двусторонним) — асимметрия лица из-за недоразвития челюстей и других прилегающих структур лёгкой и средней степени тяжести;
- односторонний черепно-челюстно-лицевой, суставной и ушной тип (редко бывает двусторонним) — выраженная асимметрия лица из-за тяжёлой степени недоразвития челюстей и прилегающий структур, отсутствия суставного отростка, головки и даже суставной ямки, атрофии подкожной клетчатки, слюнных желёз, мимических и жевательных мышц.
- Синдром первой жаберной дуги:
- односторонний нижнечелюстной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сохранением формы ушной раковины, сужением слухового прохода или свищом;
- односторонний или двусторонний нижнечелюстной и ушной тип — умеренная асимметрия лица из-за недоразвития нижней челюсти средней степени тяжести с сужением слухового прохода и аномалией ушной раковины (её опущением, уменьшением и пр.).
- Простой синдром второй жаберной дуги:
- односторонний или двусторонний ушной тип — лицо симметрично, наблюдаются аномалии ушей в сочетании с дефектом мочек и лопоухостью.
По информации европейской базы данных редких заболеваний Orphanet [4] , все клинические проявления синдрома Гольденхара можно разделить на три группы:
- Очень частые (80-99 %):
- асимметрия лица;
- недоразвитие верхней челюсти;
- нарушение слуха;
- околоушные выросты (добавочные ушные раковины);
- уплощение лицевых скул.
- Частые (30-79 %):
- аномалии внутреннего и среднего уха;
- аномалии позвонков;
- аномалии ушных раковин (чаще односторонние), вплоть до недоразвития;
- атрезия (заращение) наружного слухового прохода; ;
- нарушение грудного вскармливания;
- нарушение речи;
- расщелина нёба и/или верхней губы (заячья губа).
- Редкие (5-29 %):
- агенезия мозолистого тела (отсутствие проводящих путей между правым и левым полушариями);
- отсутствие одной или двух почек;
- аномалии гортани;
- аномалии рёбер;
- недоразвитие или отсутствие глаза, больших пальцев кистей;
- атрофия коры головного мозга; ;
- вентрикуломегалия (увеличение мозговых желудочков);
- недоразвитие лёгких;
- аномалия расположения почек;
- недоразвитие части верхнего века (колобома);
- аномалия гортани и трахеи;
- макростомия (незаращение уголка рта);
- мышечная гипотония (слабость);
- нарушение зрения;
- низкий рост;
- пороки сердца (тетрада Фалло, дефект межжелудочковой перегородки); ;
- трахеопищеводный свищ; .
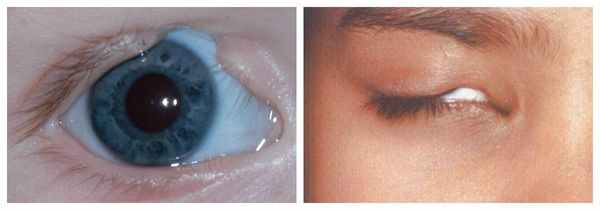
Осложнения синдрома Гольденхара
В раннем возрасте асимметрия нижней челюсти приводит к неправильному развитию и прогрессирующей деформации верхней челюсти и остальных структур лицевого скелета. Со временем ребёнку становится трудно жевать и глотать. При выраженном недоразвитии нижней челюсти у пациента могут возникнуть постоянные проблемы с дыханием, вплоть до апноэ во сне (остановки дыхания).
В целом расщелины лица и/или нёба, недоразвитие верхней и нижней челюсти, лицевых мышц, скуловой и/или височной костей способны вызывать проблемы с зубами, трудности при кормлении, нарушение речи и изменение эстетических параметров лица.
Аномалии ушных раковин в некоторых случаях сопровождаются атрезией (заращением) слухового канала либо полным его отсутствием, что приводит к нарушению слуха. Из-за этого ребёнку сложнее ориентироваться в пространстве, так как он не понимает, откуда исходит тот или иной звук.
Аномалии глаз, такие как дермоидные кисты глаз и колобомы (недоразвитие части верхнего века), способны приводить к нарушению зрительной функции вплоть до частичной или полной потери зрения [1] [4] [7] .
Диагностика синдрома Гольденхара
Как правило, диагностировать синдром Гольденхара не составляет труда. Постановка этого диагноза основана на оценке внешних признаков, клинической симптоматике и результатах дополнительных исследований — КТ, рентгенографии, МСКТ черепа, эхокардиографии и ультразвуковой диагностики. КТ, как правило, проводится для подготовки ребёнка к оперативному лечению.
Генетическое тестирование может быть предложено для подтверждения диагноза, т. е. для исключения генетических состояний, включающих аналогичные лицевые аномалии, связанные с хромосомными и моногенными нарушениями. К таким заболеваниям относятся прогрессирующая гемиатрофия лица, синдром Нагера, челюстно-лицевой дизостоз и др. Однако минимальные диагностические критерии не установлены. Имеются описания единичных случаев диагностики данного синдрома с помощью тестирования до родов.
После рождения всем детям до наступления 6 месяцев во избежание задержки психоречевого развития проводится оценка слуха. Для этого выполняется измерение слуховых вызванных потенциалов: регистрация реакции мозга на звуковые раздражители. Зачастую на поражённой стороне у детей с синдромом Гольденхара выявляется тугоухость.
Лечение синдрома Гольденхара
Для лечения пациентов с синдромом Гольденхара применяются многоэтапные хирургические вмешательства, которые проводятся в разные периоды роста и развития черепно-лицевых структур. Лечение длительное, зависит от локализации и выраженности патологии. Оно направлено на восстановление формы и размеров челюстей, ушной раковины и других структур, а также на восстановление функций слуха, жевания и улучшение эстетических параметров лица [3] [6] [8] .
Лечение проявлений синдрома Гольденхара следует начинать как можно раньше. Своевременная коррекция челюстных нарушений у ортодонта способствует успешному хирургическому лечению в последующем и сохраняет баланс лицевого скелета.
Для устранения выраженных дефектов нижней челюсти применяют индивидуально-смоделированные эндопротезы либо костно-хрящевые аутотрансплантаты из рёбер, обладающие тенденцией к росту. Для устранения дефектов ушной раковины также используются силиконовые эндопротезы либо аутотрансплантаты.
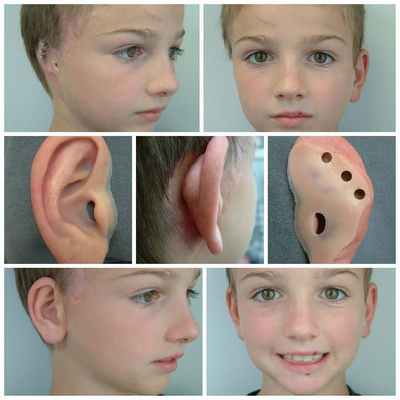
При выявлении нарушений слуха проводится слухопротезирование с помощью слуховых аппаратов либо альтернативными методами. Также необходимы регулярные занятия с сурдопедагогом и логопедом. Всё это позволяет предотвратить отставание ребёнка в речевом и общем развитии.
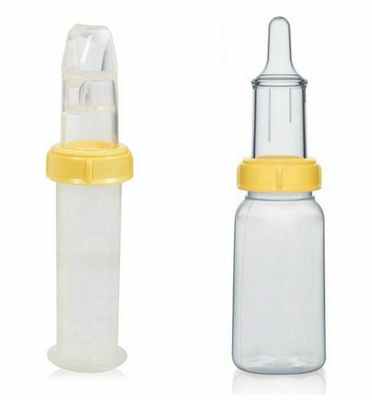
Решение проблем с кормлением заключается в применении специальных бутылочек и назогастрального зонда — трубки, которую вводят в желудок через нос.
Новообразования, локализующиеся на поверхности глазных яблок, могут быть удалены в случае нарушения зрения или при крупных размерах опухоли. У детей до 7 лет операция по удалению кисты проводится под наркозом. Врождённые пороки сердца, проблемы с почками и/или аномалии позвоночника также корректируются хирургическими методами [17] .
Прогноз. Профилактика
Прогноз жизни пациента с синдромом Гольденхара зависит от тяжести клинический проявлений, времени их диагностики и возможной коррекции. Долгосрочный прогноз предсказать сложно [13] .
Как правило, возникновение синдрома Гольденхара носит случайный, ненаследственный характер. При рождении больного ребёнка у здоровых родителей повторный генетический риск для потомства составляет не более 2-3 % [21] .
При отягощённом семейном анамнезе не исключён наследственный характер заболевания. В таком случае риск для потомства по краниофациальной микросомии повышен. Для оценки риска показано медико-генетическое консультирование. Однако отсутствие конкретного мутирующего гена, характерного для развития синдрома Гольденхара, не позволяет точно предсказать выраженность симптомов у потомства.
Первичная (массовая) профилактика синдрома Гольденхара, как и любой врождённой аномалии, заключается в информировании населения и полноценной дородовой подготовке, направленной на предупреждение возникновения заболевания.
Индивидуальная профилактика синдрома предполагает проведение медико-генетического консультирования семьи и пренатальной ультразвуковой диагностики беременной женщины в установленные сроки [12] .
Синдром голденхара. Hellp-синдром
при цьому синдромі відзначається наявність геміфаціальной мікросомії, епібульбарних тератом, преаурікулярних відростків (привушних придатків), поперечних ущелин особи, асиметрії черепа та аномалій будови хребта (дефектів сегментації хребців).
Синоніми. Окулоаурікуло-вертебральна дисплазія, синдром Гольденхара-Горліна (GoldenharGorlin), фаціоаурікуло-вертебральна дисплазія.
поширеність. Частота народження становить 0,2 на 10 000 пологів.
Етіологія. Ймовірно, захворювання виникає спорадично, з рідкісними випадками, які передбачають аутосомно-рецесивний або аутосомно-домінантний тип спадкування.
ризик рецидиву. Можливо, відсутня, якщо не відноситься до аутосомно-домінантним або аутосомно-рецесивним успадкування.
діагностика. Діагноз зазвичай встановлюється при виявленні асиметрії особи (можливо, це буде ще більш поширене з впровадженням тривимірної ехографії) або ущелини верхньої щелепи в поєднанні з односторонньою мікрофтальмія. Інші ознаки включають вади серця або мочевиводя-щей системи, а також виникнення ліпоми мозолистого тіла.
патогенез. Можливо, причиною є крововилив у ембріона в області першої та другої зябрових дуг в період, коли кровопостачання в цій галузі з стремена артерії замінюється на кровопостачання з зовнішньої сонної артерії.
поєднані аномалії. Трахеопіщеводний свищ, епібульбарной тератоми, сірінгогідромелія, затримка нервово-психічного розвитку, тетрада Фалло (Fallot), дефект міжшлуночкової перегородки, синдром аспленію, інверсія шлуночків серця з відходженням аорти та легеневого стовбура від правого шлуночка, атрезія стовбура легеневої артерії з дефектом міжшлуночкової перегородки, повний поддіафрагмальний аномальний дренаж легеневих вен, ектопічні чи злиті нирки, агенезія нирок, міхурово-сечовідний рефлюкс, обструкція мисково-сечовідного з`єднання, подвоєння сечоводів і мультикістоз нирок.
Диференціальний діагноз. Синдром Кауфмана (Kaufman) (окуло-церебро-фаціальний синдром), акрофаціальний дизостоз і асоціація Чардж (Charge).
прогноз. Крім розумової відсталості у таких пацієнтів може виникати багато ускладнень, пов`язаних з верхніми дихальними шляхами і захворюваннями хребта.
акушерська тактика. Якщо діагноз встановлено на ранніх термінах, може бути запропоновано переривання вагітності. Після народження можливе проведення косметичної корекції лицьових вад.
HELLP-синдром
Назва синдрому «HELLP»Є абревіатурою, утвореної з початкових букв станів, що виявляються при важких варіантах перебігу гестозу, який характеризується виникненням гемолізу, високим рівнем печінкових ферментів і низьким рівнем тромбоцитів (hemolys, elevated liver enzymes, and low platelets). Даний стан становить загрозу як для життя матері, так і плода. Ехографічні ознаки захворювання плода включають затримку його внутрішньоутробного розвитку, м`язову гіпотонію, зниження рухової активності, патологічні значення ДОПП-лерометріческіх показників артерій пуповини і мозкових артерій.
HELLP-синдром вражає 2-12% вагітних, у яких розвивається гестоз. При цьому велику схильність до його розвитку мають, мабуть, американки європейського типу, ніж афроамериканки.
Етіологія. HELLP-синдром є станом, специфічним для вагітності, ускладненої тяжким перебігом гестозу.
ризик рецидиву. Хоча ризик рецидиву точно невідомий, кожну жінку з проявами HELLP-синдрому при наступній вагітності слід вести як пацієнтку з підвищеним ризиком.
діагностика. Діагноз у матері встановлюється за допомогою лабораторних досліджень (явища гемолізу, підвищений рівень печінкових ферментів і тромбоцитопенія зі зменшенням кількості тромбоцитів менше 100 000, яка є найбільш постійною ознакою), а також шляхом виявлення клінічних симптомів (набряків, гіпертензії, нудоти, болю в животі внаслідок виникнення субкапсулярного крововиливу або розриву печінки). Ознаки у плода включають затримку внутрішньоутробного розвитку та зменшення обсягу навколоплідних вод, що є реакцією плода на хронічну плацентарну недостатність на тлі гіпертензії у матері. У більш важкому випадку, при мінімізації кисневого резерву, у плода можуть бути виявлені такі ознаки, як м`язова гіпотонія, зниження його рухової активності, дихальних рухів і поява патологічних доплерометричних показників кровотоку. У важких випадках нерідкі антенатальная або неонатальна загибель плода.
поєднані аномалії. Дуже важливим і небезпечним станом, що може поєднуватися з HELLP-синдромом, є синдром діссеменірованного внутрішньосудинного згортання (ДВС).
Диференціальний діагноз. Захворювання, які можуть розвиватися аналогічно, є порушення функції печінки або анемію, а також тромбоцитопенічна пурпура, тромботичну тромбоцитопенічна пурпура, гемолітичний уремічний синдром, захворювання жовчного міхура, вірусний гепатит і гострий жировий гепатоз вагітних.
прогноз. Прогноз варіює залежно від тяжкості стану як матері, так і плода. Материнська захворюваність і смертність пропорційна тяжкості системної хвороби, тоді як ті ж показники у плода в більшості випадків будуть залежати від його гестаційного віку.
акушерська тактика. Пацієнток з HELLP-синдромом слід розглядати як страждають важким гестозом. Рекомендуються госпіталізація їх в спеціалізований центр і пологи в доношенном терміні. До настання часу розродження вагітність слід вести консервативно, з інтенсивним моніторингом стану як матері, так і плода. Індукцію пологів виконують при досягненні зрілості легенів плода.
Ассоциация vater у плода. Синдром голденхара и полидактилия плода
Лучевая форма косорукости также сочетается с врожденным сколиозом. В этом случае при дифференциальной диагностике следует рассматривать три синдрома: ассоциация VATER, синдром Голденхара (Goldenhar) и синдром Klippel-Feil.
Ассоциация VATER возникает в результате нарушения мезенхимального развития в процессе эмбриогенеза в сроки до 35 дней беременности. Характерными признаками этого состояния являются сегментация позвонков (70%), атрезия ануса (80%), трахеопищеводный свищ (70%), атрезия пищевода и пороки развития лучевых костей и почек (65% и 53% соответственно). Из других аномалий отмечаются единственная артерия пуповины (35%) и врожденные пороки сердца, встречающиеся почти у 50% пациентов.
В литературе описаны случаи сочетания лучевых форм косорукости с некоторыми хромосомными аберрациями, включая трисомию 18 и 21, делецию длинного плеча хромосомы 13 и кольцевидную хромосому 4.
Некоторые заболевания характеризуются сочетанием черепно-лицевых аномалий с лучевой косорукостью. Такие синдромы возникают спорадически и имеют много общих признаков, что делает их специфическую пренатальную диагностику затруднительной. Наиболее часто всречающимися черепно-лицевыми аномалиями являются расщелины верхней губы и неба. В исследовании V. Uuspaa (3 225 наблюдений с лицевыми расщелинами) было выявлено, что в 2,8% случаев эти аномалии сочетаются с деформациями верхних конечностей.
Локтевая форма косорукости в большинстве случаев возникает как изолированная, несиндромная аномалия. Но она также может сочетаться с различными синдромами (например, с комплексом Poland).
Синдром Голденхара и полидактилия плода
Полидактилией называется наличие на конечности дополнительных пальцев. Дополнительный палец может иметь вид как мягкотканного образования, так и полностью сформированного пальца, с полноценной функцией сгибания и разгибания. Различают постаксиальную (наиболее часто встречающаяся форма), преаксиальную и центральную формы полидактилии.
При постаксиальной форме дополнительный палец локализуется на локтевой стороне кисти руки или малоберцовой стороне стопы, а при преаксиальной - на лучевой стороне кисти руки и большеберцовой стороне стопы.
В большинстве случаев полидактилия является изолированным пороком с аутосомно-доминантным типом наследования. В некоторых случаях она может входить в состав синдромов, которые обычно наследуются по аутосомно-рецессивному типу. Преаксиальная полидактилия, особенно трехфаланговый большой палец, наиболее часто бывает частью какого-либо синдрома. Центральная полидактилия состоит в наличии дополнительного пальца, который обычно располагается между III и IV пальцами.
Это поражение чаще бывает двухсторонним, наследуется по аутосомному типу и также может сочетаться с другими пороками развития верхних и нижних конечностей.
Читайте также: