Синдром Стерджа-Вебера
Добавил пользователь Morpheus Обновлено: 27.01.2026
Синдром Штурге-Вебера – это энцефалотригеминальный ангиоматоз, относящийся к факоматозам. Факоматозы (греч. Phakos - пятно) – наследственные болезни, объединенные общими звеньями патогенеза, в отличие от более широкой группы нейрокожных синдромов.
Болезнь характеризуется капиллярными или кавернозными гемангиомами, появляющимися на одной стороне лица (но не всегда) в пределах зоны кожной иннервации тройничного нерва, а также преимущественно венозными гемангиомами на мягкой мозговой оболочке над затылочной, теменной и лобной долями с той же стороны. Реже ангиомы располагаются в мозжечке, спинном мозге и во внутренних органах. Описывают отложение солей кальция в сосудах мозга – петрификаты. Выраженность ангиом может значительно варьировать. Ангиомы на коже обычно бывают уже при рождении, имеют вид «пылающего пятна» Самые частые расстройства – паралич, утрата чувствительности и стойкий дефект полей зрения – могут начинаться незаметно, медленно прогрессировать или возникать апоплектиформно. Как правило, синдром сопровождается глаукомой. Диагностируют ее у детей старше 3-5 лет чаще случайно во время профосмотров, когда выясняется значительное понижение остроты и сужение границ поля зрения, а также определяется небольшая застойная инъекция.
При исследовании внутриглазного давления (даже пальпаторно) оно оказывается повышенным, но чаще незначительно (до 30 мм рт. ст.). Наиболее явным и броским признаком ангиоматоза является сине-багровое пятно на коже лица, располагающееся по ходу тройничного нерва. Пламенные пятна могут быть также на конъюнктиве, в сосудистой оболочке и в других отделах глаза . Судороги, встречающиеся в 83% случаев, могут начаться уже в младенчестве. Они являются контралатеральными по отношению к врожденной капиллярной аномалии мягкой и паутинной оболочек головного мозга. Чаще судороги бывают локальными, реже генерализованными. Описаны гемипарезы. Судороги и умственная отсталость при синдроме Штурге-Вебера обусловлены отложениями кальция в местах сосудистых аномалий. Избыточный рост, встречающийся при синдроме Штурге-Вебера, всегда вторичен по отношению к сосудистым аномалиям.
Описаны гемигипертрофия верхней челюсти, ушных раковин. Наследуется синдром аутосомно-доминантно, однако встречаются и аутосомно-рецессивные формы. Большинство пациентов с этим заболеванием живут довольно долго (многие годы), но часто имеют резидуальный интеллектуальный дефект и гемипарез. Слабоумие сочетается с выраженными изменениями в эмоционально-волевой сфере: злопамятность, эгоцентризм, аффективность, мстительность. У больных ухудшаются память, внимание, способность усваивать новые сведения; указанные факторы значительно осложняют процесс обучения и воспитания. Эти нарушения психики отмечаются и в период между приступами. Выраженность расстройств интеллекта, особенно памяти, нарастает по мере учащения судорог. Среди умственно отсталых частота болезни Штурге-Вебера составляет 0,1%. В классическом варианте наблюдается триада симптомов: ангиоматоз кожных покровов, судорожные приступы и глаукома. Течение болезни медленно прогрессирующее; болезнь возникает, как правило, спорадически, семейные случаи являются исключительными.
Заботимся о вашем здоровье
Синдром Штурге-Вебера является спорадическим заболеванием с семейными единичными случаями. Другим названием является энцефалофациальным ангиоматозом. Основным проявлением заболевания является врождённый сосудистый невус на лице (капиллярная ангиодисплазия). Невусы, часто называемые «портвейными», являются обычно односторонними. При этом они могут быть двухсторонними, вовлекая верхнюю половину лица непосредственно вариабельно от лобной области до верхнечелюстной, ограничиваясь зачастую областями иннервации ветвей нерва тройничного.
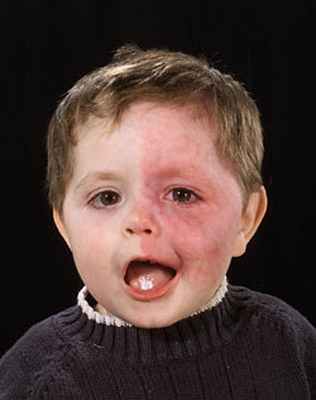
Важным свойством капиллярных ангиодисплазий является потенциальная их ассоциация с внутриглазными внутричерепными сосудистыми аномалиями. Развитие глаукомы в ипсилатеральном глазу наблюдается у пятидесяти процентов пациентов. Глаукома может быть врожденной либо развиваться к двум годам жизни. Стоит отметить, что лептоменингеальный ангиоматоз является практически во всех случаях псилатеральным в отношении лицевого невуса. Протяженность внутричерепного ангиоматоза в данном случае коррелирует с размером пятен лица.
Пораженное полушарие подвержено постепенной атрофии, включая развитие кальцификации субкортикальной пластинки. Клинические проявления синдрома Штурге-Вебера в данном случае могут характеризоваться рядом эпилептических приступов (70-90 процентов пациентов), гемипарезом, умственной отсталостью, гемианопсией, нарушениями чувствительности по гемитипу. Фармакорезистентное течение эпилепсии требовать может тотальной либо частичной гемисферэктомии.
Глаукома либо лицевые "винные пятна", а также рентгенологические или клинические признаки лептоменигеальной ангиомы:
- Признаки лептоменингеальной ангиомы в случае МРТ либо КТ;
- Задержка психического развития;
- Дефицит полей зрения;
- Судороги;
- Гемиатрофия (тела либо головного мозга);
- Гемипарез.
Классическое проявление заболевания характеризуется триадой симптомов: увеличение внутриглазного давления (глаукома), судорожные припадки, сосудистые пятна на кожном покрове лица (ангиомы). Патология часто обладает семейным характером, наследуется непосредственно ауто-сомно-доминантно. При этом встречаются аутосомно-рецессивные формы.
В случае патоморфологического исследования больных, которые страдают энцефалотригеминальным ангиоматозом, выявляются разрастания кожных сосудов, сплетений сосудов глазного яблока, мягкой мозговой оболочки. Ангиомы реже располагаются в затылочной зоне больших полушарий, во внутренних органах, спинном мозге, мозжечке. Описывают отложение кальциевых солей в мозговых сосудах - петрификаты. Выраженность ангиом способна основательно варьировать.
Ангиомы на кожном покрове могут возникать при рождении, обладая видом «пылающего пятна». В восьмидесяти процентах случаев они расположены на лице в зоне иннервации ветвей нервов. Судорожные припадки возникают в период первых лет жизни. Они обычно носят очаговый характер. Данные припадки у большинства больных заканчиваются судорожными генерализованными припадками. Имеют место бессудорожные приступы в форме мгновенных отключений сознания, застываний, вздрагиваний. У некоторых больных наблюдаются приступы головной боли с обильной рвотой (приступы мигрени).
Увеличение внутриглазного давления наблюдается с рождения либо образуется в более поздний период. Вследствие прогрессирования глаукомы снижается зрение. Возможна полная потеря зрения. Болезнь может проявляться в виде слабоумия, сопровождающегося повторяющимися приступами. При этом слабоумие может сочетаться с выраженными изменениями непосредственно в эмоционально-волевой области: мстительность, эгоцентризм, аффективность, злопамятность. У больных происходит ухудшение внимание, памяти, способности усваивать новые сведения. Вследствие данных факторов основательно усложняется процесс воспитания, обучения. Подобные психические нарушения отмечаются также между приступами. Расстройства интеллекта, памяти, увеличиваются по мере увеличения судорог. Частота заболевания Штурге — Вебера среди умственно отсталых составляет 0,1 процентов. Диагноз энцефалотригеминального ангиоматоза является подтверждённым исследованием глазного дна, внутриглазного давления, записью биотоков мозга, рентгенографией черепа.
В период лечения болезни применяются психотропные, а также противосудорожные средства, препараты, которые уменьшают внутриглазное и внутричерепное давление. Используются нейрохирургические методы лечения, а также рентгенотерапия. Дети, имеющие моносимптомную форму энцефалотригеминального ангиоматоза, обладающие исключительно косметическим дефектом в форме пылающих пятен в области лица, проходят обучение в массовой школе. В данных случаях педагогу требуется научить прочих детей не замечать у больного ребёнка косметического дефекта. У больного ребёнка в ином случае могут появляться и развиваться различные психопатологические черты характера.
В случае обучения деток с энцефалотригеминальным ангиоматозом имеет место возникновение больших трудностей, которые обусловлены увеличением психопатологических изменений, а также слабоумия, из-за основного патологического процесса.
Энцефалотригеминальный ангиоматоз (Штурге-Вебера cиндром)
Синдром Штурге (Стерджа) - Вебера — спорадический факоматоз с возникновением ангиом мягкой мозговой оболочки и кожи лица. Данный синдром к широкому спектру возможных фенотипических проявлений включающих черепно- лицевой артериовенозный метамерный синдром.
Эпидемиология
Синдром Штурге (Стерджа) - Вебера редкое заболевание с оценочной встречаемостью 1 случай на 20000-50000 [12].
Клинические проявления
Диагноз как правило очевиден за счет наличия врожденной кожной ангиомы лица (син. винное пятно, лицевой пылающий невус). Данный признак присутствует почти всегда и как правило сочетается с вовлечением лицевой ветви тройничного нерва [4]; если данная зона не поражена, синдром Штурге-Вебера маловероятен [11]. Только в 5% случаев интракраниальные изменения не сочетаются с лицевым невусом [1, 2]. В большинстве случаев (72%) невус односторонний и ипсилатерален внутричерепным изменениям.
Наиболее частым клиническим проявлением в детском возрасте являются судороги, которые встречаются в 71-89% случаев [2] и часто не поддаются лечению [1]. Они как правило появляются на первых годах жизни и сочетаются с задержкой развиния и полушарными симптомами включающими гемиплегию / гемипарезы и гемианопсию.
Приблизительно у трети пациентов встречается поражение склер и хориоидеи, что может осложнится отслоением сетчатки или глаукомой [1].
Патология
В отличие от большинства факоматозов, синдром Штурге-Вебера является спорадическим без определенного наследственного маркёра, который возможно идентифицировать [5,11]. Асоциированный ген мутации был выявлен в нуклеотидном переходе GNAQ хромосомы 9q21 [13].
Лептоменингиальная гемангиома приводит к обкрадыванию в подлежащей коре и белом веществе головного мозга с формированием локальной ишемии. В 80% случаев поражение одностороннее.
Сочетанная патология
- коарктация аорты [10]
- параганглиомы [9]
Диагностика
Рентгенография
Использовалась исторически, способна выявить гириформные кальцинаты, однако в настоящее время не играет существенной роли в диагностике данного заболевания, поскольку изменения выявляются не раньше 2-7 лет [2].
Компьютерная томография
- выявление субкортикальных кальцифинаты в сочетании с атрофией паренхимы головного мозга
- признак трамвайных рельсов за счет кортикальных и субкортикальных кальцинатов [14, 15]
- увеличение сосудистого сплетения с ипсилатеральной стороны
- в тяжелых случаях - изменения по типу синдрома Дик-Давидофф-Массона
Магнитно-резонансная томография
- Т1: cигнал в поражённой области как правило не изменен, но выявляются признаки атрофии и уменьшения объема мозговой ткани
- Т1 с парамагнетиками:
- лептоменингиальное накопление контраста в поражённой области
- увеличение в размерах ипсилатерального сосудистого сплетения
- todo see what did authors had in mind [1]
- ускоренная миелинизация у новорожденных [1]
- в старшей возрастной группе выявляются кальцинаты
- патологический дренах в глубокие вены в виде выпадения потока
Селективная ангиография
В 82% случаев при ангиограции не визуализуруются поврехностные кортикальные вены в сочетании с аномально повышеым дренажем в глубокие вены мозга [2].
Течение и прогноз
Лечение сосредоточено на контроле и предотвращении судорог, в тяжелых случаях применялась хирургическая резекция. Офтальмологическое исследование является основой для идентификации и лечения поражения глаз [4].
История и этимология
Синдром Стерджа-Вебера был описан Стерджем в 1879 году, в 1912 году Вебером и Воландом были описаны внутримозговые кальцификаты. В 1922 году Димитри впервые идентифицировал рентгенографически внутримозговые кальцификаты [2, 3].
- William Allen Sturge: английский врач (1850-1919) [3]
- Frederick Parkes Weber: английский врач (1863-1962) [3]
- Vincente Dimitri: австрийский дерматолог (1885-1955) [3]
Дифференциальный диагноз
Синонимы
- энцефалотригеминальный ангиоматоз
- невоидная аменция
- болезнь Стерджа-Вебера / Штурге-Вебера
- синдром Стерджа-Вебера-Краббе / Штурге-Вебера-Краббе
Частично использован перевод А. Николаева публикации Frank Gaillard et all [17]
Синдромом Стердж-Вебера-Краббе и врожденная глаукома (особенности клиники детского возраста и результаты лечения)
Синдром Стердж-Вебера-Краббе (энцефалоокулофасциальный гемангиоматоз, энцефалотригеминальный ангиоматоз) – врожденное, спорадически возникающее заболевание, характеризующееся ангиоматозом сосудов мозговых оболочек, капилляров лица и глаз. Заболевание редкое – 1 случай на 100 000 населения. Синдром Стердж-Вебера-Краббе относится к группе наследственно-дегенеративных заболеваний – факоматозам, или нейрокожным синдромам [1, 3, 4].
Наиболее характерный внешний признак данного системного заболевания – обширное багровое пятно на лице – пламенный невус, который располагается в зоне иннервации I и II ветвей тройничного нерва. Сосудистый невус наиболее часто – в 70% случаев – бывает односторонним, значительно реже – до 30% – двусторонним. В 40% случаев описаны ангиоматозные пятна на туловище и конечностях и в 5% описанного синдрома невус может отсутствовать. Описаны и другие проявления поражения кожных покровов: гемангиомы, врожденные или появляющиеся в первые месяцы жизни ребенка, гипертрофии и отек мягких тканей и слизистых оболочек носа, губ, десен, глотки, пятна «кофейного» цвета и участки гипопигментации на коже туловища и конечностей [1, 5, 6, 7, 10]. В основе болезни лежит наследственно обусловленная эктомезодермальная дисплазия. В этиологии синдрома в последнее время все большее значение придается наследственности [6, 10].
Врожденная глаукома является серьезным осложнением синдрома и встречается, по данным различных авторов [7, 5, 11] у 1/3 больных. Чаще развитие глаукомы описано на стороне ангиоматозного поражения век [4, 7]. Причины развития глаукомы при синдроме Стердж-Вебера-Краббе по данным различных авторов разнообразны: аномальное формирование угла передней камеры глаза; блокада угла ангиоматозными разрастаниями; повышение давления в эписклеральных сосудах, обусловленное наличием артериовенозных шунтов в эписклере и др. [7-9, 11].
Учитывая редкость синдрома, описание особенностей клиники глаукомы в детском возрасте при синдроме Стердж-Вебера-Краббе, ее лечение и результаты являются чрезвычайно актуальными.
Проанализировать клинику и результаты лечения врожденной глаукомы у детей с синдромом Стердж-Вебера-Краббе.
Материал и методы
Под наблюдением в отделе офтальмопатологии детского возраста ГУ «Института глазных болезней и тканевой терапии им. В.П. Филатова НАМН Украины» находились 13 детей с синдромом Стердж-Вебера-Краббе. Комплексное офтальмологическое обследование включало биомикроскопию, офтальмоскопию, тонометрию, тонографию, дистанционную УЗ-биометрию, УЗ-сканирование переднего и заднего отделов глаза, гониоскопию. Детям до 1 года обследование проводилось в условиях общей анестезии.
Оперативное лечение врожденной глаукомы было проведено на 7 глазах по разработанному оригинальному способу – козырьковой вискосинусотрабекулотомии [3], который заключался в первоначальном введении вискоэластика в переднюю камеру через дополнтельный парацентез с последующим введением его в конце операции под поверхностный склеральный лоскут после его шовной фиксации, а также между склерой и тенноновой капсулой в зоне вмешательства.
Результаты и обсуждение
По гендерному признаку различий нами не установлено (7 девочек и 6 мальчиков), хотя по данным литературы синдром описан чаще у лиц мужского пола [7, 9]. Возраст детей варьировал от 2 месяцев до 17 лет, семеро из них были дети раннего возраста – от 2 до 10 месяцев. Чаще – в 8 случаях – пламенный невус был расположен на одной половине лица, однако не так уж и редко в наших наблюдениях – у 5 детей – захватывал обе половины лица (рис. 1, 2). Во всех случаях гемангиома кожи лица распространялась на область век на стороне поражения. У одного ребенка дополнительно имелось гемангиоматозное поражение кожи обоих рук, а у второго сосудистые изменения были выявлены в стенке гортани на стороне поражения.
При поступлении в стационар основными жалобами родителей детей младшего возраста (2-10 месяцев) был выраженный отек роговицы. При обследовании выявлено увеличение ее диагонального размера до 14 мм по сравнению с размером роговицы парных здоровых глаз, который не превышал 11,5 мм. Имели место светобоязнь, слезотечение, беспокойное поведение ребенка – характерные для врожденного буфтальма развитой стадии. У детей старшего возраста роговичный синдром отсутствовал и причиной обращения было снижение зрения, частые боли в области виска, надбровья и головы.
В результате обследования у 11 из 13 обратившихся детей была диагностирована глаукома, при этом у 8 детей (в основном старшего возраста) была диагностирована односторонняя глаукома; у 3 детей (2, 6 месяцев и 7 лет) – бинокулярная при двухстороннем пламенном невусе лица. В одном случае с двухсторонней локализацией пламенного невуса глаукома была выявлена на одном глазу, где явления невуса были более выражены. У двоих детей (1 – с односторонней и 1 – с двусторонней локализацией сосудистого невуса) глаукома не подтвердилась.
При односторонней локализации пламенного невуса (табл. 1) у 7 детей (7 глаз) монокулярная глаукома диагностирована на стороне поражения, парные глаза – здоровы. Внутриглазное давление (ВГД) на глазах с глаукомой колебалось от 24 до 35 мм рт.ст. (в среднем 29,3±2,4 мм рт.ст.) в сравнении с 19,4±1,1 мм рт.ст. здоровых глаз. По данным УЗ-биометрии переднезадний размер глаза (ПЗО) превышал размер здоровых парных глаз и варьировал от 19,7 до 24,14 мм (в среднем 22,4±1,04 мм), при том что ПЗО здоровых глаз в среднем составил 21,4±1,6 мм. Гониоскопию удалось провести на 5 глазах, где роговица сохраняла прозрачность. Выявлялся гониодисгенез II-III степени с остатками мезодермальной ткани, на всех глазах в УПК выявлялось скопление сосудов, которые значительно прикрывали все зоны УПК, на 2 глазах визуализировались гониосинехии. Во всех случаях выявлялась патологическая экскавация зрительного нерва, отмечались извитые крупные сосуды эписклеры. На 2 глазах при проведении сканирования заднего отдела обнаружено локальное утолщение сосудистой оболочки, что свидетельствовало о возможных ангиоматозных ее изменениях, описанных в литературе [5, 7, 11].
При двусторонней локализации пламенного невуса (табл. 2) у 3 из 4 детей (6 глаз) диагностирована бинокулярная глаукома, у одного ребенка 6,5 лет глаукома была монокулярной (на стороне большей выраженности пламенного невуса), а на парном глазу имела место грубая врожденная патология – фиброз стекловидного тела, дегенерация сетчатки, гетерохромия радужки с резким снижением зрения до 0,03. Отек роговицы отмечен на 4 глазах у детей раннего возраста, у одного из них с двусторонним интенсивным центральным помутнением роговиц (рис. 1). ВГД повышено от 24 до 45 мм рт.ст. (в среднем 30,9±5,8 мм рт.ст.). Переднезадний размер (ПЗО) превышал возрастную норму и варьировал от 21,0 до 25,8 мм в среднем был равен 22,8 ±1,76 мм. Гониоскопию удалось провести на 3 глазах.Выявлялся гониодисгенез II-III степени и скопление сосудов, которые значительно прикрывали все зоны УПК, на 2 глазах корень радужки был утолщен и пронизан сосудами. Отмечалась патологическая экскавация зрительного нерва и извитые крупные сосуды эписклеры, на 2 глазах – локальное утолщение сосудистой оболочки.
Всем детям с глаукомой назначалось консервативное лечение в виде инстилляций капель стимулирующих увеосклеральный отток и снижающих продукцию внутриглазной жидкости – тимолол, бетоптик, альфаган, азопт и их сочетания. В результате консервативного лечения удалось получить компенсацию ВГД на 5 глазах (на 3-х с монокулярной глаукомой и 2-х с бинокулярной). Хотя по данным Кашинцевой Л.Т. и Кушнир В.Л. [5] медикаментозное лечение глаукомы у пациентов старшего возраста с синдромом Стердж-Вебера-Краббе не было удовлетворительным.
Оперативное вмешательство – козырьковая вискосинусотрабекулотомия – произведена на 7 глазах. При этом на 3 глазах в ходе оперативного вмешательства наблюдалось умеренное кровотечение из сосудов конъюнктивы и эписклеры, которое устранялось путем дополнительной коагуляции кровоточащих сосудов. На 2-х глазах при проведении базальной иридэктомии возникло кровотечение из сосудов корня радужки с затеканием небольших порций крови в переднюю камеру, которое было купировано введением дополнительной порции вискоэластика с вытеснением излившейся крови из передней камеры по сформированному фильтрационному каналу.
Наиболее сложное течение операции отмечалось у ребенка 17 лет, родители которого воздерживались от антиглаукоматозной операции в раннем возрасте. Зрение на этом глазу достаточно долго держалось высоким и оперативное вмешательство произведено, когда зрение снизилось до 0,1. Из-за крупного радиарного сосуда радужки, шедшего от корня на 12 часах к зрачковому краю и обильного скопления сосудов эписклеры в виде клубков, продолжающихся из зоны прикрепления верхней прямой мышцы, место оперативного вмешательства пришлось переместить кнаружи. Для снижения риска интраоперационных осложнений предварительно была выполнена задняя склерэктомия. Интенсивная поэтапная коагуляция склеры и обильное использование вискоэластика позволили устранить геморрагические осложнения, и в целом операция прошла гладко, как и послеоперационный период.
Только у одного ребенка после операции наблюдалось измельчение передней камеры, которая восстановилась после массажа зрачка, в остальных случаях послеоперационный период протекал гладко. В результате оперативного вмешательства ВГД было снижено путем формирования дополнительных путей оттока внутриглазной жидкости c образованием в послеоперационном периоде на всех глазах разлитой фильтрационной подушечки. Роговица просветлела с купированием ее отека.
У ребенка с тяжелой врожденной патологией парного глаза после оперативного лечения врожденной глаукомы при синдроме Стердж-Вебера-Краббе ВГД нормализовалось, острота зрения повысилась до 1,0. Наблюдение за этим больным на протяжении 9 лет показало, что ВГД оставалось компенсированным и глаз стал ведущим по зрению.
В отдаленные сроки наблюдения (от 3 месяцев до 9 лет) отмечалась компенсация ВГД у всех прооперированных детей. Через 5 лет после оперативного лечения у одного ребенка с односторонней локализацией невуса ВГД повысилось до 28 мм рт.ст. и было компенсировано дополнительным назначением гипотензивных капель.
Двусторонняя локализация пламенного невуса при синдроме Стердж-Вебера-Краббе по нашим наблюдениям в детском возрасте с развитием двусторонней глаукомы не является редким явлением.
При сравнительном анализе монокулярной и бинокулярной глаукомы у детей с синдромом Стердж-Вебера-Краббе можно отметить, что бинокулярная форма врожденной глаукомы отличается более агрессивным течением и чаще наблюдается у детей грудного возраста.
Консервативное лечение, включающее препараты, стимулирующие увеосклеральный отток, оказалось эффективным в основном у детей более старшего возраста с синдромом Стердж-Вебера-Краббе при слабой выраженности глаукомного процесса.
Козырьковая вискосинусотрабекулотомия, разработанная для лечения врожденного гидрофтальма, оказалась востребованной и высокоэффективной и при глаукоме у детей с синдромом Стердж-Вебера-Краббе. Наряду с описанными преимуществами (поддержка объема сформированного пространства и снижение интенсивности воспалительного ответа травмированных тканей за счет высокого содержания гиалуроновой кислоты в использованном вискоэластике), разработанный метод позволяет эффективно устранять геморрагические осложнения, неизбежные при проникающей хирургии глаукомы у пациентов с синдромом Стердж-Вебера-Краббе.
Синдром Стерджа-Вебера
Синдром Стерджа-Вебера — врожденный ангиоматоз, поражающий кожу, органы зрения и центральную нервную систему. Проявляется множественными врожденными ангиомами лицевой области, стойким эпилептическим синдромом, глаукомой, олигофренией, другими неврологическими и офтальмологическими симптомами. В ходе диагностики выполняется рентгенография черепа, КТ или МРТ церебральных структур, офтальмоскопия, измерение внутриглазного давления, гониоскопия, УЗИ глаза. Лечение включает противоэпилептическую терапию, консервативное и хирургическое лечение глаукомы, симптоматическую терапию. Прогноз во многих случаях неблагоприятный.
Общие сведения
Синдром Стерджа-Вебера — редко встречающееся врожденное ангиоматозное поражение церебральных оболочек, кожи и глаз. Распространенность находится на уровне 1 случай на 100 тыс. населения. Впервые пациента с таким синдромом описал в 1879 г. Стердж, затем в 1922 г. Вебер указал рентгенологические признаки, выявляемые при данном синдроме. В 1934 г. Краббе предположил, что у пациентов, наряду с ангиомами кожи, имеется ангиоматоз церебральных оболочек. В честь этих исследователей заболевание получило название синдром Стерджа-Вебера-Краббе. В связи с тем, что ангиомы лица локализуются в области иннервации кожи 1-ой и 2-ой ветвью тройничного нерва, в неврологии заболевание также известно под названием энцефалотригеминальный ангиоматоз. Наряду с нейрофиброматозом Реклингхаузена, синдромом Луи-Бар, туберозным склерозом, болезнью Гиппеля-Линдау и др., синдром Стерджа-Вебера входит в группу факоматозов — прогрессирующих нейрокожных заболеваний.
Причины синдрома Стерджа-Вебера
Синдром Стерджа-Вебера возникает в результате нарушений эмбрионального развития, приводящих к сбою дифференцировки экто- и мезодермальных листков. Большинство случаев составляет спорадическое появление синдрома, реже отмечается аутосомное (не сцепленное с полом) доминантное или рецессивное наследование с частичной пенетрантностью. Считается, что спорадические случаи обусловлены негативным воздействием на плод в эмбриональном периоде. Вредоносными факторами могут выступать экзо- и эндогенные интоксикации беременной, в том числе никотин, алкоголь, наркотики, различные медикаменты; внутриутробные инфекции; дисметаболические нарушения у будущей матери (например, некомпенсированный сахарный диабет или гипертиреоз).
Морфологическим субстратом заболевания является ангиоматоз — формирование и рост множественных сосудистых опухолей (ангиом), располагающихся на коже лица и в церебральных оболочках. Как правило, ангиоматоз оболочек затрагивает их конвекситальную часть, более часто наблюдается в затылочной и теменных зонах. Обычно ангиоматоз лица и ангиоматоз мозговых оболочек имеют гомолатеральное расположение, т. е. находятся на одной стороне. Однако нередко отмечается двусторонний характер поражения. В церебральных тканях, расположенных под ангиоматозно измененным участком оболочки развиваются дегенеративные процессы, происходит атрофия и избыточный рост глии, формируются кальцификаты.
Симптомы синдрома Стерджа-Вебера
Самым ярким признаком, характеризующим синдром Стерджа-Вебера, выступает ангиоматоз кожи лица. У всех пациентов сосудистое пятно является врожденным. Со временем оно может увеличиваться в размерах. Локализуется, как правило, в скуловой и подглазничной области. Бледнеет при надавливании. В начале имеет розовую окраску, затем приобретает ярко-красный или красно-вишневый оттенок. Внешний вид и распространенность ангиом различны, они могут представлять собой мелкие рассеянные очажки или сливаться в одно большое пятно, т. н. «пламенеющий невус». Ангиоматоз может охватывать полость носа, глотку, полость рта. В 70% случаев синдрома ангиоматоз является односторонним. В 40% изменения на лице сочетаются с ангиомами туловища и конечностей. Возможны и другие дерматологические симптомы: врожденные гемангиомы, локальные отеки мягких тканей, невусы, зоны гипо- и гиперпигментации. По некоторым данным в 5% случаев синдром Стерджа-Вебера протекает без характерного «пламенеющего невуса» на лице.
От 75% до 85% случаев энцефалотригеминального ангиоматоза протекают с судорожным синдромом, дебютирующим на первом году жизни. Характерны эпиприступы джексоновского типа, во время которых судороги охватывают конечности, контрлатеральные (противоположные) расположению ангиоматоза церебральных оболочек. Эпилепсия приводит к задержке психического развития, олигофрении, в отдельных случаях вплоть до идиотии. Может наблюдаться гидроцефалия, гемипарез и гемиатрофия контрлатеральных оболочечной ангиоме конечностей.
Со стороны органа зрения могут наблюдаться ангиомы сосудистой оболочки глаза, гетерохромия радужки, гемианопсия, колобомы. Примерно у трети больных диагностируется глаукома, которая вызывает помутнение роговицы, а в ряде случаев приводит к формированию гидрофтальма. В некоторых случаях синдром Стерджа-Вебера сочетается с дисплазией лицевого черепа, проявляющейся асимметрией лица; в других — с врожденным пороком сердца.
Патогномоничная триада проявлений синдрома («пламенеющий невус», нарушения зрения, неврологическая симптоматика) наблюдается лишь у пятой части больных. В остальных случаях сочетание симптомов значительно варьирует, в связи с чем на сегодняшний день выделено 10 клинических форм синдром Стерджа-Вебера. Нередки абортивные варианты синдрома, при которых клинические симптомы выражены лишь частично и в легкой степени.
Диагностика синдрома Стерджа-Вебера
Диагностировать синдром Стерджа-Вебера возможно по характерному сочетанию симптомов: наличию кожного лицевого ангиоматоза, эпиприступов и других неврологических проявлений, а также офтальмологической патологии (в первую очередь, глаукомы). Диагностический поиск проводится коллегиально неврологом, эпилептологом, офтальмологом и дерматологом.
При проведении рентгенографии черепа обнаруживаются зоны обызвествления коры, имеющие вид двойных контуров, как бы обводящих извилины в области церебрального поражения. КТ головного мозга визуализирует еще более обширные зоны кальцификации, чем показывает рентгенография. МРТ головного мозга выявляет участки дегенерации и атрофии церебрального вещества, истончения коры; позволяет исключить другие заболевания (внутримозговую опухоль, абсцесс головного мозга, церебральную кисту).
Электроэнцефалография дает возможность определить характер биоэлектрической активности мозга и диагностировать эпи-активность. Офтальмологическое обследование состоит из проверки остроты зрения, периметрии, измерения внутриглазного давления, офтальмоскопии и гониоскопии (при сохранности прозрачности роговицы), ультразвуковой биометрии глаза, АВ-сканирования.
Лечение и прогноз синдрома Стерджа-Вебера
В настоящее время синдром Стерджа-Вебера не имеет эффективного лечения. Терапия направлена на купирование основных проявлений. Проводится антиконвульсантная терапия вальпроатами, карбамазепином, леветирацетамом, топираматом.
Эписиндром зачастую оказывается резистентным к проводимому противоэпилептическому лечению, что требует перехода с монотерапии на прием комбинации из двух препаратов. В качестве одного из способов лечения применяется рентген-облучение черепа над пораженной ангиоматозом областью. По показаниям нейрохирургами может быть рассмотрен вопрос о проведении оперативного лечения эпилепсии.
Лечение глаукомы состоит в инстилляции глазных капель, снижающих секрецию внутриглазной жидкости: бримонидина, тимолола, дорзоламида, бринзоламид и пр. Однако подобная консервативная терапия зачастую оказывается малоэффективной. В таких случаях офтальмохирургами проводится хирургическое лечение глаукомы: трабекулотомия или трабекулэктомия.
К сожалению, при выраженной клинике синдром Стерджа-Вебера имеет неблагоприятный прогноз. Некупируемый эпилептический синдром приводит к выраженной олигофрении. Возможна потеря зрения, интракраниальные сосудистые нарушения, влекущие за собой вероятность развития инсульта.
Читайте также: